

Abstract
The superfamily of protein tyrosine phosphatases (PTPs) includes at least one enzyme with an RNA substrate. We recently showed that the RNA triphosphatase domain of the Caenorhabditis elegans mRNA capping enzyme is related to the PTP enzyme family by sequence similarity and mechanism. The PTP most similar in sequence to the capping enzyme triphosphatase is BVP, a dual-specificity PTP encoded by the Autographa californica nuclear polyhedrosis virus. Although BVP previously has been shown to have modest tyrosine and serine/threonine phosphatase activity, we find that it is much more potent as an RNA 5′-phosphatase. BVP sequentially removes γ and β phosphates from the 5′ end of triphosphate-terminated RNA, leaving a 5′-monophosphate end. The activity was specific for polynucleotides; nucleotide triphosphates were not hydrolyzed. A mutant protein in which the active site cysteine was replaced with serine was inactive. Three other dual-specificity PTPs (VH1, VHR, and Cdc14) did not exhibit detectable RNA phosphatase activity. Therefore, capping enzyme and BVP are members of a distinct PTP-like subfamily that can remove phosphates from RNA.
Recently, several mRNA capping enzyme genes from higher eukaryotes have been described (refs. 1–6; reviewed in ref. 7). These proteins consist of two domains, each with distinct enzymatic activity. The C-terminal guanylyltransferase domain contains motifs found in yeast and viral mRNA guanylyltransferases as well as DNA and RNA ligases (reviewed in refs. 8 and 9). Surprisingly, the N-terminal domain exhibits significant sequence similarity to the protein tyrosine phosphatase (PTP) family of enzymes but does not have appreciable PTP activity. However, it does have 5′-RNA triphosphatase activity that produces the RNA 5′-diphosphate necessary for subsequent guanylylation (1, 3, 5, 6, 10).
The PTPs are a large family of enzymes that catalyze the hydrolysis of phosphotyrosine from various proteins. They have been implicated as key players in signaling pathways controlling metabolism, cell growth, differentiation, and cytoskeletal dynamics (reviewed in refs. 11–13). All members of this family have the highly conserved HCX5R active site motif. The invariant cysteine residue within this consensus sequence acts as a nucleophile to attack the phosphate, releasing tyrosine while forming a transient phosphocysteine intermediate (reviewed in refs. 14 and 15). The cysteine of the HCX5R motif located within the N-terminal domain of the Caenorhabditis elegans mRNA capping enzyme is required for its RNA triphosphatase activity, suggesting that phosphate hydrolysis occurs by a similar mechanism (1).
The baculovirus protein phosphatase BVP (also known as BVH1 and BV-PTP) is a 19-kDa, PTP-like enzyme that can hydrolyze phosphoserine and phosphothreonine as well as phosphotyrosine residues (16–18). BVP exhibits 33% sequence identity to the N-terminal region (residues 1–174) of the CEL-1 capping enzyme triphosphatase (1, 2). Prompted by the high degree of sequence similarity to CEL-1, we tested BVP for RNA 5′-phosphatase activity.
We demonstrate here that BVP removes both γ- and β-phosphates from the 5′-triphosphate end of RNA to leave a 5′-monophosphate end. BVP can remove β-phosphate from a diphosphate-terminated RNA, indicating that the RNA triphosphatase and diphosphatase reactions are not necessarily coupled. Mutation of the active site cysteine to serine destroys both RNA phosphatase and protein phosphatase activity, suggesting that the same active site is used for both reactions. No RNA phosphatase activity was detectable with three other dual-specificity PTPs (dsPTPs) that are less closely related to CEL-1: VH1 (from vaccinia virus), VHR (the human homolog of VH1), or Cdc14 (from Saccharomyces cerevisiae). Therefore, within the PTP superfamily, CEL-1 and BVP comprise a distinct subfamily of enzymes that dephosphorylate RNA substrates.
MATERIALS AND METHODS
Recombinant Protein Production and Purification.
BVP was expressed in Escherichia coli DH5α cells as a glutathione S-transferase (GST) fusion protein and was affinity-purified by using glutathione–agarose as described (16) except that soluble extracts were treated with polyethyleneimine to remove some proteins (19) before affinity purification. GST-BVP used to determine kinetic parameters with para-nitrophenylphosphate was affinity-purified as described (16) and then subjected to MonoQ anion exchange chromatography as outlined by Taylor et al. (20). The C. elegans capping enzyme RNA triphosphatase [Cel-1(1–236)] was prepared as described (1). CDC14 was prepared as a GST fusion protein as described (20). VH1 and VHR were the gifts of John Denu (Oregon Health Sciences University).
Preparation of RNA Substrates.
