

Abstract
Cooperative interactions link the behavior of different amino acid residues within a protein molecule. As a result, the effects of chemical or physical perturbations to any given residue are propagated to other residues by an intricate network of interactions. Very often, amino acids “sense” the effects of perturbations occurring at very distant locations in the protein molecule. In these studies, we have investigated by computer simulation the structural distribution of those interactions. We show here that cooperative interactions are not intrinsically bi-directional and that different residues play different roles within the intricate network of interactions existing in a protein. The effect of a perturbation to residue j on residue k is not necessarily equal to the effect of the same perturbation to residue k on residue j. In this paper, we introduce a computer algorithm aimed at mapping the network of cooperative interactions within a protein. This algorithm exhaustively performs single site thermodynamic mutations to each residue in the protein and examines the effects of those mutations on the distribution of conformational states. The algorithm has been applied to three different proteins (λ repressor fragment 6–85, chymotrypsin inhibitor 2, and barnase). This algorithm accounts well for the observed behavior of these proteins.
Protein folding is a highly cooperative process. One of the most notable manifestations of cooperativity is that the vast majority of conformational states that are accessible to a protein have a negligible probability and never become populated to a significant extent. In most situations, the folding/unfolding equilibrium is well accounted for by a two-state process in which the population of intermediates is assumed to be zero (1). Despite this fact, there is ample evidence, particularly from hydrogen exchange data obtained under native conditions, that some partially folded conformations are always present. The observed heterogeneity in the magnitude of the hydrogen exchange protection factors measured under native conditions indicates that certain residues become exposed to the solvent as a result of local rather than global unfolding (2–13). If this is the case, cooperative interactions do not involve the entire protein molecule, and the conformational equilibrium cannot be considered as an all-or-none process in which the entire protein is either folded or unfolded. If cooperative interactions do not extend uniformly throughout the entire protein molecule, then some residues will have a more important role than others in defining cooperativity. The purpose of this paper is to identify those residues and investigate the structural distribution of cooperative interactions in proteins.
From a rigorous point of view, cooperativity originates when the partition function of a system cannot be written as the product of the individual partition functions of the constituent subsystems. This situation occurs when the interaction energy among different subsystems is not zero. In proteins, different structural elements interact with one another, establishing a hierarchical web that essentially extends throughout the entire protein. As a result, the Gibbs energy of each residue becomes a composite function of this intricate network of interactions. Deciphering this network from an analytical point of view is an enormous, if not hopeless, task. An alternative approach is to use large scale computer simulations.
To investigate the way in which cooperative interactions propagate in a protein, one ideally would set up an experiment in which the intrinsic Gibbs energy of each residue is changed one at a time, and the effects of each change on all other residues are examined. In this ideal experiment, only the energy and not the chemical nature or atomic dimensions of each residue is changed so that no structural perturbations are introduced. In the real world, this ideal experiment cannot be realized. At best, it only can be approximated by performing Ala↔Gly mutations at solvent exposed locations. With the computer, however, the energy itself can be “mutated” without the structure or the amino acid sequence of the protein being affected. We call this technique single site thermodynamic mutation (SSTM), and we show here that it can be used to identify and characterize a number of fundamental aspects of cooperativity in proteins.
MATERIALS AND METHODS
Uniformly labeled 15N λ6–85 was expressed and purified as described (14). Two NMR samples were prepared, one at pH 5.00 and one at pH 6.98. All pH values reported here have not been corrected for isotope effects regardless of deuterium content. Amide hydrogen exchange rates were determined as described (14) with the following exception: Instead of 10 mM CD3COOD, the pH 5.00 exchange buffer contained 1 mM EDTA and 20 mM CD3COOD, and the pH 6.98 buffer contained 1 mM EDTA and 20 mM D3PO4. Final protein concentrations were ≈1 mM.
