

Abstract
Protein functions have evolved in part via domain recombination events. Such events, for example, recombine structurally independent functional domains and shuffle targeting, regulatory, and/or catalytic functions. Domain recombination, however, can generate new functions, as implied by the observation of catalytic sites at interfaces of distinct folding domains. If useful to an evolving organism, such initially rudimentary functions would likely acquire greater efficiency and diversity, whereas the initially distinct folding domains would likely develop into single functional domains. This represents the probable evolution of the S1 serine protease family, whose two homologous β-barrel subdomains assemble to form the binding sites and the catalytic machinery. Among S1 family members, the contact interface and catalytic residues are highly conserved whereas surrounding surfaces are highly variable. This observation suggests a new strategy to engineer viable proteins with novel properties, by swapping folding subdomains chosen from among protein family members. Such hybrid proteins would retain properties conserved throughout the family, including folding stability as single domain proteins, while providing new surfaces amenable to directed evolution or engineering of specific new properties. We show here that recombining the N-terminal subdomain from coagulation factor X with the C-terminal subdomain from trypsin creates a potent enzyme (fXYa) with novel properties, in particular a broad substrate specificity. As shown by the 2.15-Å crystal structure, plasticity at the hydrophobic subdomain interface maintains activity, while surface loops are displaced compared with the parent subdomains. fXYa thus represents a new serine proteinase lineage with hybrid fX, trypsin, and novel properties.
Consideration of hierarchical features of protein architecture is particularly important in the design of novel enzymatic functions (1, 2). The simplest protein design problems involve the recruitment of existing evolved structural and functional elements (domains or modules) in straightforward recombination of associated properties, for example the combination of separate targeting and effector domains in immunotoxin design (3) or the shuffling of cofactor binding domains of coagulation factors (4). Many applications would benefit from the recombination of properties within a single functional domain; however, recombination of hierarchical units smaller than functional domains (referred to here as subdomains) generally risks loss of function or folding stability. A cautious attempt in this direction is the recombination of subdomains selected from homologous proteins that themselves have diversified through evolution but that retain relatively conserved catalytic machinery and subdomain interactions. The evolution of protein function via domain duplication and/or recombination has generated numerous functional domains that comprise two such subdomains (5–9). This strategy allows one to swap specific subdomain linked functions, to preserve conserved functions at the interface, and to generate new properties at nonconserved or hypervariable surfaces near the interface. Such new surface-linked properties—most prominently including binding sites—will be “unevolved” and therefore amenable to optimization for specific applications by design or directed evolution. Such an approach—the generation of new enzyme “lineages” by subdomain shuffling—was described recently in an attempt to change a hydrolase function to a transferase function by appropriate subdomain choice (10). Here, we adopt a more conservative approach and attempt to preserve efficient enzymatic function as conserved within a protein family while altering substrate specificity and other specific surface properties.
Subdomains were taken from the digestive protease trypsin and the coagulation factor Xa (fXa), both members of the S1 (chymotrypsin) family of serine proteinases (11). Because this large enzyme family is structurally and functionally well characterized, with representatives in many biotechnological and therapeutic applications, it is an interesting and potentially useful choice for novel protein design experiments. The members of the S1 family are comprised of two homologous β-barrel subdomains that pack together asymmetrically to constitute the compact catalytic domain. The subdomain interface forms the active site with the substrate binding cleft running along the subdomain boundary. Notably, the conserved catalytic tetrad residues Ser195, His57, Asp102, and Ser214 are from the C-, N-, N-, and C-terminal domains, respectively. Although the hydrophobic core structures remain conserved throughout the family (12), considerable variation is seen in the surface loops, especially surrounding the active site where they determine substrate specificities (13). Other surface features introduce cofactor binding sites (14), regulatory sites (15), etc. These variations are responsible for the large functional diversity of serine proteases that are involved in various physiological processes such as digestion (5), blood coagulation (16), fibrinolysis (17), and complement activation (18). In the case of the hybrid fXa—trypsin protein described here, swapping the fXa C-terminal hemisphere for that of trypsin decouples the hydrophobic, fXa-specific EGF2 binding surface from the fXa N-terminal hemisphere and its substrate binding sites. This particular hybrid thus was chosen as a potential first step in the engineering of a model for fXa inhibitor interactions with crystallization properties more like trypsin.