[γ-32P]- and [α-32P]ATP-terminated RNAs for the 5′-triphosphatase assay were prepared by using the DNA primase protein of bacteriophage T7 (21). This enzyme uses a DNA template to make short RNA primers (2–10 nt) that begin with the sequence pppApC (21). By using either [α-32P]- or [γ-32P]ATP, we could produce very short 5′-end labeled RNAs with high specific radioactivity. T7 primase can also incorporate ADP or AMP efficiently as the first nucleotide (22), which allowed us to make 32P-labeled oligoribonucleotides for RNA di- and monophosphatase assays. In our experience, this system is advantageous relative to other systems using E. coli or bacteriophage RNA polymerases.
The standard reaction (100 μl) contained 40 mM Tris⋅HCl (pH 7.5), 10 mM MgCl2, 10 mM DTT, 50 μg/ml BSA, 50 mM potassium glutamate, 2 mM dTTP, 2 mM CTP, 0.3 mM [γ-32P]- or [α-32P]ATP (500–1,000 cpm/pmol) (NEN/DuPont), 1 nM DNA template [5′-CCCCCGGTC(T)25-3′], and 1 μM (hexamer) T7 primase. The reaction was incubated at 37°C for 4 h, and RNA was extracted with phenol-chloroform (1:1). After extraction with ether to remove residual phenol, the reaction mixture was loaded onto a column of DEAE Sephadex A-25 (Pharmacia) pre-equilibrated with 0.2 M triethylamine-bicarbonate buffer (pH 7.6) (TEA-HCO32- buffer) in 7 M urea. Triribonucleotides (pppApCpC or pppApCpC; bold denotes position of radioactive phosphate) were eluted with a linear gradient of 0.2–1.0 M TEA-HCO32- buffer in 7 M urea. For concentration and desalting of the RNAs, the sample was diluted 10-fold with 0.2 M TEA-HCO32- buffer and was adsorbed to a small DEAE Sephadex A-25 column. RNAs were eluted with 1.0 M TEA-HCO32- buffer, lyophilized, and resuspended in water.
To prepare 5′ di- and monophosphate-terminated RNA substrates, the T7 primase reaction was performed as described above except that unlabeled CTP and radiolabeled ATP were replaced with 2 mM [α-32P]CTP and 0.3 mM ADP or AMP (22). Di- (ppApCpC) and mono- (pApCpC) phosphate-terminated triribonucleotides were purified as described above.
RNA 5′-Phosphatase Assays.
RNA 5′ -triphosphatase activity was assayed by two methods: (i) the release of [32P]Pi from [γ-32P]ATP-terminated triribonucleotides (pppApCpC; bold letter denotes labeled phosphate) or (ii) the conversion of [α-32P]ATP-terminated triribonucleotides (pppApCpC) to ADP- or AMP-terminated triribonucleotides (ppApCpC, pApCpC). The standard reaction mixture contained in 10 μl: 50 mM Tris⋅HCl (pH 7.9), 5 mM DTT, 50 μg/ml BSA, 0.5–5 μM (of termini) RNA substrate (1,000–2,000 cpm/pmol), and the enzyme preparation to be assayed. Incubation was for 5–30 min at 30°C, and the reaction was terminated by the addition of 1 M formic acid. The reaction mixture was applied to a polyethyleneimine (PEI) cellulose thin-layer plate (J. T. Baker) and chromatographed with 0.5 M KH2PO4 (pH 3.4). Radioactivity was detected by using a Fuji BasX phosphorimager and/or autoradiography. To separate monophosphate-terminated oligoribonucleotide (pApCpC) from released phosphate (Pi), the reaction products were resolved by electrophoresis on a 25% polyacrylamide—3 M urea gel. For the nucleotide phosphohydrolase assay, 5 μM [α-32P] ATP (NEN/DuPont) was used as substrate.
To test for RNA di- and monophosphatase activities, the appropriate ribonucleotide (ppApCpC, pApCpC) was incubated with enzyme fraction by using the same conditions as for the RNA triphosphatase assay. The reaction products were analyzed by TLC as described above. In some cases, electrophoresis on a 25% polyacrylamide—3 M urea gel was used to completely separate monophosphate-terminated triribonucleotide (pApCpC) from hydroxyl-terminated triribonucleotide (ApCpC).
Protein Phosphatase Assays.
Labeled protein substrates were prepared according to described procedures (16). Concentrations of protein substrates listed herein refer to the total concentration of phosphorylated residues and do not necessarily reflect the total protein concentrations because the stoichiometry of phosphorylation is not unity. Phosphatase activity with 32P-labeled protein substrates was assayed as described (20) by using 5.2 μg of GST-BVP and 3.2 μg of His7-Cel 1[1–236] or His7-Cel 1[1–236](C124S) in 30 μl of assay buffer (50 mM Tris, pH 7.9/10 mM KCl/5 mM DTT/0.1 mg/ml BSA) for 30 min at 30°C. Activity with para-nitrophenylphosphate was assayed in 50 μl of assay buffer by using 0.65 μg GST-BVP and 4.8 μg His7-Cel 1[1–236] or 4.8 μg His7-Cel 1[1–236](C124S) for 30 min at 30°C by using described procedures (20).
