

Abstract
μ opioid receptors are targets of native opioid peptides and addictive analgesic drugs. A major clinical liability of opiate drugs is their ability to cause physiological tolerance. Individual opiates, such as morphine and etorphine, differ both in their ability to promote physiological tolerance and in their effects on receptor regulation by endocytosis. Here, we demonstrate that arrestins play a fundamental role in mediating this agonist-selective regulation and that morphine-activated μ receptors fail to undergo arrestin-dependent uncoupling from cognate G proteins. Thus, highly addictive opiate drugs elude a fundamental mode of physiological regulation that is mediated by agonist-specific interaction of opioid receptors with arrestins.
Opioid receptors are G protein-coupled receptors (GPCRs) that are activated both by endogenous opioid peptides and by clinically important alkaloid analgesic drugs such as morphine. Both classes of agonist promote receptor signaling via heterotrimeric G proteins, including inhibition of adenylyl cyclase and regulation of cation channels (1, 2). Studies of knockout mice have established that morphine-induced analgesia, tolerance, and dependence are mediated by μ opioid receptors (3, 4).
After activation by agonists, μ opioid receptors are regulated by multiple mechanisms (5–11). Of these, rapid endocytosis of opioid receptors is of particular interest because it is differentially regulated by individual peptide agonists and alkaloid drugs, both in cultured cells (7, 12, 13) and native neurons (14–15). In particular, opioid peptides stimulate the internalization of μ opioid receptors within minutes while receptors fail to endocytose after prolonged activation with saturating concentrations of morphine, even though morphine strongly activates receptor-mediated signaling via heterotrimeric G proteins (7). Significant differences in the subcellular localization of opioid receptors also are observed in cultured cells after chronic treatment with morphine compared with opioid peptide, suggesting that agonist-specific differences in receptor endocytosis may have long-term physiological consequences (8). Moreover, mice treated chronically with etorphine, which stimulates receptor endocytosis to an extent similar to opioid peptide (7, 15), develop less physiological tolerance than do mice treated chronically with equieffective doses of morphine (16), further demonstrating that agonist-selective internalization could play a key role mediating the different physiological responses to opiate analgesics. Despite the probable physiological importance of this agonist-selective regulation, protein interactions that mediate the endocytosis of opioid receptors and determine their agonist selectivity have not been identified.
MATERIALS AND METHODS
Cell Culture and Immunocytochemistry.
Human embryonic kidney (HEK) 293 cells (American Type Culture Collection) were grown in DMEM supplemented with 10% fetal bovine serum (University of California, San Francisco, Cell Culture Facility). Cells were transfected by using calcium phosphate coprecipitation. For staining, cells were grown on coverslips and incubated in media containing 3.5 μg/ml M1 anti-FLAG (Kodak) antibody. Cells were treated with 5 μM agonist for 30 min, fixed in 4% formaldehyde in PBS, and then permeabilized in 0.1% Triton X-100 (Sigma). For visualization of receptor only, cells were incubated with Cy3-conjugated anti-mouse antibody. For colocalization of receptor and hemagglutination-tagged dynamin (Dyn), cells were incubated with HA-11 (Babco, Richmond, CA) and stained with two subtype-specific fluorescent-conjugated antibodies. For colocalization of arrestin and receptor, cells were incubated with anti β-arrestin antibody (a gift from Jeff Benovic, Thomas Jefferson University, Philadelphia) and then incubated with Cy3-conjugated anti-mouse and fluorescein isothiocyanate-conjugated anti-rabbit antibodies. For colocalization of GPCR kinase-2 (GRK2) and receptor, cells were incubated with anti GRK2 antibody (Santa Cruz Biotechnology) and then incubated with Cy3-conjugated anti-mouse and fluorescein isothiocyanate-conjugated anti-rabbit antibodies.
Assay of Receptor Internalization by Surface Biotinylation.