HSQC spectra were acquired on a Varian 600-MHz NMR spectrometer set at 15°C ± 0.5°C. The time between initiation of exchange and collection of the first data point was 15 min at pH 6.98 and 5 min at pH 5.00. Each 2D spectrum consisted of 4,096 data points in the 1H dimension, covering a sweep width of 8,000 Hz, and 128 points in the 15N dimension, over a sweep width of 1,350 Hz. Five initial spectra were taken at intervals of 16 minutes with 2 transients each, and all subsequent spectra consisted of 16 or 32 transients taken at minimum intervals of 2 hr. Only those peaks with heights above baseline for the first five spectra were used to calculate exchange rates. Twenty to thirty spectra were used in each experiment. Peak heights were adjusted according to the number of transients in a given spectrum. Data were processed by using a combination of nmrpipe (National Institutes of Health), felix (Biosym Technologies, Sand Diego), and kaleidagraph (Synergy Software, Reading, PA) software. Amide exchange rates were calculated by fitting peak heights versus time in kaleidagraph. Intrinsic rate constants were calculated by using the method of Bai et al. (15). All rates slower than 5 × 106 (22 residues) were determined by using the pH 6.98 data, and the remainder (33 residues) were calculated with the pH 5.00 data.
Computer Simulation of the Equilibrium Ensemble of Protein Conformations.
Previously, the corex algorithm, which generates a large number of partially folded states of a protein from the high resolution crystallographic or NMR structure, was introduced (12, 13, 16). In this algorithm, the ensemble of partially folded states of a protein is approximated with the computer by using the high resolution structure as a template. Within this framework, the entire protein is considered as being composed of different folding units. Partially folded states are generated by folding and unfolding these units in all possible combinations.
The division of the protein into a given number of folding units is called a partition. To maximize the number of distinct partially folded states, different partitions are included in the analysis. Each partition is defined by placing a block of windows over the entire sequence of the protein. The folding units are defined by the location of the windows irrespective of whether they coincide with specific secondary structure elements. By sliding the entire block of windows one residue at a time, different partitions of the protein are obtained. For two consecutive partitions, the first and last amino acids of each folding unit are shifted by one residue. This procedure is repeated until the entire set of partitions have been exhausted (see ref. 12 for details). Typically, on the order of 105 partially folded conformations are generated with the corex algorithm. For the proteins λ6–85, chymotrypsin inhibitor 2 (CI2), and barnase considered in this paper, windows of 5, 5, and 8 amino acid residues were used, resulting in 2.6 × 105, 0.4 × 105, and 1.1 × 105 partially folded conformations, respectively.
Each of the states generated by the corex algorithm is characterized by having some regions folded and some other regions unfolded. There are two basic assumptions in this algorithm: (i) The folded regions in partially folded states are native-like; and (ii) the unfolded regions are assumed to be devoid of structure. The thermodynamic quantities (ΔH, ΔS, ΔCp, and ΔG) for each state as well as the partition function and probability of each state (Pi) are evaluated by using an empirical parameterization of the energetics (17–22).
The resultant distribution of states can be used to estimate an important descriptor of the residue-specific equilibrium, the residue stability constant (κf). This quantity is the ratio of the probabilities of all states in which a residue (j) is in a folded conformation to the probabilities of all states in which that residue is in an unfolded conformation and can be expressed as:
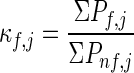 |
1 |
It has been shown, through the analysis of various protein structures, that residue specific equilibria calculated according to Eq. 1 provide quantitative agreement with those obtained experimentally from amide hydrogen exchange experiments (i.e., protection factors) (12, 13, 16). The reasonable prediction of hydrogen exchange protection factors indicates that this approach effectively captures (albeit implicitly) cooperative interactions within the protein and correctly reproduces the most probable distribution of partially folded conformations. If this is the case, the derived ensemble of partially folded conformations can be used to construct a structural map of the cooperative network within the protein.
Mapping Cooperativity: SSTM.
Within the context of the statistical approach described above, cooperativity can be examined by changing the free energy of all states in which a particular residue is folded, in essence performing a nonperturbing energy mutation of that residue. The resultant change in the statistical weight of all states in the numerator of Eq. 1 leads to a redistribution of the probabilities. As the subset of states in which a particular residue is folded (and unfolded) differs for each residue, the effect of a thermodynamic mutation will be specific for each residue in the protein. By performing individual thermodynamic mutations to each residue in the protein, it is possible to evaluate the effect of a change in each residue on all other residues. The end result of the SSTM analysis is a map from which the cooperative network of interactions within the protein can be deduced. We illustrate this analysis with the protein lambda 6–85 (λ6–85), a fragment of the lambda repressor, which contains residues 6 to 85 and which folds into a single domain (23). This protein has been well characterized from both a structural and an energetic point of view (24). The pattern of hydrogen exchange protection is predicted well by the corex algorithm as illustrated in Fig. 1. The agreement in the pattern and amplitude between predicted and experimental protection factors indicates that the algorithm correctly captures the interaction energies within λ6–85. The most significant discrepancy is with Arg 17, which, in the x-ray structure, forms a salt bridge with Asp 14 and is predicted to have a higher than observed protection. Overall, the average deviation between predicted and experimental values amounts to ±0.9 kilocalories (kcal)/mol. The experimental protection factors show variations in magnitude that reflect the existence of partially folded conformations that are within 3 kcal/mol or less from the native state. The existence of this “fine structure” in the pattern of hydrogen exchange protection defines the native state as a dynamically fluctuating subensemble of conformations.
