

Abstract
Cell fusion during yeast mating provides a model for signaling-controlled changes at the cell surface. We identified the AXL1 gene in a screen for genes required for cell fusion in both mating types during mating. AXL1 is a pheromone-inducible gene required for axial bud site selection in haploid yeast and for proteolytic maturation of a-factor. Two other bud site selection genes, RSR1, encoding a small GTPase, and BUD3, were also required for efficient cell fusion. Based on double mutant analysis, AXL1 in a MATα strain acted genetically in the same pathway with FUS2, a fusion-dedicated gene. Electron microscopy of axl1, rsr1, and fus2 prezygotes revealed similar defects in nuclear migration, vesicle accumulation, cell wall degradation, and membrane fusion during cell fusion. The axl1 and rsr1 mutants exhibited defects in pheromone-induced morphogenesis. AXL1 protease function was required in MATα strains for fusion during mating. The ability of the Rsr1p GTPase to cycle was required for efficient cell fusion, as it is for bud site selection. During conjugation, vegetative functions may be redeployed under the control of pheromone signaling for mating purposes. Since Rsr1p has been reported to physically associate with Cdc24p and Bem1p components of the pheromone response pathway, we suggest that the bud site selection genes Rsr1p and Axl1p may act to mediate pheromone control of Fus2p-based fusion events during mating.
Keywords: yeast, mating, morphogenesis, cell fusion, metalloprotease, GTPase
Cell fusion occurs in biological processes such as fertilization, viral entry into cells, myogenesis, and yeast mating (56). While the cell biology of fusion has been well described, very little is known about the molecular components and signaling events involved. Recent studies on myoblast fusion have begun to identify components that are required for this cell fusion event, which include a small GTPase, Drac1, a Drosophila member of the rho family of GTPases that mediate cytoskeleton polarization in many organisms (28). Metalloproteases, as well as proteins of unknown function, are also involved. (15, 21, 33).
Studies of the mating reaction in the yeast Saccharomyces cerevisiae have begun to identify components required for cell fusion in this system. When two haploid cells of opposite mating type encounter one another, they secrete peptide pheromones that bind to G-protein–coupled receptors on the cell surface and activate a MAP kinase signal transduction pathway (for review see reference 36). MAT a cells release a-factor and respond to α-factor, and MATα cells release α-factor and respond to a-factor. The haploid cell response to pheromone is threefold. Cells stop their progression through the cell cycle. Transcription of mating-specific genes is stimulated. Cells polarize toward each other by redirecting actin cytoskeleton assembly and secretion toward a site on the cell surface defined by external pheromone produced by the partner cell. Eventually, cell–cell contact and cell fusion occur to give rise to a diploid cell.
Cell fusion in yeast involves processes that lead to degradation of a small portion of the cell wall at the cell–cell contact region where fusion will occur (17, 36). Often, vesicles are observed poised on opposite sides of the cell–cell contact region in each cell (4, 17). In myoblast fusion, vesicles similarly align on each side of the plasma membrane in a region where localized membrane and cytoplasmic fusions will occur (15). Several yeast genes, including the fusion-specific genes FUS1 (37, 54) and FUS2 (19, 54), have been identified that when mutated lead to an accumulation of cell pairs blocked in cell fusion.
The components that define the site of cell fusion in response to mating pheromones have not been determined. The polarity establishment proteins, Bem1p, Cdc24p, and Cdc42p, which are required for bud formation during vegetative growth (29), may play a role in establishing the fusion site on the cell surface during mating since they are known to play additional roles in the pheromone signaling pathway (12, 13, 58). During budding, these proteins direct cytoskeleton polarization in response to positional information provided by a complex of bud site selection proteins that identify and decode a cortical landmark site on the cell surface. During mating, this internal budding polarity landmark is somehow moved so that cytoskeleton polarization is redirected toward the mating site, which is proposed to be marked in part by the activated pheromone receptors (11, 41).
Previously, we reported a screen to isolate cell fusion– defective yeast mutants (17). We report here that the haploid-specific bud site selection gene, AXL1, is required for morphogenesis in response to mating pheromone and cell fusion during mating. Two other haploid bud site selection genes, RSR1, a small GTPase, and BUD3, are also required. Rsr1p has known physical associations with two components of the pheromone response pathway (31, 43, 51, 58). It is possible that AXL1, RSR1, and BUD3 may function in a pathway for coupling pheromone responses to cell fusion.
Materials and Methods
Reagents, Media, and Yeast Strains
Yeast strains and plasmids are listed in Table I. Yeast rich medium (YPD), synthetic minimal medium (SD), and synthetic drop-out medium are standard media (17). 4,6-diamidino-2-phenylindole (DAPI)1 was purchased from Sigma Chemical Co. (St. Louis, MO). Affinity-purified anti-Cdc42p antibodies were a kind gift from D. Johnson (University of Vermont, Burlington, VT).
Table I.
Yeast Strains and Plasmids Used in This Study
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| LM23-3az (parental) | MAT a bar1 [FUS1–lacZ::URA3] his4 leu2 met1 trp1 ura3–52 | Ref. 35 | ||
| SRM5 | MAT a axl1 (cef3) -1 bar1 [FUS1–lacZ::URA3] his4 leu2 met1 ura3–52 | This study | ||
| LE3-11B | MAT a axl1 (cef3) -1 bar1 leu2 his4 met1 trp1 | This study | ||
| LE1B3 | MAT a axl1 (cef3) -1 bar1 met1 ura3–52 | This study | ||
| LE6B3 | MATα axl1 (cef3) -1 bar1 met1 ura3–52 | This study | ||
| LM104 | Isogenic to LM23-3az but cured of [FUS1–lacZ::URA3] | This study | ||
| LE74x | Isogenic to LM104 but axl1 -Δ1 | This study | ||
| LEab1 | Isogenic to LM104 but rsr1 Δ::URA3 | This study | ||
| LEab3 | Isogenic to LM104 but bud3 Δ::URA3 | This study | ||
| LE281 | Isogenic to LM104 but fus1 -Δ1 | This study | ||
| EYL36 | MAT a his4–34 trp1 -Δ1 ura3–52 fus2-Δ3 | Ref. 19 | ||
| LM110 | Isogenic to LM104 but Δste6::URA3 | Ref. 17 | ||
| IH1701 | MATα ade6 his2 | Ref. 17 | ||
| FC139 | MATα bar1 lys5 met1 ura3–52 | Ref. 17 | ||
| LE49a | Isogenic to FC139 but axl1 -Δ1 | This study | ||
| LE17a | Isogenic to FC139 but axl1 -Δ2 | This study | ||
| LEb1 | Isogenic to FC139 but rsr1 Δ::URA3 | This study | ||
| LEb3 | Isogenic to FC139 but bud3 Δ::URA3 | This study | ||
| LE49b1 | Isogenic to LE49a but axl1 -Δ1 rsr1 Δ::URA3 | This study | ||
| LE49b3 | Isogenic to LE49a but bud3 Δ::URA3 | This study | ||
| LEf14 | Isogenic to FC139 but fus1 Δ::URA3–1 | This study | ||
| LE272 | Isogenic to FC139 but fus2-Δ3 | This study | ||
| LE49f1-1 | Isogenic to LE49a but fus1Δ::URA3-1 | This study | ||
| LE49f2-1 | Isogenic to LE49a but fus2 Δ::URA3-1 | This study | ||
| RC757 | MATα sst2-1 his6 met1 can1 cyh2 rme | Ref. 6 | ||
| Plasmid | Description | |||
| p7-17e | YCp50 genomic library clone | This study | ||
| p7-17eΔ1 | Identical to p7-17e but axl1-Δ1 | This study | ||
| YCp17el | SalI 5.2-kb fragment containing an AXL1 allele in YCp50 | This study | ||
| pLEA7 | YIp5 containing axl1-Δ1 allele | This study | ||
| pPB181 | rsr1Δ::URA3 | Ref. 3 | ||
| pBUD3Δ#2 | bud3Δ::URA3 | Ref. 9 | ||
| pL32-GFP | RPL32 fused to GFP in pRS316 | J. Warner | ||
| pΔBA1 | axl1-Δ (848–1,142) allele in YCp50 | This study | ||
| p129 | AXL1 allele in pRS316 | Ref. 1 | ||
| p126 | axl1-H68A allele in pRS316 | Ref. 1 | ||
| p138 | axl1-E71A allele in pRS316 | Ref. 1 | ||
| YEp (RSR1) | RSR1 LEU2-2-μm based | Ref. 2 | ||
| YEp (rsr1val12) | rsr1val12 LEU2-2-μm based | Ref. 2 | ||
| pLE131 | ste6 (cef1-1) in YEp13 | Ref. 17 |
Cloning of the CEF3 Fusion Gene
CEF3 was cloned by complementation of the mating defect of the cef3 strain, LE1B3. A plasmid (p7-17) was isolated from a low-copy CEN-based S. cerevisiae genomic library (47) that when reintroduced rescued the mating and fusion defects of LE1B3. The complementing plasmid (p7-17) contained an insert of 10.7 kb. Partial sequencing data obtained from p7-17 revealed that the insert DNA corresponded to a region of Ch.XVI that contained several open reading frames (ORFs), including AXL1.
To determine if AXL1 was sufficient to rescue the mating and fusion defects of our cef3 strains (LE1B3 and LE6B3), a 5.3-kb SalI fragment containing AXL1 was subcloned into YCp50 to create YCp17e1. Introduction of YCp17e1 into LE1B3 or LE6B3 was sufficient to rescue the mating and fusion defects (data not shown). AXL1 was deleted from the complementing plasmid (p7-17) by digesting with SnaBI to release a 4.7-kb fragment containing the AXL1 ORF plus 1.0 kb of upstream and 0.08 kb of downstream flanking genomic DNA. The linearized plasmid was relegated to yield plasmid p7-17eΔ1. p7-17eΔ1 failed to restore efficient mating and cell fusion to LE1B3 or LE6B3.