Cells were grown to 80% confluency, washed with PBS, and then incubated in 3 μg/ml disulfide-cleavable biotin (Pierce) in PBS at 4°C for 30 min. Cells were washed and placed into medium for treatment. Cells labeled 100% biotinylated were left on ice in PBS. Cells were treated with 5 μM agonist for 30 min and washed with PBS, and the remaining cell surface-biotinylated receptors were stripped in 50 mM glutathione, 0.3 M NaCl, 75 mM NaOH, and 1% fetal bovine serum at 4°C for 30 min. Cells were extracted in 0.1% Triton X-100, 150 mM NaCl, 25 mM KCl, and 10 mM Tris⋅HCl, pH 7.4, and cell debris was removed by centrifugation at 10,000 × g for 10 min at 4°C. Proteins were denatured in SDS sample buffer with no reducing agent and separated by SDS/PAGE. Proteins were transferred to nitrocellulose, and biotinylated proteins were visualized by incubating with the Vectastain ABC immunoperoxidase reagent (Vector Laboratories), followed by development with ECL reagents (Amersham).
Guanine Nucleotide Exchange Assays
Membrane Preparations.
Cells were grown to 80% confluency and then pretreated with 2 μM [d-Ala2, N-McPhe4, Gly5-ol]enkephalin (DAMGO) or 2 μM morphine or left untreated. Cells were lifted in PBS + 0.04% EDTA, washed four times in 15 ml of cold PBS to remove excess agonist, and then resuspended in 5 mM Tris⋅HCl, 5 mM EDTA, 5 mM EGTA, and 0.1 mM phenylmethylsulfonyl fluoride (pH 7.5) at 4°C, and lysed by using a Polytron P10 disrupter (Kinematica, Littau, Switzerland). Membranes were recovered at 21,000 × g at 4°C for 20 min, washed, and resuspended in 50 mM Tris⋅HCl, 1 mM EDTA, 5 mM MgCl2, and 0.1 mM phenylmethylsulfonyl fluoride and frozen at −70°C.
[35S]GTPγS binding.
Methods were used as described previously (14) by using membranes prepared as above—unstimulated or stimulated with 5 μM DAMGO or morphine for 60 min at 25°C. Reactions were terminated by vacuum filtration over GF/C filters (Millipore).
Membrane Adenylyl Cyclase Assays
Membrane Preparations.
Cells were grown to 80% confluency and then pretreated with 2 μM DAMGO or morphine or left untreated. Cells were lifted in PBS + 0.04% EDTA, washed four times in 15 ml of cold PBS, and then resuspended in 1 ml of cold buffer of 25 mM MgCl2, 75 mM Tris⋅HCl, and 2 mM EDTA, pH 7.5 and pelleted at 2,000 × g for 5 min. The pellet was resuspended with a glass potter in the same buffer and assayed immediately.
Adenylyl Cyclase Assay.
Membranes were incubated in 30 mM Tris⋅HCl, 1 mM EDTA, 50 μM GTP, 0.1 mM cAMP, 40 μM ATP, 10 mM creatine phosphate, 200 units/ml creatine phosphokinase, 1 μCi [α32P]ATP, and 10 μM forskolin with or without 10 μM morphine or DAMGO at 37°C for 30 min. Reactions were stopped by addition of HCl to 1 M and applied to acidic alumina spin-columns (Pierce). Columns were washed and eluted according to the manufacturer’s instructions and eluate counted in a scintillation counter.
RESULTS AND DISCUSSION
We previously have reported functional expression of epitope-tagged versions of the μ, δ, and κ opioid receptors in HEK293 cells (7, 13, 17). Many GPCRs are internalized by clathrin-mediated endocytosis, a process that is dependent on the GTPase Dyn. The Dyn dependence of μ opioid receptor endocytosis was tested by examining the effect of K44E mutant Dyn, which inhibits endocytosis of clathrin-coated pits in a dominant negative manner (18, 19), on the endocytosis of μ opioid receptors. HEK293 cells expressing epitope-tagged μ receptors (≈1.5 × 105 receptors/cell) were examined by using an established antibody uptake assay (17). In the absence of agonist, receptors remained in the plasma membrane without detectable endocytosis for >30 min (Fig. 1A). In the presence of the alkaloid agonist etorphine, receptors were endocytosed within several minutes, as indicated by redistribution of antibody-labeled receptors from the plasma membrane to numerous endocytic vesicles visualized throughout the cytoplasm (Fig. 1B). In the presence of morphine, receptors failed to internalize (Fig. 1C) thereby demonstrating agonist-selective internalization. In cells expressing HA-tagged K44E Dyn, agonist-induced endocytosis of μ opioid receptors from the plasma membrane to endocytic vesicles was strongly inhibited. μ receptors remained almost exclusively in the plasma membrane of cells expressing high levels of K44E mutant Dyn, even in the presence of saturating concentrations (5 μM) of etorphine (Fig. 1D) or the opioid peptide DAMGO (not shown), whereas numerous receptor-containing endocytic vesicles were observed in cells that did not express mutant Dyn examined in the same field (cf. receptor staining in red in Fig. 1D with K44E staining in green in Fig. 1E; cells expressing and not expressing K44E Dyn are indicated by the open and solid arrows, respectively). Expression of HA-tagged wild-type Dyn (Fig. 1G, in green) at similar levels, in contrast, caused no detectable inhibition of μ receptor endocytosis (Fig. 1F, in red; note the localization of antibody-labeled receptors in numerous endocytic vesicles both in cells expressing and not expressing wild-type Dyn in Fig. 1 F and G, open and solid arrows, respectively). Quantitation by using the antibody uptake assay (17) (Fig. 1H) confirmed that agonist-induced endocytosis of μ opioid receptors is specifically inhibited by K44E mutant Dyn.