Figure 1.

Natural logarithm (bars) of the calculated and experimental protection factors for λ6–85. The calculated values were determined as described by Hilser and Freire (12). The solid line above the calculated values represents the residue stability constant as defined from Eq. 1. This quantity is defined for all residues independently of whether they exhibit protection or not. Shown also in the figure are the corresponding elements of secondary structure. The good agreement between calculated and experimental values indicates that the calculated ensemble captures the general features of the actual ensemble and that the network of cooperative interactions in the protein are represented accurately in this model.
Shown in Fig. 2A is the SSTM analysis of λ6–85. In this representation, the energy-mutated residue lies on the abscissa, the affected residue lies on the ordinate, and the color coding signifies the fractional effect of the mutation on that residue. Blue is seen in cases in which a mutation to the residue on the abscissa does not affect the residue on the ordinate whereas red corresponds to a large effect. The most striking and important feature of the pattern seen in Fig. 2A is that it is not symmetrical. In other words, the effect of a perturbation at residue j on residue k is not necessarily equal to the effect of the same perturbation at residue k on residue j. This result suggests that cooperativity is not intrinsically bi-directional.
Figure 2.

SSTM analysis of λ6–85 at (A) ΔGU = 8.5 kcal/mol and (B) ΔGU = 2.5 kcal/mol. Plotted is ΔΔGj,mutk (ΔGjmutk−ΔGjWT; where ΔGj = −RT ln κf,j), which represents the change in free energy at each residue j (ordinate) caused by a mutation at residue k (abscissa). For these calculations, the free energy of all states in which residue k is folded is stabilized by 1.0 kcal/mol. Red corresponds to a large effect (1.0 kcal/mol) whereas blue corresponds to a small effect (≈0.0 kcal/mol).
The directionality of cooperative behavior seen in Fig. 2A can be analyzed by considering three different effects: (i) cooperative response (i.e., the response of residue j to a mutation anywhere in the protein); (ii) donor cooperativity (i.e., the effect of a mutation in residue j on other residues); and (iii) mutual perturbation/response (i.e., the product of the effect of a mutation to residue j on k and the effect of a mutation to residue k on j, normalized by the magnitude of the perturbations;
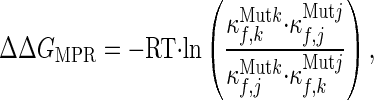 |
2 |
where ΔΔGMPR is defined as the mutual perturbation response free energy, κf,jMutk and κf,kMutj are the stability constants of residue j and k on mutation of residues k and j, and κf,jMutj and κf,kMutk are the stability constants of residues j and k on mutation of residues j and k, respectively).
Cooperative Response.
Examination of Fig. 2A reveals some general features in the pattern of protein cooperativity. The first feature is the presence of a diagonal that runs from the lower left to the upper right. This diagonal represents the trivial effect of mutating each residue on itself and the residues that are near neighbors in sequence. The second and most significant feature is that the effects of mutations do not extend to all residues in the protein; however, there is a subset of residues (in the case of λ6–85, residues 12–18 and 63–70 seen as horizontal red lines in Fig. 2A) that always are affected independently of the location of the perturbation. These residues are the most stable residues in the protein. The origin of this behavior can be explained by separating the contributions of partially folded states from those of the folded and unfolded states in Eq. 1:
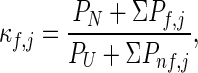 |
3A |
where the summation in the numerator includes all partially folded states in which residue j is folded, and the summation in the denominator includes all partially folded states in which residue j is not folded. In general, the residues with the highest stability constants belong to the folded regions of the most probable partially folded conformations. For those residues, ΣPf,j ≫ ΣPnf,j and also PU ≫ ΣPnf,j, and Eq. 3A essentially reduces to (PN + ΣPf,j)/PU. For these residues, the stability constants are larger in magnitude than the global unfolding constant (PN/PU); i.e., they exhibit “super-protection” as observed experimentally (11).