Deletion of Genomic AXL1, RSR1, and BUD3
AXL1 was deleted using a pop-in, pop-out strategy. A 3.0-kb SphI fragment containing the AXL1 genomic region deletion (described above) was isolated from p7-17eΔ1 and cloned into the YIp5 integration vector. The resulting plasmid, pLEA7, was linearized with XhoI to target integration at the AXL1 locus and transformed into FC139 or LM104. Transformants were cured of the URA3 marker on 5-fluoro-orotic acid (5-FOA) plates and screened for deletion of AXL1 by scoring for an altered bud site selection pattern. RSR1 and BUD3 were disrupted using plasmids pPB181 and pBUD3Δ#2, respectively (3, 9).
Construction of an AXL1 Internal Domain Deletion Allele
An axl1 allele that contained an internal deletion encompassing a domain conserved in related proteases was generated (25). YCp17e1 was digested with Bsu36I and AflII, which cut at sites 848 and 1142 within the coding region. The linearized DNA was treated with S1 nuclease to create blunt ends and relegated to generate pBA1, which contained an in frame deletion from codons 848–1142. The structure of the deletion in pBA1 was confirmed by DNA sequencing.
Quantitative Mating Assays
Quantitative matings were performed to determine the efficiency of diploid formation. The frequency of diploid formation was calculated as the number of diploids formed per total viable cells mated (52). 106 cells of the MAT a and MATα strains (exponentially growing, OD600 = 0.5–0.9) were mixed and filtered onto 0.45-μm-pore, 25-mm-diameter nitrocellulose filter discs (type BA85; Schleicher and Schuell, Keene, NH) and incubated on solid YPD medium for 4 h at 30°C. Cells were washed from the filters and diluted with SD. Dilutions were plated onto the appropriate selective medium to determine the number of diploids formed or onto YPD medium to titer the total number of viable input cells.
Quantitative Cell Fusion Assay
Filter matings were performed as described above, except that 2 × 106 cells of MAT a and MATα cells were used. Cells were washed from the filters, sonicated to break up clumps, and viewed under the microscope using phase contrast optics. The frequency of prezygote accumulation was determined as the number of cell pairs blocked in fusion per total number of cell pairs counted (prezygotes + zygotes). Cell pairs blocked in cell fusion were identified by the appearance of an intact septum/fusion bridge between the cells. Fused cell pairs (zygotes) either completely lack the septum/fusion bridge or have a partial septum/fusion bridge with an identifiable region of cytoplasmic continuity. At least 200 cell pairs were observed for each mating. The prezygote accumulation assay is very reproducible, with ∼10% variance due to environmental conditions during the mating reaction (4, 17). To visualize the mutant cell partner in mating pairs, various strains were transformed with a plasmid encoding a fusion of the L32 promoter with the gene encoding green fluorescent protein (pL32-GFP, a kind gift from J. Warner, Albert Einstein College of Medicine). Mating cell pairs were set up so that only one partner carried or lacked a GFP plasmid.
Pheromone-induced Morphological Changes
Cultures (3 ml) were grown to log phase in YPD medium at 30°C. α-Factor was added to a final concentration of 40 nM or 400 nM. After incubation for 6 h at 30°C, 16% EM-grade paraformaldehyde (Electron Microscopy Sciences, Warrington, PA) was added directly to the cultures to a final concentration of 4%. Cultures were incubated for 1 h at 25°C and then washed with PBS. Cell morphology was examined by light microscopy.
Analysis of Budding Pattern
Cells were grown to log phase in rich medium at 30°C. Budding patterns were scored as described by Chant and Herskowitz (8). Single cells were plated onto YPD medium and incubated at 30°C for approximately two cell divisions. Microcolonies at the four-cell stage were scored for an axial, bipolar, or random budding pattern.
Pheromone Response Assays
To determine sensitivity to cell cycle arrest by pheromone, ∼1 × 105 of exponentially growing cells were spread onto YPD agar, and 0.1 μg of α-factor (Sigma Chemical Co.) was spotted in duplicate onto the plates that were incubated for ∼24 h at 30°C. Zones of growth inhibition were quantitated. To measure gene induction, a plasmid (pSB234) carrying the pheromone-inducible FUS1::lacZ reporter gene fusion (54), was transformed into strains. Different concentrations of α-factor were added to cultures of exponentially growing strains, which were then incubated for 1 h at 30°C and processed to determine β-galactosidase levels (48).
FUS1 and FUS2 Disruptions
FUS1 and FUS2 disruption constructs were created by PCR. For the FUS1 disruption construct, 1.04 kb of FUS1 coding region was replaced with the URA3 gene, leaving 0.25 and 0.24 kb of flanking FUS1 region DNA. For the FUS2 disruption construct, 1.97 kb of FUS2 coding region was replaced with the URA3 gene, leaving 0.23 and 0.24 kb of flanking FUS2 genomic DNA. The fus1Δ::URA3 construct was transformed into FC139 to create FCf14 and into LE49a to create LE49f1-1. The fus2Δ::URA3 construct was transformed into LE49a to create LE49f2-1. Presence of the disruption alleles in FCf14, LE49f1-1, and LE49f2-1 was confirmed by PCR.
Immunofluorescence Microscopy
Logarithmically growing cells were treated with 5 × 10−7 M α-factor until >30% of the cells had a single mating projection (∼2.75 h at 30°C) and then fixed with 3.7% formaldehyde and prepared for staining with affinity-purified anti-Cdc42p antibodies as described by Ziman (60).
Electron Microscopy
Strains were mated on nitrocellulose filters as described above for 3.5–4 h. Cells were fixed for electron microscopy as described in reference 57. Cells were washed from the filters and resuspended in 1 ml of 2× fixative (4% glutaraldehyde, 0.2 M cacodylate buffer, pH 6.8) for 5 min at 25°C. After incubation, the cells were resuspended in 1 ml of 1× fixative (2% glutaraldehyde, 0.1 M cacodylate buffer, pH 6.8) and incubated on ice for 30 min. The fixative was removed, and the cells were washed three times in 0.1 M cacodylate buffer, pH 6.8, and then postfixed in 0.5% osmium tetroxide and 0.8% potassium ferrocyanide. Samples were then embedded and sectioned for electron microscopy.
Results
The cef3 Mutation, Complemented by AXL1, Causes Cell Fusion Defects in Two Cell Types
Previously, we reported a screen to identify mutants (Cef−) specifically defective for the cell fusion step of mating (17). Strains able to initiate mating but unable to complete cell fusion were isolated using microscopic observation of mating cell pairs as the final step of the screen. A strain, SRM5, bearing a mutation provisionally designated cef3, was chosen for further characterization.
Segregation analysis and scoring for a fusion defect revealed that 50% of the SRM5 progeny (14 fusion defective/28 total cells scored) failed to fuse when mated to a wild-type tester strain (FC139). Thus, the cef3 mutation segregated 2:2 as a single gene trait. Among the mutant progeny tested, we found that an equal number of MAT a and MATα strains displayed a fusion defect (8/14 MAT a cef3; 6/14 MATα cef3). These results suggested that the mutation could act in both haploid cell types.
A genomic clone (p7-17) complementing the cef3 mating defect was isolated from a yeast centromeric library (47). Partial sequence data was obtained from the complementing clone, and the chromosomal region to which the yeast insert DNA corresponded was identified. The insert contained several ORFs, including AXL1, which is required with its homologue STE23 for a-factor processing and mating in MAT a cells (1). AXL1 is also required for bud site selection in both MAT a and MATα cells (1, 25). Subcloning and deletion analysis of the complementing insert revealed that a 5.3-kb SalI fragment containing AXL1 coding sequence (YCp17e1) was sufficient to rescue the mating and cell fusion defects of the cef3 strains (LE1B3 and LE6B3).
Deletion of the AXL1 Gene Affects Mating Ability of a MATα Strain
Many of the effects on mating caused by an axl1 mutation when present in a MAT a strain can be explained by the defect in a-factor processing (1, 4). It has been shown that reduced pheromone production produces a cell fusion defect (4). Segregation analysis suggested that AXL1 might be required for efficient cell fusion during mating in MATα strains as well as in MAT a strains. To avoid concerns that the MATα phenotypes we observed were due to tightly linked mutations in the original cef3 strain, and to create an isogenic series of strains, we deleted AXL1 in the MATα strain FC139 by gene replacement to create strain LE49a. Because analysis of mating effects caused by an axl1 mutation is complex in a MAT a strain, most of the studies reported here concern effects in MATα strains. The Axl1p protease has been shown to play no role in processing or secretion of the α-factor pheromone in MATα strains (4, 10). Deletion of AXL1 also had no effect on production of the α-factor pheromone in our MATα strain background (data not shown). The AXL1 deletion did cause a shift from an axial to a bipolar bud site selection pattern, as has been reported in other strain backgrounds (Table II) (1, 25).
Table II.