Figure 1.

Mu opioid receptors were endocytosed in a Dyn-dependent manner. HEK293 cells expressing FLAG-epitope-tagged μ opioid receptors (μOR) were stained as described in Materials and Methods. (A) Receptors remained predominantly in the plasma membrane in the absence of agonist stimulation. (B) Cells were treated with 5 μM etorphine for 30 min at 37°C and stained. In the presence of etorphine, receptors were endocytosed as indicated by redistribution of antibody-labeled receptors from the plasma membrane to numerous endocytic vesicles visualized throughout the cytoplasm. (C) Cells were treated with 5 μM morphine for 30 min at 37°C and stained. In the presence of morphine, receptors remained in the plasma membrane. (D and E) HEK293 cells expressing the FLAG-tagged μ opioid receptor were transfected transiently with an HA-tagged dominant negative Dyn, K44E. Cells were then treated with 5 μM etorphine and stained for both receptor in red (D) and Dyn in green (E). Cells expressing K44E Dyn failed to endocytose the receptor (D and E, open arrows), whereas adjacent cells not expressing K44E Dyn did endocytose receptor (D and E, closed arrows). (F and G) Cells transiently transfected with HA-tagged wild-type Dyn also were treated and stained. HA-tagged wild-type Dyn did not affect receptor endocytosis, as cells expressing (F and G, open arrows) and not expressing (F and G, closed arrows) Dyn-endocytosed receptors. (H) Slides stained as above were coded, and receptor-containing vesicles were counted from cells expressing both wild-type and mutant Dyn as well as cells not expressing any additional Dyn.
Although both DAMGO and etorphine stimulated Dyn-dependent endocytosis of μ opioid receptors, morphine failed to induce receptor endocytosis (Fig. 1C), although morphine is an alkaloid agonist that promotes receptor-mediated inhibition of adenylyl cyclase in these cells with potency and efficacy similar to DAMGO (15). Recent studies of other GPCRs that endocytose in a Dyn-dependent manner indicate that the association of agonist-activated receptors with clathrin-coated pits is promoted by a direct protein interaction with β-arrestin (20). Furthermore, opioid receptors have been shown recently to mediate agonist-dependent recruitment of GFP-tagged β-arrestin to the plasma membrane (21). Taken together, these results suggest that arrestins may play a fundamental role in mediating opioid receptor regulation.
To examine whether β-arrestin could mediate the agonist selective internalization of μ opioid receptors, stably transfected cells expressing FLAG-tagged μ opioid receptors were transiently transfected with β-arrestin. This experimental design allowed receptor localization to be compared in adjacent cells differing only in whether they expressed additional β-arrestin, as indicated by immunostaining by using anti-β-arrestin antibody. In the absence of agonist, antibody-labeled μ receptors remained in the plasma membrane both in cells overexpressing β-arrestin and in adjacent cells not expressing additional β-arrestin (cf. receptor staining in red in Fig. 2Aa with anti-β-arrestin immunoreactivity in green in Fig. 2Ab; filled and open arrows indicate representative cells that do or do not overexpress β-arrestin, respectively). In the presence of etorphine, receptors were endocytosed both in cells expressing endogenous levels of β-arrestin and in cells overexpressing β-arrestin (Fig. 2 Ac and Ad; filled and open arrows indicate representative cells that do or do not overexpress β-arrestin, respectively). Surprisingly, overexpression of β-arrestin also promoted rapid endocytosis of μ opioid receptors in the presence of morphine, even though morphine failed to stimulate rapid endocytosis of μ receptors in adjacent cells expressing endogenous levels of arrestin. This result was indicated by the morphine-induced redistribution of receptors from the plasma membrane to endocytic vesicles that was observed exclusively in cells overexpressing β-arrestin (Fig. 2 Ae and Af, filled and open arrows indicate cells that do or do not overexpress β-arrestin, respectively).