To illustrate the effect of a mutation to any arbitrary residue, k, on the stability of residue j, the summations in Eq. 3A can be subdivided further so as to include separately those states in which residues j and k are either folded and unfolded together or individually:
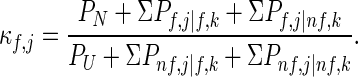 |
3B |
Upon mutation of residue k (i.e., changing the free energy of all states in which residue k is folded) by an amount, Δgf,k (=−RT⋅ln Φf,k), Eq. 3B becomes
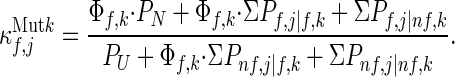 |
3C |
For the situations considered here and most situations found in the laboratory, Δgf,k is usually <2 kcal/mol, which is equivalent to a Φf,k factor as high as 30. Under those conditions, the stability constants of the most stable residues will be affected by mutations occurring anywhere in the protein because the effect of Φf,k will be seen primarily in the numerator. This is the case for residues 12–18 and 63–70 in λ6–85, as seen in Fig. 2A.
The least stable residues, on the other hand, have a relatively high probability of being unfolded in states other than the unfolded state. For these residues, ΣPnf,j ≫ PU, and Φf,k in the numerator and denominator will cancel, leaving the stability constant unaffected. In general, the least stable residues are the least affected by mutations anywhere in the protein.
From a mathematical point of view the situation can be generalized as follows: The magnitude of the cooperative response of any given residue in the protein is determined by the ratio (PU + ΣPnf,j|nf,k)/ΣPnf,j|f,k. In other words, and using the terminology of Englander and coworkers (9), under native conditions, cooperativity will be reflected in the ratio of the probability of global unfolding to the probability of local unfolding. The higher this ratio, the larger the cooperative effect. For this reason, cooperative effects increase under increasing denaturing conditions and are expected to be maximal at the transition midpoint.
Donor Cooperativity.
Unlike the cooperative response discussed in the previous section, Fig. 2A also shows that no single residue is able to affect all other residues in the protein. Mathematically, this observation is due to the fact that residues for which ΣPnf,j ≫ PU will only be affected by mutations when a large penalty exists for unfolding one residue without unfolding the other (i.e., ΣPnf,j|nf,k ≫ ΣPnf,j|f,k). Thus, only in the case in which the stability of residue k is completely coupled to every other residue in the protein will that residue be able to affect all other residues. As the complete coupling of one residue to all other residues implies a complete coupling between all residues, such a situation exists only for the case of a true two-state transition.
Mutual Perturbation/Response.
Analysis of the mutual perturbation/response of a protein according to Eq. 2 provides a qualitative picture of the bi-directionality of cooperative effects. Residues with high ΔΔGMPR values represent cases whereby mutations of residue j significantly perturb residue k, and mutations of k significantly perturb residue j. On the other hand, for low ΔΔGMPR values, mutations to j or k have less effect on the other residue. The mutual perturbation/response for λ6–85 is shown in Fig. 3 and reveals that, under conditions used to generate Fig. 2A, the highest values for ΔΔGMPR also correspond to residues 12–18 and 62–70. This result, coupled with the fact that residues 12–18 and 62–70 are affected by mutations to any residue in the protein, indicates that these residues correspond to the cooperative core of the protein. In other words, in the states with the highest probabilities, these residues are either all folded or all unfolded. Indeed, inspection of Fig. 4A reveals that these residues, while being distal in sequence, represent a contiguous cluster of structure in the core of the protein.
Figure 3.

Mutual perturbation/response analysis of λ6–85 at conditions used to generate Fig. 2A (i.e., ΔGU = 8.5 kcal/mol). Plotted is ΔΔGMPR (Eq. 2) for each pair of residues. Red corresponds to a large effect (1.0 kcal/mol) whereas blue corresponds to a small effect (0.0 kcal/mol).
Figure 4.

Ribbon diagrams of λ6–85 showing residues (colored red) that correspond to the cooperative core at (A) ΔGU = 8.5 kcal/mol and (B) ΔGU = 2.5 kcal/mol. Although distal in sequence, core residues represent a contiguous cluster in the three dimensional structure. This figure was generated by using the program molscript (32).