Role of Bud Site Selection Genes in Mating
| Relevant genotype | Prezygote accumulation* | Mating frequency‡ | Bud site selection§ (axial) | |||||
|---|---|---|---|---|---|---|---|---|
| MATα strains | % | % | % | |||||
| FC139 | Parental | 5 | 21.0 ± 5.0 | 92 | ||||
| LE49a | axl1Δ | 55 | 6.0 ± 2.0 | 37 | ||||
| LEb1 | rsr1Δ | 60 | 4.0 ± 2.0 | 14 | ||||
| LEb3 | bud3Δ | 44 | 8.0 ± 0.6 | 29 | ||||
| LE49b1 | axl1Δ rsr1Δ | 64 | 5.0 ± 0.6 | 13 | ||||
| LE49b3 | axl1Δ bud3Δ | 64 | 5.0 ± 0.1 | 16 | ||||
| LEf14 | fus1Δ | 41 | 14.0 ± 0.9 | — | ||||
| MAT a strains‖ | ||||||||
| LE74x | axl1Δ | 51 | 0.3 ± 0.1 | 40 | ||||
| LEab1 | rsr1Δ | 83 | 3.2 ± 0.4 | 17 | ||||
| LEab3 | bud3Δ | 64 | 2.6 ± 0.9 | — |
Prezygote accumulation was determined by microscopic observation as described in Materials and Methods. More than 500 mating pairs were scored for each strain pair. MAT a partner is LM23-3az.
Mating frequency was measured as the percent of prototrophic diploids formed/total viable cells after limited mating to the MAT a partner, LM23-3az, and is the average ± standard deviation of duplicate filter mating determinations.
The frequency of axial budding (not bipolar or random) was determined. Budding patterns were assayed for >100 microcolonies.
Prezygote accumulation and mating frequenices determined as above. The MATα partner for both assays was FC139.
To confirm that AXL1 was required for cell fusion in MATα cells during mating, we scored fusion by observation of mating forms microscopically. Deletion of AXL1 in the MATα strain FC139 conferred a defect in cell fusion (Fig. 1, c–f). The results of quantitation of the MATα axl1Δ fusion defect are shown in Table II. Since wild-type yeast complete cell fusion rapidly, mating intermediates rarely accumulate in normal matings. Fusion-defective strains (4, 5, 17, 18, 20, 37, 54, 55) either fail to complete mating or take longer to complete fusion steps and so accumulate mating intermediates (prezygotes) in which cells have initiated mating, but in which nuclear and cytoplasmic fusion have not occurred. The axl1 strain, LE49a, accumulated 11-fold more prezygotes than the parental strain FC139 (Table II). As a comparison, a fus1 deletion mutant (LEf14) accumulated eightfold more prezygotes than the parental strain, FC139. The mating frequency of the axl1 mutant strain was reduced to 29% of the wild-type (FC139) level. Thus, AXL1 and FUS1 had comparable effects on cell fusion under these conditions.
Figure 1.

Light microscopy of yeast mating pairs. Photomicrographs of typical mating cell pairs using Nomarski optics. (a and b) LM23-3az × FC139 [Wt × Wt]. (c–f) LM23-3az × LE49a [Wt × axl1] (two isolates). (g and h) LM23-3az × LEb1 [Wt × rsr1Δ]. (i and j) LM23-3az × LEb3 [Wt × bud3Δ].
Genetic Interaction between AXL1 and FUS2 Suggest They Act in the Same Pathway
AXL1 is a haploid-specific, pheromone-induced gene (1, 25). We wanted to determine if Axl1p acted in a pathway with either of two other known haploid-specific, pheromone-induced genes, FUS1 and FUS2, that are required only for cell fusion (37, 54). Fus1p and Fus2p are thought to act in parallel pathways leading to cell fusion (19, 37, 54). Double mutant axl1 fus1 and axl1 fus2 strains were constructed by gene replacement in the axl1 strain, LE49a (see Materials and Methods for details). The axl1 fus1 strain, LE49f1-1, exhibited defects in both cell fusion and mating assays that were greater than the single axl1 or fus1 mutant strains (Table III). LE49f1-1 accumulated 87% prezygotes, whereas prezygote accumulations of 39 and 53% were determined for the fus1 (LEf14) and axl1 (LE49a) single mutant strains, respectively. LE49f1-1 displayed a mating efficiency that was about fourfold lower than the axl1 strain (LE49a) and about sevenfold lower than the fus1 strain (LEf14) (Table III).
Table III.
Double Mutant Analysis of AXL1, FUS1, and FUS2
| MATα strain | Relevant genotype | Prezygote accumulation* | Mating frequency‡ | |||
|---|---|---|---|---|---|---|
| % | % | |||||
| FC139 | AXL1 | 7 | 21.2 ± 6.3 | |||
| LEf14 | fus1Δ | 39 | 15.9 ± 0.0 | |||
| LE272 | fus2Δ | 38 | 14.8 ± 1.1 | |||
| LE49a | axl1Δ | 53 | 8.0 ± 0.1 | |||
| LE49f1-1 | axl1Δfus1Δ | 87 | 2.4 ± 0.7 | |||
| LE49f2-1 | axl1Δfus2Δ | 46 | 8.4 ± 0.1 |
Prezygote accumulation was determined as described in Materials and Methods. More than 200 mating pairs were scored for each strain pair. MAT a partner was LM23-3az.
Mating frequency was measured as the percent of prototrophic diploids formed/total viable cells after limited mating to MAT a partner, LM24-3az. Averages ± SD of duplicate filter mating determinations.
In contrast, fus2 and axl1 did not exhibit an additive defect (Table III). The axl1 fus2 strain LE49f2-1 accumulated 46% prezygotes and exhibited an 8.4% mating efficiency. The fus2 strain accumulated 38% prezygotes and mated with an efficiency of 14.8%. The axl1 strain LE49a accumulated 53% prezygotes and mated with an efficiency of 8.0%. These results suggested that axl1 might act in the same pathway as FUS2.
Role of Other Haploid Bud Site Selection Genes in Cell Fusion
AXL1 was identified as a gene required for axial bud site selection in haploid yeast strains (1, 25). Other genes are known that also are required for this morphogenic program (7, 29). We wanted to determine if other bud site selection components were required for efficient cell fusion during mating. We tested the role of RSR1 and BUD3 in cell fusion. RSR1 encodes a ras-like GTPase that may in turn regulate the GTPase Cdc42p (3, 7, 59). BUD3 encodes a product that interacts directly or indirectly with septins and appears to act upstream of Axl1p and Rsr1p (9, 29). We constructed rsr1 and bud3 mutant strains by gene disruption in our MATα parental strain, FC139. As shown in Fig. 1, g–j, and in Table II, rsr1 (LEb1) and bud3 (LEb3) mutant strains were defective in cell fusion during mating. The MATα strain (LEb1) accumulated 60% prezygotes, and the MATα bud3 strain (LEb3) accumulated 44% prezygotes. Mating frequencies were reduced in the rsr1 and bud3 strains to an extent similar to that of the axl1 strain (Table II). The MATα rsr1 strain (LEb1) and the MATα strain (LEb3) mated to the wild-type tester strain (LM23-3az) with frequencies of 4 and 8%, respectively.
The rsr1 and bud3 strains exhibited the expected defects in bud site selection (Table II). The parental strain FC139 exhibited an axial budding pattern. This pattern was shifted in the rsr1 (LEb1) and the bud3 (LEb3) mutant strains. The rsr1 mutant exhibited a random pattern of bud site selection, whereas the bud3 mutant exhibited a bipolar pattern as described by others (1, 8, 9, 25). MAT a rsr1 (LEab1) and MAT a bud3 (LEab3) mutants exhibited mating defects similar to the MATα mutant strains, indicating that RSR1 and BUD3 play a role in mating in both cell types (Table II).
Since several components required for proper haploid bud site selection appeared to be required for fusion steps in mating, we wanted to determine if RSR1 and BUD3 might be acting in the same pathway as AXL1. We constructed an axl1 rsr1 strain (LE49b1) and an axl1 bud3 strain (LE49b3) and mated them to the tester strain LM23-3az. The strain LE49b1 mated at 24% of the wild type level and accumulated 13-fold more prezygotes, similar to what was determined for the single axl1 and rsr1 mutants (Table II). The strain LE49b3 also mated at 24% of the wild-type level and accumulated 13-fold more prezygotes. Thus, it appeared that RSR1, BUD3, and AXL1 might act in the same pathway to promote cell fusion during mating as they do to promote bud site selection during vegetative growth, though the results were less clear cut than the interaction of AXL1 and FUS2.
Electron Microscopy of [Wt × Wt] Prezygotes
To characterize the mating steps at which fusion-specific genes might act on an ultrastructural level, we performed electron microscopy to identify potential mating intermediates (prezygotes) (42). Wild-type (Wt) intermediates were characterized first. Prezygote intermediates of wild-type strains have been observed but are rare and remain poorly characterized (17). We sought to characterize wild-type fusion forms to better define the steps that might be defective in our mutant strains (Fig. 2, A–F). Several features were clear in these wild-type prezygotes. The cell walls of the mating cell pair were knitted to form a smooth, nearly seamless structure resembling the walls observed in zygotes, suggesting that outer cell wall fusion may precede septum degradation. Nuclei were oriented generally close to the fusion zone, as previously described (17, 19, 30) (Fig. 2 D). In a few of the prezygotes, we observed what appeared to be streaming of electron-dense material across the cell–cell contact region at a localized point between the two cells (Fig. 2 D). The [Wt × Wt] prezygotes at some stages had an overall morphological appearance similar to our previously described [ste6 (cef1) × Wt] prezygotes, indicating that mutations that block cell fusion do not always disrupt prezygotic structure (17).
Figure 2.


Electron microscopy of yeast mating pairs. Electron micrographs of mating cell pairs after 3.5 h are shown. (A–F) [LM23-3az (Wt) × FC139 (Wt)]; (G and H) [LM23-3az (Wt) × LE272 (fus2)]; (I–L) [LM23-3az (Wt) × LE17a (axl1)]; (M and N) [LM23-3az (Wt) × LEb1 (rsr1)]; (O and P) LEab1 [(rsr1) × (rsr1)]. cw, cell wall; v, vesicles; n, nucleus; ed, electron-dense material. Cell wall margins not visible in some panels. Bars, 1 μm.