RESULTS
BVP Releases the γ-Phosphate from 5′-Triphosphate-Terminated RNA.
To test for RNA 5′-phosphatase activity, recombinant BVP protein was incubated with an RNA trinucleotide labeled at the γ position of its 5′-triphosphate end (pppApCpC). TLC of the reaction products showed that BVP released the labeled phosphate and therefore possesses RNA 5′-triphosphatase activity (Fig. 1, lanes 2–4). The conserved cysteine at position 119 of BVP is required for dsPTP activity (16), so we tested whether this residue is also essential for RNA triphosphatase activity. A mutant BVP enzyme in which the cysteine had been changed to serine (C119S) had no detectable RNA triphosphatase activity (Fig. 1, lanes 5–7). Therefore, BVP has RNA 5′-triphosphatase activity that requires the same nucleophilic cysteine as the protein tyrosine phosphatase activity.
Figure 1.

BVP releases γ-phosphate from triphosphate-terminated RNA. [γ-32P]ATP-terminated trinucleotide RNA was incubated for 30 min at 30°C either with buffer (lane 1), the indicated amounts of wild-type BVP (lanes 2–4), or mutant BVP(C119S) (lanes 5–7). The reaction mixture was analyzed by TLC on PEI–cellulose plates. The position of the 32P label is designated by the asterisk. The position of phosphate was determined by treating the substrate with CIP (data not shown). Occasionally, some fluctuation in the background of free phosphate (lane 7) was observed; this was not due to the BVP protein.
BVP Leaves a 5′-Monophosphate End on RNA.
To determine whether BVP leaves a 5′-diphosphate end (as does capping enzyme), the RNA triphosphatase assay was next performed with an RNA whose 5′-triphosphate end was labeled at the α-phosphate (pppApCpC) (Fig. 2). As previously observed with longer RNA transcripts (1), CEL-1 RNA triphosphatase left a diphosphate 5′ end (Fig. 2A, lanes 9–11). No activity was detected with a mutant CEL-1 in which the nucleophilic cysteine was mutated to serine (C124S; Fig. 2A, lanes 12–14). In contrast to the capping enzyme triphosphatase, BVP produced RNA that migrated faster than diphosphate-terminated RNA on a PEI cellulose plate (Fig. 2A, lanes 2–4) This species migrated almost as fast as monophosphate released by calf intestinal phosphatase (CIP) (Fig. 2A, lane 8). The BVP mutant C119S did not shift RNA (Fig. 2A, lanes 5–7).
Figure 2.
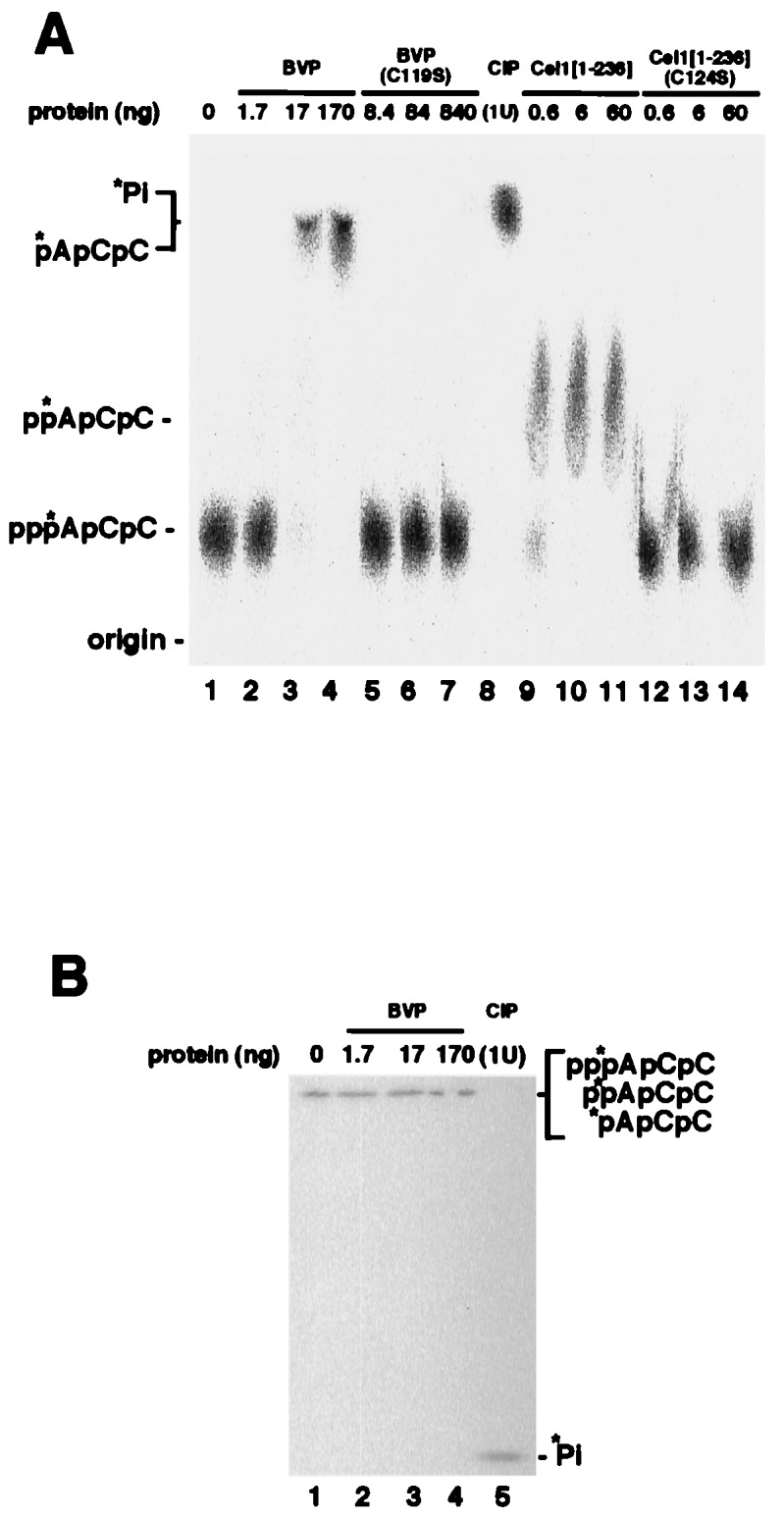