MATERIALS AND METHODS
Materials.
Enterokinase and enzymes used for DNA manipulation were from Boehringer Mannheim. Chromogenic substrates were from Boehringer Mannheim and Chromogenix (Moelndal, Sweden). 4-NPGB was from Merck (Darmstadt, Germany), Benzamidine Sepharose 6B was from Pharmacia, and PPACK was purchased from Bachem.
Construction of Recombinant Proteins.
The rfX-expression plasmid was constructed as described (19). The DNA fragment encoding rtrypsinogen was amplified by PCR on human pancreas cDNA constructed in the phage vector Lambda ZAP II (Stratagene) by using the oligonucleotides 5′-AAAAAACCATGGATGATGATGACAAGATCGTTGGG-3′ and 5′AAAAAAAAGCTTCATTAGCTATTGGCAGCTATGGTGTTC-3′ and cloned into rfX-expression plasmid. The expression vector for the fX/trypsinogen-hybrid (rfXY) was constructed by cloning the DNA fragment of the rfX-part into the rtrypsinogen-expression plasmid. The rfX—DNA fragment was amplified by PCR on the rfX plasmid by using the oligonucleotides 5′-AAAAAAATGCATCACCACCACGACGATGACGACAAGATCGTGGGAGGCTACAACTGCAAGGACGGGGAGGTACCCTGGCAGGCCCTGCTCATC-3′ and 5′-AAAAAACCAGTGGCTGGAGGGGCGGTGGGCAGAGAGGCAGGCGCCACGTTCATGCG-3′. It encodes residues 16–121 (chymotrypsin numbering) carrying the trypsinogen sequence at residues 20, 21, and 27 to account for a different disulfide bond between fX and trypsinogen.
Expression, Folding, and Purification.
rfX, rtrypsinogen, and rfXY were expressed, folded, and purified as described (19). rTrypsinogen and rfXY were activated with enterokinase in 20 mM Tris⋅HCl (pH 8.0), 50 mM NaCl at 37°C at a concentration of 2 mg/ml recombinant protein, and 2 μg/ml enterokinase and further purified by affinity chromatography on benzamidine Sepharose (20). The active site concentration was determined by titration with 4-NPGB (21).
Amidolytic Assays.
Assays were carried out in 50 mM Tris, 200 mM NaCl, 5 mM CaCl2, and 0.1% polyethylene glycol 8000 (pH 8.0) at 25°C as described (19).
Structure Determination.
Purified rfXYa was inhibited with a fivefold excess of PPACK and crystallized in 100 mM Hepes (pH 7.8), 15% polyethylene glycol 6000, and 15 mg/ml rfXYa at 4°C in sitting drops by using the vapor diffusion technique. Reflections (28,062) were collected on a MAR imaging plate system and processed with mosflm (22) and programs from the ccp4 Suite (23). The space group was C2 with one molecule in the asymmetric unit, and cell constants were a = 65.5 Å, b = 48.9 Å, c = 75.5 Å, and β = 111.5°. The structure was solved by molecular replacement by using a search model of rfXYa constructed by combining coordinates from fXa (PDB ID code 1hcg) (24) and trypsin (PDB ID code 1trn) (25). Rotational and translational search was done with amore (26) by using data from 15 to 3.5 Å. A single solution was found with a correlation factor of 51.8% and a R-factor of 38.7%. Crystallographic refinement was performed with x-plor (27) and main (28) by using a total of 10,714 reflections in the range of 8.0–2.15 Å. The data used for refinement were 89.3% complete to 2.15 Å (91.6% completeness in the 2.25–2.15 Å shell) with an R-merge of 7.2%. Refinement included rigid body minimization, overall temperature refinement, positional refinement, individual temperature factor refinement, and manual model building against 2Fo-Fc and Fo-Fc electron density maps. The model was refined to an Rcryst of 18.5% (Rfree 24.1%) and contains 82 water molecules. The quality of the final model was evaluated with procheck (29) with 85.6% of the residues in the core region and 14.4.% in the allowed region of the Ramachandran plot. The rms deviation of bond lengths was 0.005 Å and that of bond angles was 1.2°. Atomic coordinates have been deposited in the Protein Data Bank.