Electron Microscopy of [axl1Δ × Wt] Prezygotes
The [axl1Δ × Wt] prezygotes had a very different appearance from the [Wt × Wt] prezygotes (Fig. 2, I–L). In many of the cell pairs observed, we often found that one of the two nuclei was not positioned near the fusion zone and appeared misaligned with respect to the partner nucleus. We observed a similar nuclear positioning defect in [fus2Δ × Wt] prezygotes (Fig. 2 G). Nuclear misalignment has been previously reported for fus2 mutant strains (19). Of 18 cell pairs in which nuclear position of both cells could be observed in [axl1Δ × Wt] crosses, 11% had both nuclei properly aligned, as opposed to 100% of nuclei aligned for [Wt × Wt] crosses. In these electron micrographs, we could not unambiguously determine if it was the mutant or wild-type partner that contained a misaligned nucleus. We determined the frequency of nuclear alignment by staining [mutant × mutant] prezygotes with DAPI to visualize the nuclei and counting the number of cell pairs with aligned nuclei vs. misaligned nuclei (Table IV). [fus1Δ × fus1Δ] prezygotes exhibited aligned nuclei with a frequency of 97%. In contrast, [fus2Δ × fus2Δ] and [axl1Δ × axl1Δ] prezygotes exhibited aligned nuclei with a frequency of 16 and 8%, respectively.
Table IV.
Quantitation of Nuclear Positioning by DAPI Staining
| Mating pair* | Relevant genotype | Nuclei aligned‡ | ||
|---|---|---|---|---|
| % | ||||
| LE281/FCf14 | fus1Δ/fus1Δ | 97 | ||
| EYL36/FC272 | fus2Δ/fus2Δ | 16 | ||
| LE74x/LE17a | axl1Δ/axl1Δ | 8 | ||
| LEab1/LEb1 | rsr1Δ/rsr1Δ | 22 |
MAT a and MATα cells were mated on filters for 3.5 h at 30°C (refer to Materials and Methods). Mating pairs were: LE281 (MAT a fus1-Δ1)/FCf14 (MATα fus1Δ::URA3), EYL36 (MAT a fus2-Δ3)/LE272 (MATα fus2-Δ1), LE74x (MAT a axl1-Δ1)/LE17a (MATα axl1-Δ2), and LEab1 (MAT a rsr1Δ::URA3)/LEb1 (MATα rsr1Δ::URA3).
Mating mixtures were stained with DAPI to visualize nuclei (refer to Materials and Methods). More than 30 mating pairs were scored for each strain combination.
In all of the cell pairs observed by electron microscopy, the cell that harbored the misaligned nucleus exhibited a distorted, swollen appearance. The swollen cell often appeared to bulge over each side of the cell–cell contact region, giving the appearance of engulfing its partner (Fig. 2 I). In addition, we observed pockets of electron-dense material within the cell wall space on either side of the cell– cell contact region in many of the [axl1Δ × Wt] prezygotes (Fig. 2 L). Based on light microscopy experiments described below (Fig. 3), we believe that the swollen partner cells are the axl1 mutants. In [Wt × Wt] matings, a portion of the cell wall separating the mating pair underwent breakdown without apparent alterations of other portions of the cell wall, which might indicate that axl1 mutants have a defect in control of cell wall changes in mating.
Figure 3.

Expression of GFP in mating cell pairs. (A, light image, and B, fluorescent image) LM104 (Wt) × FC139 (Wt)/pL32-GFP. (C, light image, and D, fluorescent image) LM104 (Wt)/ pL32-GFP × FC139 (Wt). (E, light image, and F, fluorescent image) LM104 (Wt) × LE17a (axl1)/ pL32-GFP. (G, light image, and H, fluorescent image) LM104 (Wt)/pL32-GFP × LE17a (axl1). (I, light image, and J, fluorescent image) LM104 (Wt)/pL32-GFP × LEb1 (rsr1). (K, light image, and L, fluorescent image) LM110/p131 ste6 (cef1) × FC139/pL32-GFP. Matings were done for 3.5 h at 30°C. Light image, phase-contrast; fluorescent image, FITC filter.
Electron Microscopy of Prezygotes of [rsr1Δ × Wt]
The overall structure of [rsr1Δ × Wt] prezygotes was similar to the [axl1Δ × Wt] prezygotes (Fig. 2, M and N). In cell pairs where nuclei were visible, we often observed that one of the nuclei failed to align with the fusion septum. However, the nuclear misalignment observed in [rsr1Δ × Wt] prezygotes was not as severe as what we observed in [axl1Δ × Wt] prezygotes. DAPI staining of [rsr1Δ × rsr1Δ] prezygotes revealed that nuclei were aligned with a frequency of 22%, which was greater than the frequencies observed for [fus2Δ × fus2Δ] or [axl1Δ × axl1Δ] (Table IV) but less than wild-type. As with the [axl1Δ × Wt] prezygotes, we often observed that one of the cells in the [rsr1Δ × Wt] prezygotes bulged out over each edge of the cell–cell contact region. The distorted cell in [rsr1Δ × Wt] pairs often contained the misaligned nucleus. Overall, the rsr1 defects resembled those of the axl1 cells but appeared less severe.
Accumulation of Vesicles in Prezygotes
We noticed that vesicles often accumulated in prezygotes formed by the axl1, rsr1, and fus2 mutant strains (Fig. 2). In [Wt × Wt] prezygotes, apparent vesicles ∼100 nm in size were occasionally observed poised on either side of the cell–cell contact region, suggesting that they might play a role in fusion processes (4, 17). In many of the [axl1Δ × Wt] prezygotes, we observed that vesicles had accumulated to a larger extent in the presumptive (swollen) axl1 partner than in the presumptive wild-type partner or in [Wt × Wt] prezygotes (Fig. 2, A–F). It was interesting that in the partner with fewer vesicles in [axl1Δ × Wt] prezygotes, we observed what appeared to be vesicles fusing with the plasma membrane. However, in the partner cell with excess vesicles, we never observed a presumptive vesicle fusion event (Fig. 2). A similar vesicle over-accumulation pattern was observed in [fus2Δ × Wt] and in [rsr1Δ × Wt] prezygotes.
We determined the relative vesicle accumulation in the prezygotes (Table V). To quantitate vesicle accumulation in [mutant × Wt] crosses, we calculated a ratio of the (highest vesicle number)/(lowest vesicle number) for cell pairs. We found that vesicles were present at a ratio of 1.4 in [Wt × Wt] prezygotes. Thus, in [Wt × Wt] prezygotes both cells tended to have a similar number of vesicles in EM slices in which vesicles were visible. A similar ratio of 1.7 was measured for the [Wt × ste6(cef1)] prezygotes (17), which resembled [Wt × Wt] prezygotes in structure. In contrast, the [axl1Δ × Wt] prezygotes accumulated vesicles at a ratio of 4.0, and the [rsr1Δ × Wt] prezygotes accumulated vesicles at a ratio of 4.6. The [fus2Δ × Wt] prezygotes accumulated vesicles at a ratio of 2.7. Thus, some but not all of the fusion mutants led to an asymmetry of vesicle accumulation. The cells studied did not represent a large enough pool for us to be conclusive on this point, though the difference did appear striking. Also, an assumption of this type of calculation is that the lowest number of vesicles represents wild-type and the highest number of vesicles represents the mutant. We did not attempt to determine which was wild-type and which was mutant in this analysis. Vesicle quantitation also is made difficult by the fact that not every EM slice contained vesicles.
Table V.
Ratio of Vesicle Accumulation Observed by Electron Microscopy
| Cross | Relevant genotype | Vesicle accumulation differential* | ||
|---|---|---|---|---|
| LM23-3az × FC139 | Wt× Wt | 1.4 | ||
| LM23-3az × LM110/p131 | Wt × cef1-1 | 1.7 | ||
| LM23-3az × FC272 | Wt × fus2-Δ1 | 2.7 | ||
| LM23-3az × LE17a | Wt × axl1-Δ2 | 4.0 | ||
| LM23-3az × LEb1 | Wt × rsr1Δ::URA3 | 4.6 |
Ratio of partner with largest number of vesicles/partner, with smaller number of vesicles. The number of mating cell pairs scored for each cross were: Wt × Wt, 5; Wt × cef1-1, 1; Wt × fus2-Δ1, 4; Wt × axl1-Δ2, 5; Wt × rsr1Δ::URA3, 5.
Light Microscopy of [axl1Δ × Wt] and [rsr1Δ × Wt] Prezygotes
Prezygotes of both [axl1Δ × Wt] and [rsr1Δ × Wt] were characterized by light microscopy. To distinguish mutant and wild-type cells in prezygotes, we rendered one of the partner strains fluorescent. A construct that expressed GFP fused to a constitutively expressed L32 ribosomal gene promoter was introduced into one partner. Prezygotes were identified as cell pairs containing only one fluorescent cell (16). Partial fusion, if it occurred, or full fusion could also be identified because the GFP marker is localized to the cytoplasm and thus provided a means to monitor a breach in the septum leading to cytoplasmic mixing and staining of the whole zygote.