Figure 2.
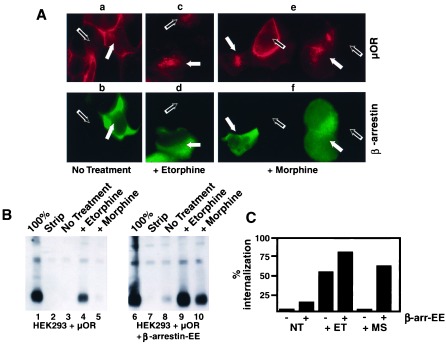
Overexpression of β-arrestin could facilitate μ opioid receptor internalization in the presence of morphine. (A) HEK293 cells stably expressing FLAG-epitope-tagged μ opioid receptors were transiently transfected with a plasmid overexpressing β-arrestin and stained for both receptor in red (μOR) and arrestin in green. In the absence of agonist, β-arrestin overexpression failed to stimulate endocytosis of μ opioid receptors (Aa and Ab). In the presence of 5 μM etorphine, receptors were endocytosed in both cells overexpressing (Ac and Ad, closed arrows) and those expressing only endogenous arrestins (Ac and Ad, open arrows). Cells expressing endogenous arrestins and treated with 5 μM morphine (Ae and Af, open arrows) failed to endocytose the receptor. However, receptors were endocytosed in the presence of morphine in cells overexpressing β-arrestin (Ae and Af, closed arrows). (B) HEK293 cells stably expressing FLAG-epitope-tagged μ opioid receptors and stably overexpressing an EE-tagged version of β-arrestin were used to quantitate receptor internalization. Surface-biotinylated μ opioid receptors (lanes 1 and 6) were cleaved in the presence glutathione (lanes 2 and 7). In the absence of agonist, little endocytosis of receptors was observed in cells expressing endogenous levels of β-arrestin (lane 3) or in cells overexpressing β-arrestin (lane 8). Etorphine-induced endocytosis of μ opioid receptors was observed readily in both cell lines (lanes 4 and 9). Although morphine caused no detectable stimulation of receptor endocytosis in cells expressing β-arrestin at endogenous levels (cf. lanes 3 and 5), a substantial amount of morphine-induced endocytosis of receptors was observed in cells overexpressing β-arrestin (cf. lanes 8 and 10). (C) Quantitation of these effects demonstrated that etorphine-induced (ET) endocytosis observed at steady state (30 min) was <2-fold enhanced in cells overexpressing β-arrestin (bars 3 and 4). Overexpression of β-arrestin enhanced morphine (MS)-induced endocytosis of μ opioid receptors by >40-fold (bars 5 and 6), far in excess of the effect of β-arrestin on either agonist-independent or etorphine-induced endocytosis.
The effect of β-arrestin on endocytosis of μ opioid receptors was examined further in stably transfected cells expressing both FLAG-tagged μ opioid receptors and EE-tagged β-arrestin (22). Cell clones were selected that expressed opioid receptors at levels similar to the stably transfected cells described above that do not overexpress β-arrestin. Western blot analysis indicated that EE-tagged β-arrestin was expressed in these cells at levels ≥30-fold higher than the endogenous levels of β-arrestin protein (not shown). The epitope-tagged version of β-arrestin used to make this double stable cell line also facilitated morphine-induced internalization of μ opioid receptors as indicated by the redistribution of receptors from the plasma membrane to endocytic vesicles upon morphine stimulation (not shown). To quantitate this effect, cell surface biotinylation was used to assay the endocytosis of μ opioid receptors in the cells overexpressing EE-tagged β-arrestin compared with that in cells expressing similar amounts of μ opioid receptor but expressing endogenous levels of β-arrestin.