RESULTS
Design of the Hybrid Enzyme.
To engineer the fXa/trypsin-chimera (rfXYa), a gene was constructed by combining the DNA encoding the N-terminal β-barrel of fXa with the DNA of the C-terminal β-barrel of trypsin. The boundary of the subdomains was chosen by graphical inspection of the crystal structures of fXa (24) and trypsin (25) to be residue 122 (Fig. 1), located in a linker region between the N- and C-terminal β-barrel. A problem arose near the N terminus of the molecule, where Cys22 forms disulfide bridges with cysteine partners that differ between fXa (Cys27) and trypsin (Cys157). The former is located within the N-terminal subdomain, whereas the latter bridges the two subdomains (Fig. 2). Initially, both variants were constructed in parallel. To preserve packing, additional mutations were introduced in the vicinity of the disulfide bridge: In the case of Cys22-Cys27 (fXa-variant), we introduced Cys157Met; in the case of Cys22-Cys157 (trypsin-variant), we introduced Gln20Tyr, Glu21Asn, and Cys27Val. Both hybrid proteins were expressed in Escherichia coli as insoluble protein aggregates (inclusion bodies) and successfully folded in vitro. The folding efficiency of ≈20–40% correctly folded material per starting material was in the range of that of rtrypsin and rfX (data not shown). Preliminary characterization of both disulfide bridge variants revealed no significant difference with respect to folding and formation of activity, and we focused on the trypsin variant for structural and enzymatic characterization.
Figure 1.

Primary structure of the fXa/trypsin hybrid. The primary structure of rfXYa is shown in an amino acid sequence alignment (single letter code) of the catalytic domain of human coagulation factor Xa and human trypsin 1. Conserved residues (38% sequence identity) are boxed and shaded blue. Segments taken from fXa are colored yellow; those from trypsin are colored red. Three residues at the N terminus (Y20,E21 and V27) were taken from trypsin (red) to account for a different disulfide bridge in fXa (C22-C27) and trypsin (C22-C157). The trypsin disulfide bridge C22-C157 was used. Both subdomains contribute to the catalytic tetrad (denoted by asterisk).
Figure 2.

Crystal structure of the fXa/trypsin hybrid. Ribbon plot of the crystal structure of fXYa. The N-terminal subdomain is shown in red, and the C-terminal subdomain is shown in yellow. Both subdomains adopt a β-barrel fold and assemble asymmetrically to generate the fold typical of the chymotrypsin family. Disulfide bridges are depicted in green (the N-terminal bridge discussed in the text is located behind the C-terminal barrel). The D-Phe-Pro-Arg inhibitor is shown with magenta sticks. It is bound to the active site, which is formed at the subdomain interface. The catalytic triad residue side chains are displayed explicitly as sticks.
Overall Structure.
To study the structural effects of the subdomain shuffling and to confirm correct folding, we determined the crystal structure of the hybrid in complex with the covalently bound d-Phe-Pro-Arg-chloromethylketone (PPACK) inhibitor. The 2.15-Å crystal structure (Fig. 2) confirmed the formation of the general structural features of S1 family serine proteinases: Both subdomains adopted the six-stranded antiparallel β-barrel fold with native disulfide bridges and assembled asymmetrically to form the substrate binding cleft and the catalytic tetrad; the N terminus inserted in the C-terminal barrel to form the activating buried salt bridge with Asp194; the C terminus formed a helix across the N-terminal β-barrel.