The prezygotic structures formed in LE49a/LM104 [axl1Δ × Wt] mating mixtures appeared to be distorted as expected from the ultrastructural analysis (Fig. 3, E–H). The mutant cell tagged with GFP was swollen in size relative to the wild-type partner. Often we observed in [axl1Δ × Wt] prezygotes a bulging of the mutant partner cell on either side of the cell contact region, as observed by electron microscopy. It sometimes appeared that the mutant was “engulfing” the wild-type partner cell. In addition, the cell–cell contact region in [axl1Δ × Wt] prezygotes appeared to be wider than the contact region observed in [ste6(cef1) × Wt] prezygotes (Fig. 3, K and L), which were similar to [Wt × Wt] prezygotes. We observed that the vacuole was swollen in mating axl1 mutant cells but not in wild-type or ste6(cef1) cells. When the L32–GFP fusion protein was expressed in the wild-type partner cells rather that the mutant, the axl1 partner was still larger. Thus, the altered morphology of the GFP-tagged axl1Δ cells was not an artifact due to the presence of GFP. The rsr1 partner (LE1b) in [rsr1Δ × Wt] crosses appeared, like axl1, to be the larger of the two cells in mating pairs as determined by the GFP assay (Fig. 3, I and J).
An axl1Δ Strain Exhibits a Partial Defect in Morphogenic Response to α-Factor
Since axl1 cells in mating cell pairs appeared to have an aberrant morphological appearance, we wished to determine if the mutant cells exhibited morphological defects when treated with pheromone in the absence of a partner. We found that differing conditions of α-factor treatment led to the formation of cells with widely differing morphologies. Under some of the conditions used, we observed that axl1 cells formed mating projections that were similar to projections formed by the wild-type cells. However, treatment of either axl1 (LE1B3) or wild-type (LM23-3az) cells with 40 nM α-factor for 6 h led to a distinct difference in the appearance of mating projections. The axl1 cells formed a heterogeneous mixture of mating projections that appeared overall to be broader than normal (“dumpy”) with a mixture of other morphological forms, including undeformed cells and a small number of cells displaying normal mating projections (Fig. 4 A). Under these treatment conditions, the wild-type cells formed a more uniform population of mating projections, giving cells the classic pear-shaped morphology. At a higher pheromone concentration (400 nM, 4 h), the axl1 morphogenic defect persisted but was more subtle. The axl1 cells formed mating projections that were shorter and had rounder tips than wild-type shmoos (Fig. 4 B).
Figure 4.
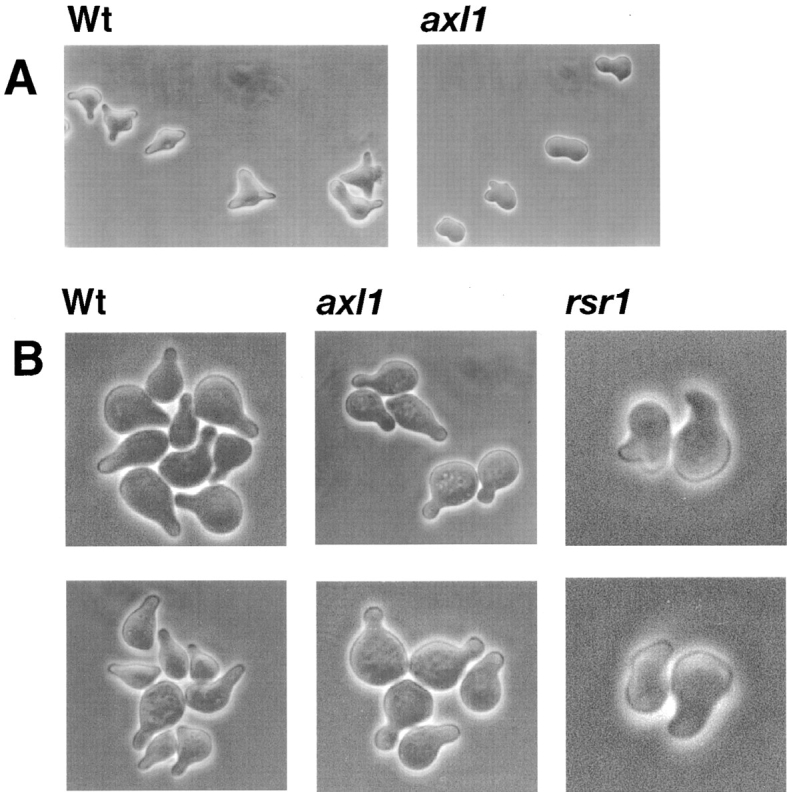
Pheromone-induced cell morphogenesis. MAT a strains were treated with α-factor and viewed by phase-contrast microscopy. (A) Cells treated with 40 nM α-factor for 6 h. (B) Cell treated with 400 nM α-factor for 4 h. The strains are: (A, left) LM23-3az (Wt); (right) LE1B3 [axl(cef3-1)]. (B, left) LM23-3az (Wt), (middle) LE1B3 [axl(cef3-1)]; (right) LEab1 (rsr1).
Using a quantitative growth inhibition assay, we found that an axl1 strain had an α-factor sensitivity that was equivalent to that of the isogenic wild-type strain (data not shown). The altered morphology observed for the axl1 strain did not appear to be due to perturbation of the underlying actin cytoskeleton, as visualized by staining actin with rhodamine-conjugated phalloidin (data not shown). Also, the axl1 strain appeared to orient properly in a micropipette assay designed to measure chemotropic responses towards a source of pheromone (data not shown) (50).
An rsr1 strain also appeared to form mating projections that were altered in shape compared with mating projections formed by wild-type cells and appeared more defective than mating projections formed by axl cells (Fig. 4 B). The axl1 and rsr1 morphogenic defects appeared to be similar to, but much less extreme than that reported for spa2, pea2, and tpm1 mutants (27, 32, 55).
Axl1p Protease Activity Is Required for Cell Fusion and Morphogenesis
AXL1 encodes a metalloprotease with homology to insulin-degrading enzymes (1, 25). Axl1p protease activity is required for a-factor pheromone processing but appears dispensable or required at a lower level for axial bud site selection functions (1). The role of the Axl1p protease in cell fusion during mating was determined. We created a novel protease-defective allele of Axl1p by an in-frame deletion spanning codon 848 to codon 1142. The deletion encompassed a region that lies outside of the presumed metal-binding domain, but which has high homology to mammalian and Drosophila insulinases (1, 25). We first tested the deletion allele for in vivo protease function by assaying for the ability to promote a-factor pheromone processing in MAT a axl1 cells. The construct expressing the protease deletion allele, pBA1, failed to rescue the a-factor production defect of the MAT a axl1 strain (LE1B3; data not shown). When axl1Δ (848–1142) was expressed in either the MAT a strain, LE1B3 (not shown), or in the MATα axl1Δ strain, LE17a, axial bud site selection was restored to a level similar to that provided by the wild-type AXL1 gene (Table VI). By these assays, we could not distinguish a complete protease defect from a severe but partial defect. As a comparison, we tested previously characterized point substitution mutations axl1-H68A and axl1-E71A of AXL1 that inactivate a metal-binding site required for protease activity (1). The axl1 metal-binding site mutants complemented the bud site selection defect conferred by axl1 mutations in our strain background as reported by others. These substituted proteins have been shown to accumulate at steady-state levels that were indistinguishable from wild-type levels in the cell (1).
Table VI.
Role of AXL1 Protease Functions in Cell Fusion
| Plasmid | Allele | a-Factor production* | Prezygote accumulation‡ | Bud site selection§ (axial) | ||||
|---|---|---|---|---|---|---|---|---|
| % | % | |||||||
| YCp17e1 | pAXL1 | + | 8 | 96 | ||||
| YCp50 | Vector | − | 60 | 42 | ||||
| p126 | pAxl1-H68A | − | 76 | 91 | ||||
| p138 | pAxl1-E71A | − | 60 | 99 | ||||
| pΔBA1 | pAxl1-Δ (848–1,142) | − | 61 | 92 |
a-Factor production determined by halo assay (1, 17, 38) with plasmids in a MATα axl1 strain (LE1B).
Plasmids were assayed in the MATα axl1-Δ1 strain, LE49a, mated to LM23-3az. More than 200 mating pairs were counted per cross.
The frequency of axial (neither bipolar nor random) budding was determined. Budding patterns were assayed for >100 microcolonies.
We tested the AXL1 mutant alleles with apparent protease deficiencies for the ability to promote cell fusion in the MATα axl1Δ strain, LE17a. The axl1Δ (848–1142), axl1(H68A), or axl1(E71A) protease-defective alleles failed to rescue the cell fusion defect. The cell fusion defect of LE17a was rescued by wild-type AXL1 expressed from a CEN-based plasmid (Table VI).
The ability of protease-defective alleles of axl1 to complement the morphogenic defects of an axl1 strain was determined. Mutant axl1 strains expressing the protease-defective alleles (LE1B3/pBA1 [not shown] or p126 [axl1-H68A]; Fig. 5 B) were exposed to 40 nM α-factor for 6 h. The protease-defective alleles of AXL1 failed to promote normal morphogenesis in response to α-factor. Thus, Axl1p protease activity appears to be required for efficient morphogenesis and cell fusion during mating.
Figure 5.

Pheromone-induced morphologies of a strain defective for Axl1p protease activity. LE1B3 [axl1Δ] with various plasmids derived from p129 treated with α-factor (40 nM) for 6 h. (A) LE1B3/p129 (pAXL1); (B) LE1B3/p126 (paxl1-H68A); (C) LE1B3/pRS316 (pVector).