Surface-biotinylated μ opioid receptors (Fig. 2B, lanes 1 and 6) were cleaved in the presence of the membrane-impermeant reducing agent glutathione (Fig. 2B, lanes 2 and 7), thereby allowing only endocytosed receptors to be detected specifically by their resistance to cleavage by extracellular glutathione. In the absence of agonist, little endocytosis of μ opioid receptors was observed in cells expressing endogenous levels of β-arrestin (Fig. 2B, lane 3) or in cells overexpressing β-arrestin (Figs. 2B, lane 8). Etorphine-induced endocytosis of μ opioid receptors was observed readily in both cell lines (lanes 4 and 9). Quantitation of this effect demonstrated that etorphine-induced endocytosis observed at steady-state (30 min) was moderately (<2-fold) enhanced in cells overexpressing β-arrestin (Fig. 2C, bars 3 and 4). In addition, these experiments confirmed that overexpression of β-arrestin profoundly influenced the agonist selectivity of receptor endocytosis. Although morphine caused essentially no detectable stimulation of receptor endocytosis in cells expressing β-arrestin at endogenous levels (Figs. 2B, cf. lanes 3 and 5), a substantial amount of morphine-induced endocytosis of μ opioid receptors was observed in cells overexpressing β-arrestin (Figs. 2B, lanes 8 and 10). Quantitation of these results indicated that overexpression of β-arrestin enhanced morphine-induced endocytosis of μ opioid receptors by >40-fold (Fig. 2C, bars 5 and 6), far in excess of the effect of β-arrestin on either agonist-independent or etorphine-induced endocytosis. Thus β-arrestin had two effects on the regulation of opioid receptors by Dyn-dependent endocytosis, a moderate enhancement of the endocytic efficacy of agonists that normally promote endocytosis, as well as a much more profound effect on the agonist selectivity of opioid receptor endocytosis.
It is well established that arrestin interaction with agonist-activated receptors modulates signaling by uncoupling the activated receptor from cognate heterotrimeric G proteins (23–25). Therefore, we examined whether morphine-activated receptors were resistant to arrestin-dependent uncoupling. Cells expressing μ opioid receptor were treated for 5 min with DAMGO or morphine or left untreated, and residual agonist was washed from the cells. Membranes were prepared from these three cell lines, and the ability of the μ receptors to mediate GTP exchange on G proteins was measured. Cells that were not pretreated with agonist stimulated in vitro GTP exchange efficiently after receptor activation by both morphine and DAMGO stimulation (Fig. 3A, open bars), demonstrating that, in our system, DAMGO and morphine were both good agonists for GTP exchange. After pretreatment of cells with DAMGO, receptor-mediated GTP exchange upon rechallenge with agonist almost was completely abolished (Fig. 3A, black bars), indicating that DAMGO caused efficient uncoupling of opioid receptors from heterotrimeric G proteins during the pretreatment. This effect could not be explained by endocytosis of receptors because only partial receptor endocytosis is observed after 5 min of agonist stimulation (15), yet uncoupling from G proteins was virtually complete at this time point. In contrast, receptors pretreated with morphine under identical conditions still mediated agonist-dependent GTP exchange upon rechallenge with DAMGO at levels virtually indistinguishable from those observed without agonist pretreatment (Fig. 3A, shaded bars), indicating that pretreatment with morphine failed to uncouple the receptors from cognate G proteins. To determine whether overexpression of β-arrestin could facilitate morphine-induced receptor-G protein uncoupling, identical experiments were performed by using cells overexpressing β-arrestin. In marked contrast to results obtained by using cells expressing endogenous levels of arrestins, morphine pretreatment of cells overexpressing β-arrestin caused substantial receptor-G protein uncoupling (Fig. 3B, cf. solid and hatched bars). The differential effects of morphine and DAMGO on receptor desensitization also were observed at a downstream effector assayed by using a membrane adenylyl cyclase assay.
Figure 3.