Comparison to fXa and Trypsin.
The active site of rfXYa retains the structures conserved between the respective parent molecules (Fig. 3), including the catalytic tetrad, the oxyanion hole, and other catalytic elements at the subdomain boundary. Small changes in side chain orientations may be attributed to the presence of the covalently bound PPACK inhibitor. Successful subdomain assembly and particularly activity confirms the complementarity of the hydrophobic 3,000 Å2 contact face. Approximately 30% of residues making up the interface contacts differ between trypsin and fXa, presenting the possibility of unfavorable mismatches. Although these mismatches occur throughout the core elements of the interface, they are more abundant at surface segments of the interface (Fig. 4). Consistent, however, with the activity and overall stability of the hybrid, the hydrophobic core elements generally retained their structure with minor plastic adjustments to accommodate mismatched residues. In contrast, several surface loops showed large divergence from their structure observed in the parent molecule (Fig. 4).
Figure 3.

Comparison of the active site of fXYa to fXa and trypsin. Final model of fXYa showing the active site (thick sticks using the color code of Fig. 2) with representative 1.0 σ contoured 2Fc-Fo electron density for the catalytic tetrad residues His52, Asp102, Ser195, and Ser214 as well as for the PPACK inhibitor. The N-terminal subdomain of fXYa (red) is superimposed with the N-terminal subdomain of fXa (blue); the C-terminal subdomain (yellow) is superimposed with the C-terminal subdomain of trypsin (green). The structure shows a well conserved catalytic triad and specificity pocket. Some side chain adjustments in substrate binding sites (S1-S3) presumably originate from interaction with PPACK.
Figure 4.
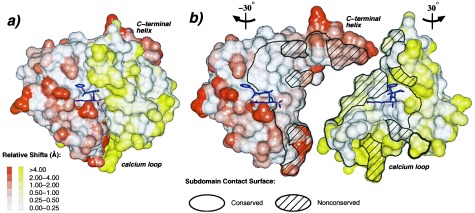
Plasticity of the domain interface. (A) Connolly surface representation of rfXYa using the orientation and colors of Fig. 2. The surface was calculated for each subdomain separately. The extent of displacement of rfXYa surface atoms relative to the parent molecules after Ca superposition of the corresponding subdomains is color coded (Inset). PPACK (magenta) is shown as stick model. (B) Same as A but with N- and C-terminal subdomains rotated −30° and +30° about the vertical axis and separated to display the subunit interface. A stick model of the inhibitor (magenta) is displayed with each subdomain to clarify the respective orientation and to mark the location of the active site. The black bounded area denotes the contact surface of both subdomains; the shaded areas indicate surfaces of residues that differ between fX and trypsin. Core elements of the interface are structurally conserved. Surface patches of the interface are often deformed, especially at positions where nonconserved residues from both subdomains contact each other, such as the C-terminal helix and the calcium loop. These deviations involve both main chain and side chain geometry.
Inhibitor Binding.
The tripeptide inhibitor PPACK was bound to the active site formed at the subdomain interface (Fig. 5). PPACK interacted with the hybrid enzyme as a substrate analog as seen in other serine protease–PPACK complexes such as thrombin (30), coagulation factor IXa (fIXa; ref. 31), and protein C (32). P1-Arg was bound covalently to His57 and Ser195 with its side chain forming a salt bridge with Asp194 at the base of the S1-Pocket. The main chain of PPACK formed the “canonical” antiparallel β-sheet with the main chain residues Ser214, Trp215, and Gly216 located at the rim of the S1-pocket. The conformations of the P1-arginine and the P2-proline strongly resembled those found in the other serine protease–PPACK complexes. The side chain of P3-D-Phe, however, was found not to occupy the S3/S4 pocket as in thrombin and protein C but instead was rotated away from the active site in a conformation similar to that in PPACK-fIXa.
Figure 5.