Constitutive Activation of RSR1 Inhibits Cell Fusion in Wild-Type Cells
RSR1 encodes a small GTPase that plays a role in bud site selection. It has been proposed that Rsr1p must cycle between an active and inactive state in order to carry out its role in bud site selection (2). Constitutively activated alleles of RSR1 fail to function in bud site selection. We wanted to determine if Rsr1p functioned in a similar manner during cell fusion, or whether constitutive Rsr1p activation was sufficient for its function in mating. We expressed a constitutively activated allele of RSR1 (rsr1val12) in the MAT a rsr1 strain, LEab1 (2). The rsr1val12 strain accumulated 51% prezygotes when mated to a parental tester strain (FC139). LEab1 expressing a wild-type copy of RSR1 accumulated only 7% prezygotes under these conditions (Table VII). This observation indicated that the rsr1val12 allele of RSR1 was unable to provide a Rsr1p function needed in cell fusion. In addition, the activated allele acted in a dominant-negative fashion in a wild-type strain (LM104). The RSR1/rsr1val12 strain LM104(pPB264) accumulated 41% prezygotes when crossed to the wild-type partner strain, FC139. The rsr1val12-activated allele also caused randomization of the bud site selection pattern in the wild-type transformed strain and failed to suppress the budding defect of the rsr1Δ strain, LEab1 (Table VII), as previously reported (2).
Table VII.
Ability of the Activated Allele of RSR1, rsr1val12, to Function in Cell Fusion and Bud Site Selection
| MAT a strain/plasmid | RSR1 allele | Prezygote accumulation* | Bud site selection‡ | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Axial | Bipolar | Random | ||||||||
| % | % | % | % | |||||||
| LEab1/pPB161 | wt | 7.2 | 84.0 | 7.0 | 9.0 | |||||
| LEab1/pPB264 | rsr1val12 | 51.4 | 15.6 | 12.8 | 71.6 | |||||
| LM104/pPB161 | wt, wt | 3.8 | 88.3 | 9.8 | 1.9 | |||||
| LM104/pPB264 | wt, rsr1val12 | 41.0 | 24.4 | 46.5 | 29.1 | |||||
Prezygote accumulation was determined by microscopic observation as described in Materials and Methods. More than 200 mating pairs were scored for each strain pair. The MATα partner was FC139.
Budding patterns were observed for >100 microcolonies.
Subcellular Localization of Cdc42p in axl1 and rsr1 Cells
We wanted to determine if either Axl1p or Rsr1p was involved in localizing Cdc42p during mating. During budding, Cdc42p acts with the bud site selection components, which include Rsr1p, to orient the actin cytoskeleton toward a specific site on the cell surface that will give rise to a bud. During mating, Cdc42p is required to polarize the cytoskeleton in response to pheromone, resulting in pear-shaped cells. Axl1p or Rsr1p might be involved in recruiting Cdc42p to the site on the cell surface where cell fusion would occur. We looked at the subcellular localization of Cdc42p in MAT a axl1 (LE74x) and MAT a rsr1 (LEab1) mutant strains by indirect immunofluorescence microscopy using anti-Cdc42p antibodies (60). Cdc42p was localized to the mating projection tip of α-factor–treated axl1 and rsr1 mutant cells (Fig. 6). Localization of Cdc42p in the axl1 and rsr1 mutant strains was equivalent to that observed in the parent cells (LM23-3az) and also to what had been described previously by Ziman et al. (60). Occasionally, we observed that the Cdc42p cap staining patterns were slightly offset in the axl1 and rsr1 mutant strains, but the significance, if any, is unclear. Axl1p and Rsr1p appear to be not required for the proper polarization of Cdc42p to the mating projection tip formed in response to pheromone.
Figure 6.

Localization of Cdc42p. MAT a cells were treated with α-factor for 2.75 h. Cdc42p was visualized by indirect immunofluorescence. Isogenic strains were (top) LM23-3az [Wt], (middle) LE74x [axl1], (bottom) LEab1 [rsr1].
Discussion
We have found that Axl1p, Rsr1p, and Bud3p components of haploid bud site selection processes in yeast are required for efficient cell morphogenesis and fusion during mating. Axl1p protease activity was required for mediating morphological responses during mating and cell fusion. Rsr1p might mediate a connection to pheromone signaling since it physically interacts with Cdc24p and Bem1p, which in turn interact with the β subunit of G protein coupled to pheromone receptors and the Ste20p (PAK) kinase controlling polarity responses, respectively (31, 43, 51, 58). The group of products that function together for bud site selection in vegetative cells could serve, under pheromone receptor control, to mediate a cell fusion program during mating. AXL1 genetically acted in the same pathway as FUS2, and an axl1 defect produced many of the same ultrastructural defects during mating observed in fus2 strains. We propose that AXL1 and other bud site selection genes may act in conjunction with components of the pheromone response pathway and FUS2 to establish a fusion zone at the cell surface.
Our work may point to control of cell fusion by pheromone signaling at the site of cell–cell contact. Previous speculation on control of cell fusion by the pheromone response has focused solely on transcriptional control of expression of genes like FUS1 and FUS2 (19, 54). Additional control of fusion at the cell surface is likely. The haploid bud site selection machinery appears to be associated with the cell fusion site (as described below). Bud site selection proteins in turn associate with components of the pheromone pathway and may be capable of transmitting signals that control fusion processes. It makes sense that spatial definition of the point of cell–cell contact transmitted by cell surface pheromone receptors might act to control the activity of the fusion machinery. We envision that in addition to transcriptional processes, localized pheromone-activated signals control regulated secretion, cell wall changes, and cytoskeletal changes needed for cell fusion. Future studies will be required to determine whether the connection between Fus2p function and localized activation of receptors is as simple as this model suggests. The reason that strains underexpressing pheromones exhibit a mating fusion defect could be that the local activation of fusion processes that we propose requires a higher level of receptor occupancy than is required for initiation of early mating responses (39).
Analysis of axl1 fus1 and axl1 fus2 double mutant strains indicated that AXL1 functioned in the same cell fusion pathway as FUS2 but not FUS1. FUS2 encodes a novel protein that, along with FUS1, is dedicated to cell fusion during mating. FUS1 and FUS2 appear to act in distinct cell fusion pathways with some overlapping aspects (19, 37, 54). Both FUS1 and FUS2 mutant strains are defective in completing cell wall degradation before membrane fusion (37, 54). The identification of additional components acting in one of these established pathways will greatly help the elucidation of the mechanism of fusion since many genes involved in fusion appear to act independently of one another (26). Localization studies of the Fus2p indicated that it may be associated with the cytoskeleton, the contact region between mating cells, and with vesicles localized to the mating projection tip (19). In addition, Fus2p is required for proper nuclear alignment during mating (19). Our electron microscopy analysis revealed that axl1 conferred an overall phenotype that was similar to a fus2 mutant strain. We found that like a fus2 mutant strain, our axl1 mutant strain displayed a defect in nuclear alignment. In addition, both axl1 and fus2 mutant strains often accumulated vesicles at the presumptive fusion site, suggesting a defect in vesicle fusion. In wild-type prezygotes, vesicles were localized to, but not grossly accumulated at, a spatially restricted site within the cell–cell contact region. It may be that AXL1, RSR1, and FUS2 are involved in a process that includes marking the fusion site for microtubule capture and for vesicle fusion.
The vesicle accumulation and cell wall defects observed in the axl1 and fus2 mutants are reminiscent of defects observed for some of the sec mutants. A mutation in SEC3 led to vesicle accumulation in vegetative cells (23). In addition, sec3 mutant cells often accumulated electron dense material in the mother–bud septum region, suggesting that faulty secretion may lead to cell wall defects (23). We observed in several of the [axl1Δ × Wt] prezygotes an accumulation of electron-dense material within the cell wall at the periphery of the cell–cell contact region. In addition, in the light micrographs of [axl1Δ × Wt] prezygotes, the presumed axl1 cell appeared to be “engulfing” the partner cell, which might reflect a change in cell wall plasticity. We believe that these cell wall phenotypes could be the result of misdirected vesicles carrying cell wall degradative enzymes. Our wild-type prezygotes arrested with a smoothly fused, contiguous cell wall and appeared to restrict degradation of the cell wall to one point within the cell–cell contact zone. Whether Axl1p is required for properly localized secretion during mating remains to be tested. It is possible that, like Sec3p, Axl1p plays a role in defining a site on the cell surface where vesicle fusion is directed. Axl1p plays several, apparently independent roles in the cell and hence may not be localized solely to the cell surface (1, 7, 10). The NH2-terminal half of FUS2, like SEC2, contains homology to myosin (19, 40), and Fus2p appears partly to be localized on vesicles. Perhaps an alternative secretory pathway is activated during mating.
Axl1p is a member of a large family of metalloendoproteases that include insulinase and N-arginine dibasic convertase (1, 25). We found that an Axl1p protease function is required for morphogenesis and cell fusion during mating. It is interesting to note that N-arginine dibasic convertase has been proposed to be involved in spermatid morphogenesis (14). Proteases of this family usually process small peptides or clip the NH2 terminus of larger proteins (10, 24, 44). It is possible that Axl1p is required to proteolitically process the NH2 terminus of a larger protein involved in morphogenesis or cell fusion. One possible fusion candidate is Fus2p. Fus2p contains a potential site for Axl1p cleavage between residues 12 and 13, a region with homology to myosin (Elia, L., and L. Marsh, unpublished observation). Two-hybrid analysis suggests that Axl1p could be part of a larger complex that includes Fus2p (Boone, C., personal communication). Members of the PAK family of kinases that include Ste20p and Cla4p, which are involved in pheromone signaling and cytoskeletal control, can be activated by proteolysis and could be candidate morphogenesis targets (45).