BVP leaves a 5′-monophosphate end. The RNA triphosphatase assay was performed with [α-32P]ATP-terminated trinucleotide RNA (label is indicated by an asterisk). After incubation for 10 min at 30°C, the reaction mixture was analyzed by (A) PEI–cellulose TLC or (B) 25% polyacrylamide gel electrophoresis. The positions of ADP- and AMP-terminated RNA trinucleotides were determined by using ADP- and AMP-terminated trimers as standards (not shown). In A, RNA substrate was incubated with the following: buffer (lane 1), the indicated amount of BVP (BVP, lanes 2–4), an active site mutant of BVP [BVP(C119S), lanes 5–7], 1 unit of CIP (lane 8), the C. elegans capping enzyme triphosphatase (Cel-1[1–236], lanes 9–11), or the corresponding active site mutant [Cel-1[1–236](C124S), lanes 12–14]. Reactions in lanes 1–5 of B were identical to those in lanes 1–4 and 8 of A.
Because monophosphate-terminated trimeric RNA and free phosphate migrated similarly in the TLC analysis (Fig. 2A, lanes 3, 4, and 8), we also analyzed the reaction products on a 25% polyacrylamide gel (Fig. 2B). From this experiment, it was clear that [32P]α-phosphate was not released by BVP (Fig. 2B, lane 2–5). The reaction products also were digested with RNase T2, and BVP-treated RNA released pAp while CIP released Ap (data not shown). Therefore, we conclude that BVP leaves a 5′-monophosphate end on RNA.
We also tested BVP for RNA di- and monophosphatase activity by using trimers carrying two or one 5′ phosphate (ppApCpC, pApCpC) (Fig. 3). CEL-1 RNA triphosphatase did not show any RNA di- or monophosphatase activity (data not shown). The wild-type BVP converted diphosphate-terminated RNA to a species (Fig. 3A, lanes 1–4) that migrated similarly to 5′ hydroxyl-terminated RNA (ApCpC) produced with CIP (Fig. 3A, lane 8). The mutant C119S had no activity (Fig. 3A, lanes 5–7). To distinguish 5′-monophosphate-ended trimer from 5′-hydroxyl-ended trimer, the same reactions were resolved by electrophoresis on a 25% polyacrylamide gel (Fig. 3B). CIP was used to produce hydroxyl-terminated RNA (Fig. 3B, lane 5). In contrast to CIP, BVP did not produce any 5′-hydroxyl ends on either the diphosphate- (Fig. 3B, lanes 2–4) or monophosphate- (data not shown) terminated RNA. Furthermore, RNase T2 digestion of reaction products showed that BVP-treated RNA released only pAp from the 5′-end (data not shown).
Figure 3.