Inhibitor binding. The active site of PPACK-rfXYa (colored sticks using the code of Fig. 2) superimposed with the active site of PPACK-thrombin (grey sticks; PDB ID code 1ppb). PPACK binds in a substrate-like binding mode: P1-arginine extends into the S1-pocket; the backbone forms a β-sheet with the enzyme backbone at positions 214–216; and P2-proline occupies the S2-pocket. Characteristic hydrogen bonds of the transition state–serine proteinase interaction are formed (dashed lines). The binding mode is identical to that observed in PPACK thrombin except for the orientation of the P3 sidechain: in thrombin, the phenyl moiety of P3 fits into the S3/S4 site whereas in rfXYa it is rotated toward the bulk solvent.
Amidolytic Activity and Specificity.
As an engineered protein, the hybrid molecule displayed a surprisingly high catalytic activity toward a variety of synthetic substrates (Table 1). In fact, its amidolytic activity was similar to that of rtrypsin and rfXa [and also to the respective native proteins (19, 33)], in accord with the well formed active site in the crystal structure.
Table 1.
Kinetic parameter for hydrolysis of several synthetic trypsin/fXa-substrates by rfXa, rfXYa, and rtrypsin*
| rfXa
|
rfXYa
|
rtrypsin
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| kcat, s−1 | Km μM | kcat/Km | kcat, s−1 | Km, μM | kcat/Km | kcat, s−1 | Km, μM | kcat/Km | |
| Pyro-Glu-Gly-Arg-pNA | 83 | 830 | 0.1 | 41 | 15 | 2.7 | 176 | 85 | 2.1 |
| Bz-Ile-Glu-Gly-Arg-pNA | 118 | 167 | 0.7 | 39 | 17 | 2.3 | 128 | 43 | 3.0 |
| MS-D-Phe-Gly-Arg-pNA | 138 | 275 | 0.5 | 44 | 15 | 2.9 | 145 | 30 | 4.8 |
| MOC-D-Nle-Gly-Arg-pNA | 215 | 199 | 1.1 | 52 | 22 | 2.4 | 153 | 43 | 3.6 |
| Bz-β-Ala-Gly-Arg-pNA | 66 | 134 | 0.5 | 68 | 49 | 1.4 | 225 | 208 | 1.1 |
| CB-Val-Gly-Arg-pNA | 91 | 525 | 0.2 | 36 | 13 | 2.7 | 95 | 32 | 3.0 |
| Bz-Pro-Phe-Arg-pNA | 53 | 265 | 0.2 | 119 | 115 | 1.0 | 38 | 17 | 2.2 |
| D-Val-CHA-Arg-pNA | 95 | 1480 | 0.1 | 91 | 68 | 1.3 | 46 | 129 | 0.4 |
| Tos-Gly-Pro-Arg-pNA | 107 | 149 | 0.7 | 39 | 23 | 1.7 | 95 | 13 | 7.3 |
| Tos-Gly-Pro-Lys-pNA | 44 | 540 | 0.06 | 55 | 67 | 0.8 | 26 | 56 | 0.5 |
The parameters of preferred substrates for each enzyme are emphasized in bold. Substrates were chosen according to commercial availability and variation of P1, P2, and P3.
The SE is 5–15%.
Bz, benzyl; CB, carbobenzoxy; CHA, cyclohexylalanyl; MOC, methoxycarbonyl; MS, methylsulfonyl; pNA, p-nitroanilin; Tos; tosyl.
rfXYa exhibited a molecular recognition profile similar to that of rtrypsin, with Km values more typical of rtrypsin than rfXa. This is presumably because of the concentration of substrate binding interactions predominantly by the trypsin subdomain. More specifically, the S1 subsite is formed by the C-terminal subdomain (trypsin part), whereas S2 and S3 are formed by both subdomains. However, substrates containing a large residue in P2 are hydrolyzed quite differently than by rtrypsin, presumably because of the contribution of the 99-loop (fXa subdomain) considered to be responsible for fXa’s preference for glycine residues in P2 (34). A plot of the transition state stabilization energy differences for all substrates revealed a considerably broadened substrate selectivity of rfXYa compared with rtrypsin and rfXa (Fig. 6). Although both rtrypsin and rfXa displayed ≈2 kcal/mol transition state stabilization difference between the most favorable and least favorable substrate tested, the chimera displayed <1 kcal/mol.