AXL1 expression is limited to haploid, mating-competent cells and is induced in response to mating pheromones in both MAT a and MATα cells (1, 25) (Boone, C., personal communication). These features are commonly found in genes involved in mating (19, 20, 54). Axl1p was previously shown to be required for efficient mating of MAT a cells via its role in production of the a-factor mating pheromone (4). The role of AXL1 in MATα cells appears to be limited to pheromone response rather than pheromone production. We, like others, found that Axl1p activity is not required for α-factor processing or secretion (4). Though the axl1 strains exhibited morphogenic defects in mating, the wild-type and axl1 strains appeared equally responsive to the growth arrest activity of α-factor, suggesting that Axl1p does not affect mating via effects on Fus3p (MAP kinase) activation. The appearance of axl1 strains in the absence of pheromone was normal. In addition to AXL1, we found a requirement for two other bud site selection genes, RSR1and BUD3, in cell fusion during mating. These genes are believed to act together with AXL1 during vegetative growth, and they may play mechanistically related roles during cell fusion.
Prior evidence suggested that the bud site selection gene products remain functional during mating. First, cells exposed to pheromone and then allowed to resume vegetative growth have been observed to site the first bud at the mating projection tip, suggesting that bud site selection components can be diverted away from the previous bud site and redirected toward the site of concentrated pheromone signaling (53). Second, it has been shown that mutations in RSR1, BUD2, BUD3, and BUD4 cause cells to alter the site of projection formation from an axial to a random placement in response to a uniform concentration of pheromone, suggesting that they have the ability to influence the direction of pheromone-induced cytoskeletal polarization (34). Third, mutations in the diploid bud site selection genes, BNI1 (22), SPA2 (27), PEA2 (13, 55), and BUD6/AIP3 (22), also have been shown to influence projection formation and mating. Finally, special alleles of CDC24 have been isolated that cause defects in mating projection formation and mating cell orientation but not in budding during vegetative growth (13, 41). When present in mating cells, these CDC24 alleles appear to disconnect pheromone responses from cell polarization, causing the cells to form mating projections next to the bud site (default locus) rather than toward the source of pheromone. Thus, in normal mating cells, bud site selection products may be recruited to the site of cell fusion. Down regulation of two bud site selection components, Bud4p and Axl2p, during mating has been previously reported (46, 49). These observations may indicate that bud site selection is replaced by a distinct but mechanistically related process during mating. It is intriguing to speculate that pheromone-induced proteins such as Fus2p or Fus1p might replace the function of the down-regulated products and reconfigure the bud site selection machinery for a novel fusion purpose.
Our genetic findings suggest that RSR1 and BUD3 are required for cell fusion in both haploid cell types and that they may function in the same pathway as AXL1. We showed that Rsr1p must cycle between active and inactive states to carry out its fusion function. A similar requirement is observed in vegetative bud site selection. Like Axl1p, Rsr1p appears to be required for full morphological changes in response to pheromone and during mating. The fact that we have found a requirement for the haploid bud site selection genes in mating is surprising given that in the past these genes have been reported dispensable for mating (7, 16, 29). It is possible that genetic redundancy and strain differences can account for this discrepancy. FUS3 was found in a background that was missing the redundant MAP kinase, KSS1 (18). In a strain with wild-type KSS1, loss of FUS3 does not greatly reduce mating (18). In a parallel fashion, it is possible that STE23, a homologue of AXL1, could supply AXL1 mating functions (1). We have not tested whether our strains are functional for STE23 or not. Some of the bipolar bud site selection genes may serve redundant mating functions since RSR1, a dual haploid/diploid bud site gene, was required for cell fusion. Mutations in the diploid bipolar bud genes BNI1, SPA2, PEA2, and RVS161 have been previously shown to lead to defects in cell fusion during mating (16, 55). It will be of interest to see if they also act in the FUS2 pathway. It is likely that overlapping polarity determinants function in mating and budding (27).
Our studies support the idea that yeast mating and budding share common components. Proteins that play roles in controlling cell wall changes, secretion, and cytoskeleton polarization during budding have been shown to play roles as well in cell fusion during mating (16, 55). However, unlike budding, mating is under control of extracellular signals. It will be interesting to see how complex processes in the cell are brought under the control of signaling pathways.
Acknowledgments
We are grateful to Alan Bender, John Chant, Charlie Boone, Jonathan Warner, Elaine Elion, Joshua Trueheart, Ira Herskowitz, and Fred Chang for strains and plasmids; Doug Johnson for anti-Cdc42 antibodies; Neil Adames and Charlie Boone for communication of results before publication; Jeffrey Segall for help with cell orientation assays; Frank Macaluso and Leslie Gunther for expert technical assistance with electron microscopy; and Anjani Shah for helpful comments on the manuscript.
This work was supported by the Cancer Core Support Grant P30CA13330 from the National Cancer Institute.
Abbreviations used in this paper
- DAPI
4,6-diamidino-2-phenylindole
- GFP
green fluorescent protein
- ORF
open reading frame
- Wt
wild-type
Footnotes
Address all correspondence to Lorraine Marsh, Department of Cell Biology, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461. Tel.: (718) 430-2841. Fax: (718) 430-8574. E-mail: marsh@aecom.yu.edu
References
- 1.Adames N, Blundell K, Ashby MN, Boone C. Role of yeast insulin-degrading enzyme homologs in propheromone processing and bud site selection. Science. 1995;270:464–467. doi: 10.1126/science.270.5235.464. [DOI] [PubMed] [Google Scholar]
- 2.Bender A. Genetic evidence for the roles of the bud-site-selection genes BUD5 and BUD2in control of the Rsr1p (Bud1p) GTPase in yeast. Proc Natl Acad Sci USA. 1993;90:9926–9929. doi: 10.1073/pnas.90.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bender A, Pringle JR. Multicopy suppression of the CDC24 budding defect in yeast by CDC42 and three newly identified genes including the res-related gene RSR1. . Proc Natl Acad Sci USA. 1989;86:9976–9980. doi: 10.1073/pnas.86.24.9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brizzio V, Gammie AE, Nijbroek G, Michaelis S, Rose MD. Cell fusion during yeast mating requires high levels of a-factor mating pheromone. J Cell Biol. 1996;135:1727–1739. doi: 10.1083/jcb.135.6.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brizzio V, Gammie AE, Rose MD. Rvs161p interacts with Fus2p to promote cell fusion in Saccharomyces cerevisiae. . J Cell Biol. 1998;141:567–584. doi: 10.1083/jcb.141.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan RK, Otte CA. Isolation and genetic analysis of Saccharomyces cerevisiae mutants supersensitive to G1 arrest by afactor and α factor pheromones. Mol Cell Biol. 1982;2:11–20. doi: 10.1128/mcb.2.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chant J. Generation of cell polarity in yeast. Curr Opin Cell Biol. 1996;8:557–565. doi: 10.1016/s0955-0674(96)80035-4. [DOI] [PubMed] [Google Scholar]
- 8.Chant J, Herskowitz I. Genetic control of bud site selection in yeast by a set of gene products that constitute a morphogenetic pathway. Cell. 1991;65:1203–1212. doi: 10.1016/0092-8674(91)90015-q. [DOI] [PubMed] [Google Scholar]
- 9.Chant J, Mischke M, Mitchell E, Herskowitz I, Pringle JR. Role of Bud3 in producing the axial budding pattern of yeast. J Cell Biol. 1995;129:767–778. doi: 10.1083/jcb.129.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen P, Sapperstein SK, Choi JD, Michaelis S. Biogenesis of the Saccharomyces cerevisiae mating pheromone a-factor. J Cell Biol. 1997;136:251–269. doi: 10.1083/jcb.136.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chenevert J. Cell polarization directed by extracellular cues in yeast. Mol Biol Cell. 1994;5:1169–1175. doi: 10.1091/mbc.5.11.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chenevert J, Corrado K, Bender A, Pringle J, Herskowitz I. A yeast gene (BEM1)necessary for cell polarization whose product contains two SH3 domains. Nature. 1992;356:77–79. doi: 10.1038/356077a0. [DOI] [PubMed] [Google Scholar]
- 13.Chenevert J, Valtz N, Herskowitz I. Identification of genes required for normal pheromone-induced cell polarization in Saccharomyces cerevisiae. . Genetics. 1994;136:1287–1297. doi: 10.1093/genetics/136.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chesneau V, Prat A, Segretain D, Hospital V, Dupaix A, Foulon T, Jegou B, Cohen P. NRD convertase: a putative processing endoprotease associated with the axoneme and the manchette in late spermatids. J Cell Sci. 1996;109:2737–2745. doi: 10.1242/jcs.109.11.2737. [DOI] [PubMed] [Google Scholar]
- 15.Doberstein SK, Fetter RD, Mehta AY, Goodman CS. Genetic analysis of myoblast fusion: blown fuse is required for progression beyond the prefusion complex. J Cell Biol. 1997;136:1249–1261. doi: 10.1083/jcb.136.6.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorer R, Boone C, Kimbrough T, Kim J, Hartwell LH. Genetic analysis of default mating behavior in Saccharomyces cerevisiae. . Genetics. 1997;146:39–55. doi: 10.1093/genetics/146.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elia L, Marsh L. Role of the ABC transporter Ste6 in cell fusion during yeast conjugation. J Cell Biol. 1996;135:741–751. doi: 10.1083/jcb.135.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elion E, Grisafi PL, Fink GR. FUS3 encodes a cdc2 + /CDC28-related kinase required for the transition from mitosis into conjugation. Cell. 1990;60:649–664. doi: 10.1016/0092-8674(90)90668-5. [DOI] [PubMed] [Google Scholar]