BVP has independent RNA 5′-diphosphatase activity. The RNA diphosphatase assay was performed with 5′-diphosphate-terminated RNA trinucleotide that was labeled with 32P at internal phosphates (designated by asterisks). RNA substrate was incubated for 10 min at 30°C with buffer, the indicated amounts of wild-type BVP, active site mutant BVP(C119S), or 1 unit of CIP. The reaction mixtures were analyzed by PEI–cellulose TLC (A) or 25% polyacrylamide gel electrophoresis (B).
Reactions done at low concentrations of BVP, or taken at early time points, showed clearly that diphosphate ends appear before formation of monophosphate ends (data not shown). The results presented above demonstrate that BVP has independent RNA tri- and diphosphatase activities, and leaves a 5′-monophosphate on RNA. Because both of these activities depend on the same active site cysteine, we believe that BVP sequentially removes the γ and β phosphate groups from RNA.
Polynucleotide Specificity of BVP.
The RNA triphosphatase activity of eukaryotic capping enzymes is specific for polynucleotide RNA (1, 23–25). In contrast, the vaccinia virus capping enzyme has a weak nucleotide phosphohydrolase (NTPase) activity (26–28). To determine whether BVP has NTPase activity, we incubated [α-32P]ATP with BVP or CEL-1 (Fig. 4). In contrast to CIP (Fig. 4, lane 8), BVP and CEL-1 did not release phosphate and therefore have no NTPase activity.
Figure 4.

BVP does not have nucleotide phosphohydrolase activity. [α-32P]ATP was incubated for 10 min at 30°C either with buffer (lane 1), the indicated amounts of BVP (lanes 2–4), C. elegans capping enzyme triphosphatase (Cel-1[1–236], lanes 5–7), or 1 unit of CIP (lane 8). The reaction mixtures were analyzed by TLC on PEI–cellulose plates. The positions of authentic ADP and AMP markers were detected by UV light.
Relative Activity of BVP on Protein and RNA.
The ability of BVP to remove phosphate from various nucleotide and artificial protein substrates was quantitated and compared with that of the RNA triphosphatase from the C. elegans capping enzyme CEL-1 and the bona fide protein tyrosine phosphatase CDC14 (Table 1, activities are expressed as nanomoles of substrate produced per minute per milligram of enzyme). Both CEL-1 and BVP hydrolyzed para-nitrophenylphosphate. The kcat/Km values of 0.4 and 17.4 M−1 s−1, respectively, indicate that BVP is a 40-fold more efficient catalyst of this reaction than CEL-1. As previously reported (16–18), BVP dephosphorylated both protein tyrosine and serine/threonine residues. However, the activity of BVP with identical substrate concentrations was two to three orders of magnitude lower than that of Cdc14, a dual specificity phosphatase from budding yeast (Table 1). CEL-1 was capable of hydrolyzing phosphotyrosine-containing protein substrates, albeit with rates that are 10- to 20-fold lower than those of BVP and about four orders of magnitude lower than those of Cdc14. CEL-1 was essentially inactive with phosphoserine/threonine-containing substrates. No activity toward any substrate was detected for the Cel-1 (C124S) mutant (data not shown). These in vitro results indicate that both CEL-1 and BVP are relatively inefficient protein phosphatases.
Table 1.
Substrate specificity of BVP
| Substrate | Specific activity, nmol min−1 mg−1
|
|||
|---|---|---|---|---|
| Conc., μM | GST-Cdc14p* | Cel-1(1-236) | GST-BVP | |
| pppACC | 3 | ND | 1000 | 200 |
| ppACC | 5 | ND | ND | 600 |
| PNPP | 50,000 | 1200 | 34.6 | 204 |
| pY-MBP | 5 | 31 | 0.006 | 0.13 |
| pY-casein | 2.5 | 4 | 0.024 | 0.18 |
| pY-RCML | 5 | — | 0.012 | 0.22 |
| pS/T-MBP | 5 | 8 | ND | ND |
| pS-casein | 5 | 7 | ND | 0.003 |
| pS/T-histone | 5 | 1 | 0.001 | 0.006 |
| pS/T-RCML | 5 | ND | ND | 0.001 |
Values taken from Taylor et al. (20)
ND, no detectable phosphate was released (the limit of detection for protein substrate assays was ≈0.05–0.1 pmol of phosphate released); —, this combination was not tested.
By comparison, both CEL-1 and BVP exhibited very potent RNA phosphatase activity. BVP and CEL-1 dephosphorylated RNA with specific activities that were three and four orders of magnitude greater, respectively, than those obtained by using comparable concentrations of the best phosphoprotein substrates. To determine whether RNA 5′-phosphatase activity is a common feature of dual specificity PTPs, three other dsPTPs were tested. Vaccinia virus VH1 (29), human VHR (30), and Cdc14 (20) exhibited no detectable phosphatase activity against either tri-, di-, or monophosphate-terminated RNAs even at very high enzyme or substrate concentrations (Table 1 and data not shown). Therefore, we conclude that BVP is much more similar in substrate preference to the capping enzyme triphosphatase than it is to other dsPTPs.