Figure 6.

Plot of the transition state stabilization free energy ΔG‡ = −RTln(kcat/Km/knon) (35) for hydrolysis of substrates listed in Table 1 by rfXa (•), rfXYa (■), and rtrypsin (▴). The rate of the noncatalyzed reaction knon is independent of the enzyme used and was set to 1 for the purpose of the comparison. The SEs are shown as bars. To indicate selectivity, the substrates are sorted according the increasing ΔG‡ values for each of the enzymes separately.
DISCUSSION
In an alternate subdomain swapping approach that attempted also to mix different enzymatic reactions, Nixon et al. (10) succeeded in supplementing a hydrolase activity with some transferase activity by introducing a new subdomain linked substrate binding site. However, the combination of subdomains catalyzing similar but different reactions lead to an overall loss of catalytic efficiency by a factor of ≈10−4. Our more conservative approach, combining subdomains from enzymes catalyzing the same reaction but with different substrate specificity profiles, led to no significant loss of activity. This might be considered surprising because the catalytic machinery and the substrate binding cleft are formed with equal contributions from each subdomain. As established by the crystal structure and the kinetic analysis, the homologous but diversified subdomains from trypsin and fXa, when recombined, not only retain their subdomain specific features but also join to form a highly active proteolytic complex with novel features.
The relative preservation of structures in rfXYa compared with the parent molecules resembles the pattern of structural variability and conservation within the chymotrypsin family (12), that is, core elements remain conserved while surface elements are variable, with hypervariability at domain interface regions. This is reflected also in the kinetic data of rfXYa: Specific features of rtrypsin and rfXa substrate specificities are recognizable, whereas overall specificity is broadened. In general, the hybrid specificity seems derived from three structural elements: the N-terminal subdomain 99-loop contributes a fXa-like preference for small P2 residues; the C-terminal subdomain S1 site and parts of P3/P4 have overall trypsin-like substrate affinities; and, loop rearrangements in the intersubdomain boundary lead to a broadened specificity (Fig. 4).
Experiments with variants of chymotrypsin (36) and unrelated proteins (37) have demonstrated the high sensitivity of enzyme specificity and activity to seemingly minor or distant mutations. In the case of fXYa, the functional origin of the broad substrate specificity seems to be the compensation of high Km values (an estimate of the substrate binding for amide substrates) by high kcat values (an estimate of the acylation rate for amide substrates). By comparison, lower Km values with rfXa and rtrypsin correlate with higher kcat values. This indicates that rtrypsin and rfXa bind favorable substrates (those with low Km values) in a form susceptible for cleavage, while rfXYa binds favorable substrates in a form less susceptible for cleavage. Taking the tripeptide inhibitor PPACK as a model for such a transition state (38), the structure shows that the PPACK P1–arginine and P2–proline bind the hybrid with conformations and hydrogen bonds as observed for other serine proteinases (30–32). The conformation of P3-d-phenylalanine, however, resembled that of PPACK–fIXa in that the phenyl moiety is not inserted into the S3/S4 but rather points toward solvent (Fig. 5). This binding mode disables specific P3 recognition by S3/S4, reflecting the broad P3 specificity of the hybrid. This feature is a consequence of the subdomain contact at S3/S4, formed by the 170 loop and 99 loop, which is more restricted than in the parent enzymes. Good fXa and trypsin substrates, those with large hydrophobic D-amino acids in P3 (Table 1), are consequently less favorable, flattening the specificity profile. In addition, substrate interactions in S3 also were shown to influence the S1 specificity (36). Although all hydrogen bonds characteristic for the family are formed in PPACK–fXYa (Fig. 5), small distortions caused by unfavorable P3 recognition can broaden the S1 specificity by influencing the loop formed by residues 216–220 (39).