The extent of the cooperative core, as identified above, is a function of the stability of the protein. Under strongly native conditions (i.e., conditions used to generate Fig. 2A; ΔGU = 8.5 kcal/mol), the cooperative core includes only a relatively small fraction of the total number of residues as seen in Fig. 4A. Eq. 3 indicates that, as the stability of the protein is decreased (i.e., PU is increased), more residues will begin to have their denominators dominated by PU. The end result is a cooperative core that increases in size as the stability of the protein decreases. Fig. 2B shows the SSTM analysis of λ6–85 under more destabilizing conditions than those used to generate Fig. 2A (i.e., ΔGU = 2.5 kcal/mol). Evident from this analysis is the increase in the number of residues that are affected by a mutation to any residue in the protein. Likewise, inspection of Fig. 4B reveals that these residues represent a contiguous cluster in the three dimensional structure. These results are consistent with what has been observed experimentally in a number of cases (7, 9, 11, 25–27). Namely, when amide hydrogen exchange experiments are performed on a protein as a function of temperature, pH, or denaturant, the protection factors for the various residues converge to a common value as the stability of the protein is decreased. In fact, the formalism presented here predicts just such a behavior, as demonstrated (12, 13, 16, 27).
Analysis of CI2.
CI2 is a 64-residue protein that folds in a two-state fashion into a single structural domain (28). As seen in Fig. 5A, CI2 consists of a central β-sheet that is sandwiched between an α-helix on one side and the reactive site loop on the other. An interesting feature is evident in the mutual perturbation/response of CI2 (Eq. 2) as shown in Fig. 5B. Namely, the general pattern suggests that residues in the reactive site loop (residues 53–62) behave in a mutually cooperative manner and are influenced much less by residues outside of the loop. Similarly, residues outside the reactive site affect each other to a greater extent than they affect residues in the reactive site. Further, Arg 67 and Leu 68 (highlighted in Fig. 5A) show relatively high mutual perturbation/response with all residues in the protein. Thus, this analysis suggests that, although being a single domain protein, CI2 can be viewed as consisting of two substructures, the structural core of the protein that contains the β-sheet and the α-helix, and the reactive site loop. Within this context, residues 67 and 68 represent the key linchpin residues, energetically linking the two substructures. In other words, the effects of perturbations in either substructure are propagated to the other mainly through residues 67 and 68. Consistent with these results, residue 68 has been implicated as being a major contributor to the structural stability of CI2 (29) and is among the most conserved residues in chymotrypsin inhibitors.
Figure 5.

(A) Ribbon diagram of CI2 showing the binding loop (residues 53–62) highlighted in yellow. Side chains for the key linchpin residues R67 and L68 are shown. Both residues are part of the central β-strand; R67 projects into the binding loop, and L68 projects into the hydrophobic core formed by the β-sheet and the α-helix. This figure was generated by using molscript. (B) Mutual perturbation/response analysis of CI2 under native conditions. Plotted is ΔΔGMPR (Eq. 2) for each pair of residues, highlighting the difference between large (red), intermediate (blue), and small (purple) effects. Labeled are those residues belonging to the binding loop.
Analysis of Barnase.
Barnase is a 110-residue ribonuclease that folds into the structure shown in Fig. 6A (30). The native structure of barnase has three helices and a five-stranded β-sheet and is characterized by a deep active site cleft that divides a portion of the protein into two lobes. Inspection of the mutual perturbation/response for barnase (Eq. 2) as shown in Fig. 6B reveals that the contiguous residues 23–51 behave in a mutually cooperative manner with themselves and with residues 74–82 and are influenced less by other residues. A similar situation exists for residues 50–74. The significant feature of this observation is that these two groups of residues correspond to the two lobe structures described above as shown in Fig. 6A. Further, as was the case with CI2, key cooperative residues exist in barnase (i.e., Arg 87, Ile 88, Leu 89, Tyr 90, and Ser 91) that show relatively high mutual perturbation/response with all residues in the protein. Also similar to CI2, these linchpin residues occupy a centralized location within the structure of the protein. Thus, this analysis suggests that barnase, while being a single domain protein, can be viewed as consisting of a core structure with two lobes on either side of the binding cleft. Under strongly native conditions and in the absence of substrate, these lobe structures behave in a relatively independent fashion from each other and from the rest of the barnase structure. Residues 87–91 represent the key linchpin residues in this protein, energetically linking the two lobe structures with each other and with the remainder of the molecule.