DISCUSSION
Baculovirus BVP (also known as BVH1 or BV-PTP) has been shown to be a dual specificity protein phosphatase that can remove phosphate from tyrosine, serine, and threonine residues (16–18). We show here that BVP is much more potent as an RNA 5′-tri- and diphosphatase. BVP was tested for RNA phosphatase activity because of its sequence similarity to the C. elegans mRNA capping enzyme (CEL-1; see refs. 1 and 2). Like the capping enzyme RNA triphosphatase, BVP can efficiently release phosphates from the 5′-triphosphate end of polynucleotide RNA. Both BVP and capping enzyme are active on RNA that is at least trinucleotide length, but they do not hydrolyze mononucleotides. The similarity of BVP to capping enzyme triphosphatase in sequence, activity, and substrate preference argues strongly that its physiological substrate is RNA.
BVP and capping enzyme differ in one important respect. Capping enzyme removes only the γ-phosphate from triphosphate-terminated RNA, leaving a diphosphate end that is joined with GMP to form the cap (1, 3, 5, 6, 10). In contrast, BVP releases both γ- and β-phosphates to produce a monophosphate end. BVP also can release β-phosphate from 5′-diphosphate termini, indicating that the triphosphatase and diphosphatase reactions are not linked necessarily. Our kinetic experiments indicate that the removal of the γ- and β-phosphates occurs sequentially. Both RNA phosphatase activities were destroyed by mutation of the active site cysteine to serine (Figs. 2 and 3). The PTP activity of BVP also was destroyed completely by this mutation (16), suggesting that the same active site is used for all of the phosphorylated substrates.
On the basis of function, structure, and sequence, the PTPs have been grouped into four main families: (i) the tyrosine-specific phosphatases, (ii) the dsPTPs that can hydrolyze phosphoserine and phosphothreonine as well as phosphotyrosine, (iii) the cdc25 phosphatases, and (iv) the low molecular weight phosphatases (reviewed in ref. 15). Three other dsPTPs tested (VH1, VHR, and CDC14) did not have any detectable RNA 5′-phosphatase activity (Table 1 and data not shown), and their sequences are much less similar to capping enzyme than BVP. These criteria suggest that BVP and the CEL-1 capping enzyme constitute a distinct subfamily of PTP-like enzymes. The GenBank database contains several other ORFs that are closely related to BVP and capping enzymes (1, 2). Furthermore, we recently have isolated a cDNA encoding a human homolog of BVP (T. Deshpande, L. Hao, and H. Charbonneau, unpublished data) suggesting that there are additional proteins within this subfamily. It will be important to test these enzymes for RNA phosphatase activities.
The physiological substrate and biological function of BVP remain unknown. Baculoviruses are circular double-strand DNA viruses that replicate in the nucleus of the insect host cell. Expression of the BVP gene occurs late in infection, with protein appearing in both the nucleus and cytoplasm as well as the viral particle (18, 31). BVP is not essential for viral replication, but a virus carrying a BVP deletion has reduced titers (by 50% or more) and is inefficient at forming viral occlusion bodies in Sf-21 cells (31, 32). The relevance of these results is not completely clear because the deletion virus appears to have normal infectivity in insect larvae.
If RNA is the physiological substrate for BVP, what role might the enzyme play in the viral life cycle? We present some speculative ideas for future testing. RNA 5′-triphosphate ends are known to occur in two places: gene transcripts produced by RNA polymerases and the RNA primers laid down by primases at origins of DNA replication. The finding that BVP deletion viruses are viable might suggest that it is not involved in either of these essential processes, but it is also possible that there are cellular enzymes that can at least partially compensate for the loss of BVP activity.
In all cases studied to date, short RNA primers are used to initiate replication of DNA. This RNA must be removed from the heteroduplex and the resulting gap filled in and ligated. It is unknown whether the RNA 5′-triphosphate end would create a barrier to the viral or cellular enzyme that removes the primer. Endonucleolytic enzymes would probably not be affected, but a 5′ to 3′ exonuclease might have specificity for a particular 5′ end configuration. One possible function for BVP could be in processing the 5′ end of the replication primers.
Early baculovirus gene expression is thought to be carried out by the host cell RNA polymerase II, and viral mRNA is presumably capped by the host cell enzyme (33). Late in infection, viral gene expression becomes resistant to α-amanitin and levels of host transcripts drop dramatically (reviewed in ref. 34). BVP might be involved in a viral-specific mRNA capping reaction. The capping enzymes of some rhabdoviruses (a class of nonsegmented, cytoplasmic RNA virus) remove two phosphates from the transcript 5′ end and then transfer GDP to the 5′-monophosphate end of an RNA chain (35–38). Another possibility is that BVP generates a 5′-monophosphate end that can be used as a substrate in some sort of RNA ligation, a reaction that is related mechanistically to capping. One final possibility is that BVP represses host mRNA capping and/or splicing by interfering with the 5′ ends of mRNA and/or snRNAs. RNAs lacking a cap will be destabilized, inefficiently spliced, and poorly translated (39, 40).