The hybrid of two serine proteinase halves (subdomains), chosen from two divergent members of the S1 chymotrypsin family with an overall sequence identity of roughly 40%, demonstrates what might be generally expected from subdomain hybrids: Subdomain-linked properties as well as general enzyme family properties are preserved, whereas novel properties associated with the periphery of the subunit interface arise. Although the preservation of properties enables rational engineering of some functional characteristics, the unpredictable generation of new properties is simultaneously prohibitive and fortuitous for design. Opportunity lies in the aptitude of these properties for subsequent optimization, for example by directed evolution methods. This supports our expectation that the generation of new subfamily “lineages” is likely to be of general applicability to protein design problems.
Acknowledgments
We thank Dr. Hans Brandstetter and Dr. Martin Renatus for stimulating discussions and Dr. Natascha Rieder and Annette Karcher for help with cloning and expression of proteins. K.P.H. is grateful for a stipend from the Studienstiftung des deutschen Volkes e.V. (German Scholar Foundation).
ABBREVIATIONS
- fX
coagulation factor Xa
- PPACK
D-Phe-Pro-Arg-chloromethylketone
- rfX
recombinant desGla-desEGF1 factor X
- rfXY
fX(16–121)/trypsin(122–246)-hybrid
- rtrypsin
recombinant human trypsin
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (PDB ID code 1fxy).
References
- 1.Bryson J W, Betz S F, Lu H S, Suich D J, Zhou H X, O’Neil K T, DeGrado W F. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 2.Lesk A M, Rose D R. Proc Natl Acad Sci USA. 1981;78:4304–4308. doi: 10.1073/pnas.78.7.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brinkmann U. Mol Med Today. 1996;2:439–446. doi: 10.1016/1357-4310(96)84848-9. [DOI] [PubMed] [Google Scholar]
- 4.Chang J Y, Stafford D W, Straight D L. Biochemistry. 1995;34:12227–12232. doi: 10.1021/bi00038a017. [DOI] [PubMed] [Google Scholar]
- 5.Neurath H. Science. 1984;224:350–357. doi: 10.1126/science.6369538. [DOI] [PubMed] [Google Scholar]
- 6.McLachlan A D. Cold Spring Harbor Symp Quant Biol. 1987;52:411–420. doi: 10.1101/sqb.1987.052.01.048. [DOI] [PubMed] [Google Scholar]
- 7.Heringa J, Taylor W R. Curr Opin Struct Biol. 1997;7:416–421. doi: 10.1016/s0959-440x(97)80060-7. [DOI] [PubMed] [Google Scholar]
- 8.Doolittle R F, Bork P. Sci Am. 1993;269:50–56. doi: 10.1038/scientificamerican1093-50. [DOI] [PubMed] [Google Scholar]
- 9.Liepinsh E, Kitamura M, Murakami T, Nakaya T, Otting G. Nat Struct Biol. 1997;4:975–979. doi: 10.1038/nsb1297-975. [DOI] [PubMed] [Google Scholar]
- 10.Nixon A E, Warren M S, Benkovic S J. Proc Natl Acad Sci USA. 1997;94:1069–1073. doi: 10.1073/pnas.94.4.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rawlings N D, Barrett A J. Methods Enzymol. 1994;244:19–61. doi: 10.1016/0076-6879(94)44004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lesk A M, Fordham W D. J Mol Biol. 1996;258:501–537. doi: 10.1006/jmbi.1996.0264. [DOI] [PubMed] [Google Scholar]
- 13.Perona J J, Craik C S. Prot Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banner D W, D’Arcy A, Chene C, Winkler F K, Guha A, Königsberg W H, Nemerson Y, Kirchhofer D. Nature (London) 1996;380:41–46. doi: 10.1038/380041a0. [DOI] [PubMed] [Google Scholar]