Morphine stimulated GTP exchange but failed to promote uncoupling of receptor from G protein and failed to desensitize adenylyl cyclase activity. (A) Cells expressing μ opioid receptor were pretreated for either 5 or 30 min with DAMGO (DG) or morphine (MS) or left untreated and residual agonist washed from cells. Membranes were prepared from these cells and the ability of the μ receptors in these membranes to activate GTP exchange on G proteins in vitro was measured. Receptors from cells that were not pretreated with agonist stimulated GTP exchange efficiently with both morphine and DAMGO (open bars). Receptors pretreated with DAMGO for either 5 or 30 min were very inefficient at promoting GTP exchange upon DAMGO stimulation (black bars), indicating that the μ opioid receptors in these cells had become uncoupled from their G proteins during the DAMGO pretreatment. Receptors pretreated with morphine were still as effective at stimulating GTP exchange upon DAMGO stimulation (shaded bars) indicating that pretreatment with morphine failed to uncouple the receptors from their G proteins. (B) Cells expressing both μ opioid receptor and EE-tagged β-arrestin were pretreated with morphine or left untreated and membranes prepared as above. Receptors from cells overexpressing arrestin (hatched bars) pretreated with morphine were significantly impaired in their ability to stimulate GTP exchange, differing markedly from cells expressing endogenous levels of arrestin (solid bars), demonstrating that overexpression of arrestin facilitated functional uncoupling of morphine-bound opioid receptors. These assays were done in triplicate three times with comparable results. (C) Cells expressing μ opioid receptor were pretreated for 5 min with DAMGO (DG) or morphine (MS) or left untreated, and residual agonist washed from cells. Membranes were prepared from these cells, and the ability of the μ receptors in these membranes to inhibit forskolin-stimulated adenylyl cyclase activity was measured. Receptors from cells that were not pretreated with agonist inhibited adenylyl cyclase efficiently with both morphine and DAMGO stimulation (white bars). Receptors from cells pretreated with DAMGO for 5 min were significantly impaired in their ability to inhibit adenylyl cyclase activity upon DAMGO stimulation (black bar). Receptors from cells pretreated with morphine were still as effective at inhibiting adenylyl cyclase upon DAMGO (shaded bar). (D) Cells expressing μ opioid receptor and overexpressing β-arrestin-EE were pretreated for 5 min with morphine (MS) or left untreated, and residual agonist washed from cells. Membranes were prepared and the ability of the μ receptors to inhibit forskolin-stimulated adenylyl cyclase activity was measured. Receptors from cells that were not pretreated with agonist inhibited adenylyl cyclase as efficiently as cells expressing endogenous arrestins. Cells overexpressing arrestin that were pretreated with morphine were significantly less efficient at inhibiting adenylyl cyclase than cells expressing endogenous levels of arrestins (cf. hatched and unhatched shaded bars). These assays were repeated twice in triplicate with similar results.
As above, cells expressing the μ opioid receptor were pretreated with either DAMGO or morphine for 5 min or left untreated and residual agonist was washed from cells. Membranes were prepared from these cells and the ability of the μ receptors to inhibit forskolin-stimulated adenylyl cyclase activity upon DAMGO rechallenge was assayed to measure receptor-mediated signal transduction through Gi. With untreated membranes, both DAMGO and morphine inhibited adenylyl cyclase activity with equivalent efficiency (Fig. 3C, white bars), demonstrating that in our system both are good agonists for receptor-mediated inhibition of adenylyl cyclase. Membranes prepared from cells pretreated with DAMGO were impaired significantly in their ability to inhibit adenylyl cyclase activity (Fig. 3C, black bar), consistent with desensitization caused by functional uncoupling of receptors. However, membranes prepared from cells pretreated with exhibited efficient receptor-mediated inhibition of adenylyl cyclase activity upon DAMGO rechallenge at levels virtually indistinguishable from those of membranes from untreated cells (Fig. 3C, shaded bar). To determine whether overexpression of β-arrestin could facilitate morphine-induced desensitization, identical experiments were performed by using cells overexpressing β-arrestin. Again, in contrast to results obtained by using cells expressing endogenous levels of arrestins, morphine pretreatment of cells overexpressing β-arrestin caused substantial receptor desensitization (Fig. 3D, cf. solid and hatched bars). Consequently, morphine-activated opioid receptors elude two arrestin-dependent mechanisms of regulation, functional uncoupling from heterotrimeric G proteins and Dyn-dependent endocytosis, suggesting strongly that the different effects of morphine and native peptides on opioid receptor regulation are mediated directly by agonist-specific differences in receptor interaction with arrestins. Thus, we propose that individual agonists induce distinct activated conformations of μ opioid receptor, which have similar effects on the activation of cognate heterotrimeric G proteins but differ significantly in their ability to interact with arrestins.