Figure 6.

(A) Ribbon diagram of barnase showing the active site cleft. Shown in blue are residues comprising lobe A (residues 23–51 and 74–82). Shown in yellow are residues comprising lobe B (residues 50–73). The linchpin residues R87-S91 are part of one of the central β strands and are shown in red. This figure was generated by using molscript. (B)Mutual perturbation/response analysis of barnase under native conditions. Plotted is ΔΔGMPR (Eq. 2) for each pair of residues. Red corresponds to a large effect, purple corresponds to a small effect, and blue corresponds to an intermediate effect. Labeled are those residues belonging to lobe A (23–51 and 74–82) and lobe B (50–73).
CONCLUSIONS
The analysis presented here suggests that a recently developed statistical approach to macromolecular equilibrium can be used to probe cooperativity. The intricate array of interactions in proteins can be dissected by analyzing the way in which single-site “energy mutations” are manifested. As a result of this analysis, a number of important properties can be identified:
The effect of a change at residue j on residue k is not necessarily equal to the effect of the same change at residue k on residue j. Thus, cooperativity in proteins is not intrinsically bi-directional.
The number of residues involved in the cooperative core of the protein is minimal under conditions of highest stability. In general, cooperative effects in proteins increase as the population of the unfolded state increases and are maximal at the midpoint of the folding/unfolding transition. It is known that, under native conditions, protection factors in proteins differ significantly from each other; however, they converge to a common value under increasing denaturing conditions, thus supporting this finding.
Only in a true two-state transition will a mutation in one residue affect all other residues. Thus, the observed residue-specific variation in response to mutations provides unequivocal evidence for the non-two-state nature of the equilibrium under native conditions.
The results presented here and those previously obtained from the analysis of hydrogen exchange protection factors (12, 13, 16) provide insights into the nature of the native state and rationalize previous observations (6, 7, 9, 31). The native state can be considered as a statistical subensemble of conformations that originate from the existence of local unfolding transitions throughout most of the protein molecule. These local transitions essentially are uncorrelated with one another, demonstrating the absence of long range cooperativity. Within such a system, the effects of perturbations to individual residues are propagated only minimally to other residues. The only residues that do show significant cooperativity are those for which the probability of local unfolding is lower than the probability of global unfolding (i.e., for those in which PU dominates the denominator of Eq. 3C). Such residues define the cooperative core of the protein.
From the point of view of protein folding, it is evident that the extent of cooperative interactions is maximal under conditions close to the midpoint of the unfolding transition, as reflected in the pattern of hydrogen protection factors of all proteins that have been studied at various stabilities (7, 9, 11, 25, 26). The observation that the dimensions of the cooperative core are minimal under native conditions is indicative of the energetic independence of the various substructures in the protein, a condition that may facilitate the occurrence of local conformational changes necessary to function.
Acknowledgments
This work was supported by grants from the National Institutes of Health (Grants GM51362 and GM57144).
ABBREVIATIONS
- SSTM
single site thermodynamic mutation
- CI2
chymotrypsin inhibitor 2
- kcal
kilocalorie
References
- 1.Lumry R, Biltonen R, Brandts J F. Biopolymers. 1966;4:917–944. doi: 10.1002/bip.1966.360040808. [DOI] [PubMed] [Google Scholar]