- 15.Dang Q D, Di Cera E. Proc Natl Acad Sci USA. 1996;93:10653–10656. doi: 10.1073/pnas.93.20.10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davie E W, Fujikawa K, Kisiel W. Biochemistry. 1991;30:10363–10370. doi: 10.1021/bi00107a001. [DOI] [PubMed] [Google Scholar]
- 17.Patthy L. Fibrinolysis. 1990;1:153–166. [PubMed] [Google Scholar]
- 18.Goldberger G, Bruns G A P, Rits M, Edge M D, Kwiatkowski D J. J Biol Chem. 1987;262:10065–10071. [PubMed] [Google Scholar]
- 19.Hopfner K-P, Brandstetter H, Karcher A, Kopetzki E, Huber R, Engh R A, Bode W. EMBO J. 1997;16:6626–6635. doi: 10.1093/emboj/16.22.6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnaswamy S, Church W S, Nesheim M E, Mann K G. J Biol Chem. 1987;262:3291–3299. [PubMed] [Google Scholar]
- 21.Fiedler F, Seemüller U, Fritz H. In: Methods of Enzymatic Analysis. Bergmeyer H, editor. Weinheim, Germany: Verlag Chemie GmbH; 1984. [Google Scholar]
- 22.Leslie A G W. Mosflm User Guide, Mosflm Version 5.1. Cambridge, U.K.: MRC Laboratory of Molecular Biology; 1994. [Google Scholar]
- 23.Collaborative Computational Project, No. 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 24.Padmanabhan K, Padmanabhan K P, Tulinsky A, Park C H, Bode W, Huber R, Blankenship D T, Cardin A D, Kisiel W. J Mol Biol. 1993;232:947–966. doi: 10.1006/jmbi.1993.1441. [DOI] [PubMed] [Google Scholar]
- 25.Gaboriaud C, Serre L, Guy-Crotte O, Forest E, Fontecilla-Camps J C. J Mol Biol. 1996;259:995–1010. doi: 10.1006/jmbi.1996.0376. [DOI] [PubMed] [Google Scholar]
- 26.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 27.Brünger A T. x-plor, Version 3.1. A System for X-Ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. [Google Scholar]
- 28.Turk D. Ph.D. thesis. München: Technische Universität; 1992. [Google Scholar]
- 29.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 30.Bode W, Mayr I, Baumann U, Huber R, Stone S R, Hofsteenge J. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brandstetter H, Bauer M, Huber R, Lollar P, Bode W. Proc Natl Acad Sci USA. 1995;92:9796–9800. doi: 10.1073/pnas.92.21.9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mather T, Oganessyan V, Hof P, Huber R, Foundling S, Esmon C, Bode W. EMBO J. 1996;15:6822–6831. [PMC free article] [PubMed] [Google Scholar]
- 33.Lottenberg R, Christensen U, Jackson C M, Coleman P L. Methods Enzymol. 1981;80:341–361. doi: 10.1016/s0076-6879(81)80030-4. [DOI] [PubMed] [Google Scholar]
- 34.Brandstetter H, Kühne A, Bode W, Huber R, von der Saal W, Wirthensohn K, Engh R. J Biol Chem. 1996;271:29988–29992. doi: 10.1074/jbc.271.47.29988. [DOI] [PubMed] [Google Scholar]
- 35.Cannon W R, Singleton S F, Benkovich S J. Nat Struct Biol. 1996;3:821–833. doi: 10.1038/nsb1096-821. [DOI] [PubMed] [Google Scholar]
- 36.Hedstrom L, Szilagyi L, Rutter W J. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 37.Mesecar A D, Stoddard B L, Koshland D E., Jr Science. 1997;277:202–206. doi: 10.1126/science.277.5323.202. [DOI] [PubMed] [Google Scholar]
- 38.Tsilikounas E, Rao T, Gutheil W G, Bachovchin W W. Biochemistry. 1996;35:2437–2444. doi: 10.1021/bi9513968. [DOI] [PubMed] [Google Scholar]
- 39.Perona J J, Craik C S. J Biol Chem. 1997;272:29987–29990. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]