- 19.Elion EA, Trueheart J, Fink GR. Fus2 localizes near the site of cell fusion and is required for both cell fusion and nuclear alignment during zygote formation. J Cell Biol. 1995;130:1283–1296. doi: 10.1083/jcb.130.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erdman S, Lin L, Malczynski M, Snyder M. Pheromone-regulated genes required for yeast mating differentiation. J Cell Biol. 1998;140:461–483. doi: 10.1083/jcb.140.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erickson MRS, Galletta BJ, Abmayr SM. Drosophila myoblast cityencodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J Cell Biol. 1997;138:589–603. doi: 10.1083/jcb.138.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evangelista M, Blundell K, Lontine MS, Chow CJ, Adames N, Pringle JR, Peter M, Boone C. Bni1p, a yeast formin linking cdc42p and the actin cytoskeleton during polarized morphogenesis. Science. 1997;276:118–122. doi: 10.1126/science.276.5309.118. [DOI] [PubMed] [Google Scholar]
- 23.Finger FP, Hughes TE, Novick P. Sec3p is a spatial landmark for polarized secretion in budding yeast. Cell. 1998;92:559–571. doi: 10.1016/s0092-8674(00)80948-4. [DOI] [PubMed] [Google Scholar]
- 24.Fricke B, Betz R, Friebe S. A periplasmic insulin-cleaving proteinase (ICP) from Acinetobacter calcoaceticus sharing properties with protease III from Escherichia coliand IDE from eucaryotes. J Basic Microbiol. 1995;35:21–31. doi: 10.1002/jobm.3620350107. [DOI] [PubMed] [Google Scholar]
- 25.Fujita A, Oka C, Arikawa Y, Katagai T, Tonouchi A, Kuhara S, Misumi Y. A yeast gene necessary for bud-site selection encodes a protein similar to insulin-degrading enzyme. Nature. 1994;372:567–570. doi: 10.1038/372567a0. [DOI] [PubMed] [Google Scholar]
- 26.Gammie AE, Brizzio V, Rose MD. Distinct morphological phenotypes of cell fusion mutants. Mol Biol Cell. 1998;9:1395–1410. doi: 10.1091/mbc.9.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gehrung S, Snyder M. The SPA2 gene of Saccharomyces cerevisiaeis important for pheromone-induced morphogenesis and efficient mating. J Cell Biol. 1990;111:1451–1464. doi: 10.1083/jcb.111.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hall A. Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu Rev Cell Biol. 1994;10:31–54. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- 29.Herskowitz I, Park HO, Sanders SL, Valtz N, Peter M. Programming of cell polarity in budding yeast by endogenous and exogenous signals. Cold Spring Harbor Symp Quant Biol. 1995;60:717–727. doi: 10.1101/sqb.1995.060.01.078. [DOI] [PubMed] [Google Scholar]
- 30.Kurihara LJ, Beh CT, Latterich M, Schekman R, Rose MD. Nuclear congression and membrane fusion: two distinct events in the yeast karyogamy pathway. J Cell Biol. 1994;126:911–924. doi: 10.1083/jcb.126.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leeuw T, Fourest-Lieuvin A, Wu C, Chenevert J, Clark K, Whiteway M, Thomas DY, Leberer E. Pheromone response in yeast: association of Bem1p with proteins of the MAP kinase cascade and actin. Science. 1995;270:1210–1213. doi: 10.1126/science.270.5239.1210. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Bretscher A. Characterization of TPM1disrupted yeast cells indicates an involvement of tropomyosin in directed vesicular transport. J Cell Biol. 1992;118:285–299. doi: 10.1083/jcb.118.2.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo L, Liao Y, Jan L, Jan Y. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 1994;8:1787–1802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- 34.Madden K, Snyder M. Specification of sites for polarized growth in Saccharomyces cerevisiaeand the influence of external factors on site selection. Mol Biol Cell. 1992;3:1025–1035. doi: 10.1091/mbc.3.9.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marsh L. Substitutions in the hydrophobic core of the α-factor receptor of Saccharomyces cerevisiae permit response to Saccharomyces kluyveriα-factor and to antagonist. Mol Cell Biol. 1992;12:3959–3966. doi: 10.1128/mcb.12.9.3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marsh, L., and M.D. Rose. 1997. The pathway of cell and nuclear fusion during mating in S. cerevisiae. In The Molecular and Cellular Biology of the Yeast Saccharomyces: Life Cycle and Cell Biology. J.R. Pringle, J.R. Broach, and E.W. Jones, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 827–888.
- 37.McCaffrey G, Clay F, Kelsey K, Sprague GF., Jr Identification and regulation of a gene required for cell fusion during mating of the yeast Saccharomyces cerevisiae. . Mol Cell Biol. 1987;7:2680–2690. doi: 10.1128/mcb.7.8.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michaelis S, Herskowitz I. The a-factor pheromone of Saccharomyces cerevisiaeis essential for mating. Mol Cell Biol. 1988;8:1309–1318. doi: 10.1128/mcb.8.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore SA. Comparison of dose-response curves for α factor-induced cell division arrest, agglutination, and projection formation of yeast cells. J Biol Chem. 1983;258:13849–13856. [PubMed] [Google Scholar]
- 40.Nair J, Muller H, Peterson M, Novick P. Sec2 protein contains a coiled-coil domain essential for vesicular transport and a dispensable carboxy-terminal domain. J Cell Biol. 1990;110:1897–1909. doi: 10.1083/jcb.110.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nern A, Arkowitz RA. A GTP-exchange factor required for cell orientation. Nature. 1998;391:195–198. doi: 10.1038/34458. [DOI] [PubMed] [Google Scholar]
- 42.Osumi M, Shimoda C, Yanagishima N. Mating reaction in Saccharomyces cerevisiae. Changes in the fine structure during the mating reaction. Arch Microbiol. 1974;97:27–38. [PubMed] [Google Scholar]
- 43.Park HO, Bi E, Pringle JR, Herskowitz I. Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc Natl Acad Sci USA. 1997;94:4463–4468. doi: 10.1073/pnas.94.9.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pierotti AR, Prat A, Chesneau V, Gaudoux F, Leseney AM, Foulon T, Cohen P. N-arginine dibasic convertase, a metalloendopeptidase as a prototype of a class of processing enzymes. Proc Natl Acad Sci USA. 1994;91:6078–6082. doi: 10.1073/pnas.91.13.6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramos E, Wysolmerski R, Masaracchia R. Myosin phosphorylation by human cdc42-dependent S6/H4 kinase/gammaPAK from placenta and lymphoid cells. Recept Signal Transduct. 1997;7:99–110. [PubMed] [Google Scholar]
- 46.Roemer T, Madden K, Chang J, Snyder M. Selection of axial growth sites in yeast requires Axl2p, a novel plasma membrane glycoprotein. Genes Dev. 1996;10:777–793. doi: 10.1101/gad.10.7.777. [DOI] [PubMed] [Google Scholar]
- 47.Rose MD, Novick P, Thomas JH, Botstein D, Fink GR. A Saccharomyces cerevisiaegenomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–243. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- 48.Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 198 pp.
- 49.Sanders SL, Herskowitz I. The BUD4protein of yeast, required for axial budding, is localized to the mother/BUD neck in a cell cycle–dependent manner. J Cell Biol. 1996;134:413–427. doi: 10.1083/jcb.134.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Segall JE. Polarization of yeast cells in spatial gradients of a mating factor. Proc Natl Acad Sci USA. 1993;90:8332–8336. doi: 10.1073/pnas.90.18.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simon MN, De Virgilio C, Souza B, Pringle JR, Abo A, Reed SI. Role for the Rho-family GTPase Cdc42 in yeast mating-pheromone signal pathway. Nature. 1995;376:702–705. doi: 10.1038/376702a0. [DOI] [PubMed] [Google Scholar]
- 52.Sprague GF., Jr Assay of yeast mating reaction. Methods Enzymol. 1991;194:77–93. doi: 10.1016/0076-6879(91)94008-z. [DOI] [PubMed] [Google Scholar]
- 53.Tkacz JS, MacKay VL. Sexual conjugation in yeast: cell surface changes in response to the action of mating hormones. J Cell Biol. 1979;80:326–333. doi: 10.1083/jcb.80.2.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trueheart J, Boeke JD, Fink GR. Two genes required for cell fusion during yeast conjugation: evidence for a pheromone-induced surface protein. Mol Cell Biol. 1987;7:2316–2328. doi: 10.1128/mcb.7.7.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valtz N, Herskowitz I. Pea2 protein of yeast is localized to sites of polarized growth and is required for efficient mating and bipolar budding. J Cell Biol. 1996;135:725–739. doi: 10.1083/jcb.135.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.White JM. Membrane fusion. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- 57.Wright R, Basson M, D'Ari L, Rine J. Increased amounts of HMG-CoA reductase induce “karmellae”: a proliferation of stacked membrane pairs surrounding the yeast nucleus. J Cell Biol. 1988;107:101–114. doi: 10.1083/jcb.107.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao ZS, Leung T, Manser E, Lim L. Pheromone signalling in Saccharomyces cerevisiae requires the small GTP-binding protein Cdc42p and its activator CDC24. . Mol Cell Biol. 1995;15:5246–5257. doi: 10.1128/mcb.15.10.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng Y, Bender A, Cerione RA. Interactions among proteins involved in bud-site selection and bud-site assembly in Saccharomyces cerevisiae. . J Biol Chem. 1995;270:626–630. doi: 10.1074/jbc.270.2.626. [DOI] [PubMed] [Google Scholar]
- 60.Ziman M, Preuss D, Mulholland J, O'Brien JM, Botstein D, Johnson DI. Subcellular localization of Cdc42p, a Saccharomyces cerevisiaeGTP-binding protein involved in the control of cell polarity. Mol Biol Cell. 1993;4:1307–1316. doi: 10.1091/mbc.4.12.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]