- 2.Clarke J, Fersht A R. Fold Des. 1996;1:243–254. doi: 10.1016/S1359-0278(96)00038-7. [DOI] [PubMed] [Google Scholar]
- 3.Jeng M-F, Englander S W. J Mol Biol. 1991;221:1045–1061. doi: 10.1016/0022-2836(91)80191-v. [DOI] [PubMed] [Google Scholar]
- 4.Radford S E, Buck M, Topping K D, Dobson C M, Evans P A. Proteins. 1992;14:237–248. doi: 10.1002/prot.340140210. [DOI] [PubMed] [Google Scholar]
- 5.Loh S N, Prehoda K E, Wang J, Markley J L. Biochemistry. 1993;32:11022–11028. doi: 10.1021/bi00092a011. [DOI] [PubMed] [Google Scholar]
- 6.Kim K-S, Fuchs J A, Woodward C K. Biochemistry. 1993;32:9600–9608. doi: 10.1021/bi00088a012. [DOI] [PubMed] [Google Scholar]
- 7.Kim K-S, Woodward C. Biochemistry. 1993;32:9609–9613. doi: 10.1021/bi00088a013. [DOI] [PubMed] [Google Scholar]
- 8.Woodward C. Trends Biochem Sci. 1993;18:359–360. doi: 10.1016/0968-0004(93)90086-3. [DOI] [PubMed] [Google Scholar]
- 9.Bai Y, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulman B A, Redfield C, Peng Z, Dobson C M, Kim P S. J Mol Biol. 1995;253:651–657. doi: 10.1006/jmbi.1995.0579. [DOI] [PubMed] [Google Scholar]
- 11.Swint-Kruse L, Robertson A D. Biochemistry. 1996;35:171–180. doi: 10.1021/bi9517603. [DOI] [PubMed] [Google Scholar]
- 12.Hilser V J, Freire E. J Mol Biol. 1996;262:756–772. doi: 10.1006/jmbi.1996.0550. [DOI] [PubMed] [Google Scholar]
- 13.Hilser V J, Freire E. Proteins. 1997;27:171–183. doi: 10.1002/(sici)1097-0134(199702)27:2<171::aid-prot3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 14.Huang G S, Oas T G. Biochemistry. 1995;34:3884–3892. doi: 10.1021/bi00012a003. [DOI] [PubMed] [Google Scholar]
- 15.Bai Y, Milne J S, Mayne L, Englander S W. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilser V J, Townsend B D, Freire E. Biophys Chem. 1997;64:69–79. doi: 10.1016/s0301-4622(96)02220-x. [DOI] [PubMed] [Google Scholar]
- 17.Murphy K P, Freire E. Adv Protein Chem. 1992;43:313–361. doi: 10.1016/s0065-3233(08)60556-2. [DOI] [PubMed] [Google Scholar]
- 18.Gomez J, Hilser J V, Xie D, Freire E. Proteins Struct Funct Genet. 1995;22:404–412. doi: 10.1002/prot.340220410. [DOI] [PubMed] [Google Scholar]
- 19.Hilser V J, Gomez J, Freire E. Proteins. 1996;26:123–133. doi: 10.1002/(SICI)1097-0134(199610)26:2<123::AID-PROT2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 20.Lee K H, Xie D, Freire E, Amzel L M. Proteins Struct Funci Genet. 1994;20:68–84. doi: 10.1002/prot.340200108. [DOI] [PubMed] [Google Scholar]
- 21.D’Aquino J A, Gómez J, Hilser V J, Lee K H, Amzel L M, Freire E. Proteins. 1996;25:143–156. doi: 10.1002/(SICI)1097-0134(199606)25:2<143::AID-PROT1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 22.Luque I, Mayorga O, Freire E. Biochemistry. 1996;35:13681–13688. doi: 10.1021/bi961319s. [DOI] [PubMed] [Google Scholar]
- 23.Pabo C O, Lewis M. Nature (London) 1982;298:443–447. doi: 10.1038/298443a0. [DOI] [PubMed] [Google Scholar]
- 24.Huang G S, Oas T G. Biochemistry. 1995;34:3884–3892. doi: 10.1021/bi00012a003. [DOI] [PubMed] [Google Scholar]
- 25.Chamberlain A K, Handel T M, Marqusee S. Nat Struct Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 26.Mayo S L, Baldwin R L. Science. 1993;262:873–876. doi: 10.1126/science.8235609. [DOI] [PubMed] [Google Scholar]
- 27.Roder H. Methods Enzymol. 1989;176:446–473. doi: 10.1016/0076-6879(89)76024-9. [DOI] [PubMed] [Google Scholar]
- 28.McPhalen C A, James M N G. Biochemistry. 1987;26:261–269. doi: 10.1021/bi00375a036. [DOI] [PubMed] [Google Scholar]
- 29.Shakhnovich E, Abkevich V, Ptitsyn O. Nature (London) 1996;379:96–98. doi: 10.1038/379096a0. [DOI] [PubMed] [Google Scholar]
- 30.Baudet S, Janin J. J Mol Biol. 1991;219:123–131. doi: 10.1016/0022-2836(91)90862-z. [DOI] [PubMed] [Google Scholar]
- 31.Swint-Kruse L, Robertson A D. Biochemistry. 1995;34:4724–4732. doi: 10.1021/bi00014a029. [DOI] [PubMed] [Google Scholar]
- 32.Kraulis P J. J Appl Crystallog. 1991;24:946–950. [Google Scholar]