Several distinct mechanisms appear to be used to remove phosphates from the 5′ end of RNAs. The mRNA capping enzyme triphosphatases from higher eukaryotes, yeast, and vaccinia virus all have similar activities but share no apparent sequence similarity (1–6, 25, 41). The mechanism of the higher eukaryotic capping enzymes and BVP appear closely related to that of protein tyrosine phosphatases, but the yeast and vaccinia triphosphatase mechanisms are not yet understood. It also remains to be determined whether RNA phosphatases are involved in cellular processes other than capping. Future studies of BVP may provide clues as to what those processes might be.
Acknowledgments
We are grateful to J. Denu (Oregon Health Sciences University) for the gifts of VHR and VH1 proteins, H. Matsuo (Harvard Medical School) for valuable suggestions about purification of RNA, C. R. Moore (Harvard Medical School) for critical reading of the manuscript, and M. Saper (University of Michigan), C. Walsh, and C. Richardson (Harvard Medical School) for helpful discussions. Funding for this study came from a grant from the Council for Tobacco Research (to S.B.). S.B. also is supported by a Pew Biomedical Scholar Award and a Junior Faculty Research Award from the American Cancer Society. H.C. is supported by National Institutes of Health Grant CA59935 and a Junior Faculty Research Award from the American Cancer Society.
ABBREVIATIONS
- PTPs
protein tyrosine phosphatases
- dsPTPs
dual-specificity PTPs
- GST
glutathione S-transferase
- PEI
polyethyleneimine
- CIP
calf intestinal phosphatase
References
- 1.Takagi T, Moore C R, Diehn F, Buratowski S. Cell. 1997;89:867–873. doi: 10.1016/s0092-8674(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 2.Wang S P, Deng L, Ho C K, Shuman S. Proc Natl Acad Sci USA. 1997;94:9573–9578. doi: 10.1073/pnas.94.18.9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yue Z, Maldonado E, Pillutla R, Cho H, Reinberg D, Shatkin A J. Proc Natl Acad Sci USA. 1997;94:12898–12903. doi: 10.1073/pnas.94.24.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCracken S, Fong N, Rosonina E, Yankulov K, Brothers G, Siderovski D, Hessel A, Foster S, Shuman S, Bentley D L. Genes Dev. 1997;11:3306–3318. doi: 10.1101/gad.11.24.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsukamoto T, Shibagaki Y, Murakoshi T, Suzuki M, Nakamura A, Gotoh H, Mizumoto K. Biochem Biophys Res Commun. 1998;243:101–108. doi: 10.1006/bbrc.1997.8038. [DOI] [PubMed] [Google Scholar]
- 6.Yamada-Okabe T, Doi R, Shimmi O, Arisawa M, Yamada-Okabe H. Nucleic Acid Res. 1998;26:1700–1706. doi: 10.1093/nar/26.7.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shuman S. Proc Natl Acad Sci USA. 1997;94:12758–12760. doi: 10.1073/pnas.94.24.12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shuman S. Prog Nucleic Acids Res Mol Biol. 1995;50:101–129. doi: 10.1016/s0079-6603(08)60812-0. [DOI] [PubMed] [Google Scholar]
- 9.Shuman S, Schwer B. Mol Microbiol. 1995;17:405–410. doi: 10.1111/j.1365-2958.1995.mmi_17030405.x. [DOI] [PubMed] [Google Scholar]
- 10.Ho C K, Sriskanda V, McCracken S, Bentley D, Schwer B, Shuman S. J Biol Chem. 1998;273:9577–9585. doi: 10.1074/jbc.273.16.9577. [DOI] [PubMed] [Google Scholar]
- 11.Neel B, Tonks N K. Curr Opin Cell Biol. 1997;9:193–204. doi: 10.1016/s0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 12.Charbonneau H, Tonks N K. Annu Rev Cell Biol. 1992;8:462–493. doi: 10.1146/annurev.cb.08.110192.002335. [DOI] [PubMed] [Google Scholar]
- 13.Walton K M, Dixon J E. Annu Rev Biochem. 1993;62:101–120. doi: 10.1146/annurev.bi.62.070193.000533. [DOI] [PubMed] [Google Scholar]
- 14.Denu J M, Stuckey J K, Saper M A, Dixon J E. Cell. 1996;87:361–364. doi: 10.1016/s0092-8674(00)81356-2. [DOI] [PubMed] [Google Scholar]
- 15.Fauman E B, Saper M A. Trends Biochem Sci. 1996;21:413–417. doi: 10.1016/s0968-0004(96)10059-1. [DOI] [PubMed] [Google Scholar]
- 16.Sheng Z, Charbonneau H. J Biol Chem. 1993;268:4728–4733. [PubMed] [Google Scholar]
- 17.Hakes D J, Martell K J, Zhao W-G, Massung R F, Esposito J J, Dixon J E. Proc Natl Acad Sci USA. 1993;90:4017–4021. doi: 10.1073/pnas.90.9.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D, Weaver R F. Virology. 1993;195:587–595. doi: 10.1006/viro.1993.1410. [DOI] [PubMed] [Google Scholar]
- 19.Burgess R R. Methods Enzymol. 1991;208:3–10. doi: 10.1016/0076-6879(91)08003-z. [DOI] [PubMed] [Google Scholar]
- 20.Taylor G S, Liu Y, Baskerville C, Charbonneau H. J Biol Chem. 1997;272:24054–24063. doi: 10.1074/jbc.272.38.24054. [DOI] [PubMed] [Google Scholar]
- 21.Kusakabe T, Richardson C C. J Biol Chem. 1997;272:5943–5951. doi: 10.1074/jbc.272.9.5943. [DOI] [PubMed] [Google Scholar]
- 22.Kusakabe T, Richardson C C. J Biol Chem. 1997;272:12446–12453. doi: 10.1074/jbc.272.19.12446. [DOI] [PubMed] [Google Scholar]
- 23.Yagi Y, Mizumoto K, Kaziro Y. EMBO J. 1983;2:611–615. doi: 10.1002/j.1460-2075.1983.tb01471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh N, Mizumoto K, Kaziro Y. J Biol Chem. 1984;259:13923–13929. [PubMed] [Google Scholar]
- 25.Tsukamoto T, Shibagaki Y, Imajoh-Ohmi S, Murakoshi T, Suzuki M, Nakamura A, Gotoh H, Mizumoto K. Biochem Biophys Res Commun. 1997;239:116–122. doi: 10.1006/bbrc.1997.7439. [DOI] [PubMed] [Google Scholar]
- 26.Shuman S, Surks M, Furneaux H, Hurwitz J. J Biol Chem. 1980;255:11588–11598. [PubMed] [Google Scholar]
- 27.Myette J R, Niles E G. J Biol Chem. 1996;271:11945–11952. doi: 10.1074/jbc.271.20.11945. [DOI] [PubMed] [Google Scholar]
- 28.Yu L, Shuman S. J Virol. 1996;70:6162–6168. doi: 10.1128/jvi.70.9.6162-6168.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan K, Broyles S S, Dixon J E. Nature (London) 1991;350:359–362. doi: 10.1038/350359a0. [DOI] [PubMed] [Google Scholar]
- 30.Ishibashi T, Bottaro D P, Chan A, Miki T, Aaronson S A. Proc Natl Acad Sci USA. 1992;89:12170–12174. doi: 10.1073/pnas.89.24.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Miller L K. J Gen Virol. 1995;76:2941–2948. doi: 10.1099/0022-1317-76-12-2941. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Miller L K. J Virol. 1995;69:4533–4537. doi: 10.1128/jvi.69.7.4533-4537.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jun-Chuan Q, Weaver R F. J Virol. 1982;43:234–240. doi: 10.1128/jvi.43.1.234-240.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Reilly D R, Miller L K, Luckow V A. Baculovirus Expression Vectors: A Laboratory Manual. New York: Oxford Univ. Press; 1994. pp. 19–25. [Google Scholar]
- 35.Abraham G, Rhodes D P, Banerjee A K. Cell. 1975;5:51–58. doi: 10.1016/0092-8674(75)90091-4. [DOI] [PubMed] [Google Scholar]
- 36.Gupta K C, Roy P. J Virol. 1980;33:292–303. doi: 10.1128/jvi.33.1.292-303.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shuman S. Virology. 1997;227:1–6. doi: 10.1006/viro.1996.8305. [DOI] [PubMed] [Google Scholar]
- 38.Bisalliom M, Lemay G. Virology. 1997;236:1–7. doi: 10.1006/viro.1997.8698. [DOI] [PubMed] [Google Scholar]
- 39.Fresco L D, Buratowski S. RNA. 1996;2:584–596. [PMC free article] [PubMed] [Google Scholar]
- 40.Lo H J, Huang H, Donahue T F. Mol Cell Biol. 1998;18:665–675. doi: 10.1128/mcb.18.2.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niles E G, Condit R C, Caro P, Davidson K, Matusick L, Seto J. Virology. 1986;153:96–112. doi: 10.1016/0042-6822(86)90011-5. [DOI] [PubMed] [Google Scholar]