The interaction of many GPCRs with arrestins has been shown to be mediated both by an agonist induced conformational change of the receptor and by phosphorylation of the agonist-activated receptor by GRKs (24, 25). We thus examined whether differences in GRK phosphorylation of the μ opioid contribute to differences in the ability of morphine and DAMGO-activated receptors to interact with arrestins. Cells stably expressing the μ opioid receptor were transfected transiently with a plasmid overexpressing GRK2 (βARK1). Dual label fluorescence microscopy allowed receptor localization to be compared in adjacent cells differing only in whether they expressed additional GRK2. In the absence of agonist, antibody-labeled μ receptors remained in the plasma membrane both in cells overexpressing GRK2 and in adjacent cells not expressing additional GRK2 (Fig. 4, cf. receptor staining in A with GRK2 immunoreactivity in B; open and filled arrows indicate representative cells that do or do not overexpress GRK2, respectively). Overexpression of GRK2 promoted rapid endocytosis of μ opioid receptors in the presence of morphine, however, even though morphine failed to stimulate rapid endocytosis of μ receptors in adjacent cells expressing endogenous levels of GRK2. (Fig. 4 C and D, open and filled arrows indicate cells that do or do not overexpress GRK2, respectively). These results suggest that differences in GRK-mediated phosphorylation may account, at least in part, for agonist selective differences in the ability of opioid receptors to interact with arrestins.
Figure 4.

Overexpression of GRK2 facilitated morphine-induced receptor internalization. Cells stably transfected with FLAG-epitope-tagged μ opioid receptor were transiently transfected with GRK2. Cells were treated with morphine (C and D) or left untreated (A and B) and stained as for receptor (A and C) and GRK2 (B and D). In the absence of agonist, GRK2 overexpression failed to stimulate receptor endocytosis (A and B, open arrows). However, in the presence of morphine, overexpression of GRK2 facilitated receptor internalization (C and D, open arrows), even though adjacent cells expressing endogenous GRKs failed to endocytose receptor (C and D, closed arrows ).
As a model, we propose that a dynamic cycle between agonist-induced activation, desensitization, and internalization of opioid receptors is critical for normal physiological signaling. In our model, tolerance to morphine develops not because cells have down-regulated receptor expression but because receptors are activated in a way that prevents their normal physiological cycling. This model is consistent with observations that long-term treatment of rats with high doses of morphine fails to cause significant down-regulation of μ opioid receptors, although these animals develop profound physiological tolerance to opiate agonists (26, 27). This model also is consistent with observations that equieffective doses of etorphine and morphine differ substantially in their propensity to induce physiological tolerance (16). By analogy with certain other GPCRs (20, 21), we propose that agonist-dependent arrestin binding to μ opioid receptors therefore serves a dual role. First, arrestin binding rapidly desensitizes cells to the presence of agonist by uncoupling receptor from G protein. Second, by mediating association of the receptor with clathrin-coated pits, arrestin targets receptors to recycling endosomes from where they are sent back to the cell surface to resensitize the cell to agonist. Thus the ability of morphine to activate opioid receptors without promoting these arrestin-mediated regulatory mechanism is possibly expected to cause important disturbances of physiological homeostasis, contributing to the pathophysiology of opiate tolerance and dependence.
In conclusion, we have elucidated a distinct molecular mechanism that mediates the agonist specificity of opioid receptor regulation and distinguishes the physiological actions of analgesic drugs observed both in vivo and in cultured cells. By establishing that morphine-activated μ opioid receptors strongly activate heterotrimeric G proteins but evade arrestin-mediated receptor regulation, our results suggest a precise molecular mechanism to explain the different physiological actions of opioid peptides and alkaloid analgesic drugs, such as morphine, on opioid receptor function. This mechanism may play a critical role in the biology of opiate drug tolerance and dependence, as morphine and etorphine differ significantly in their ability to promote both internalization of receptor and the development of tolerance in vivo (16). In addition to their importance to the effects of opiate drugs on the endogenous opioid system, these studies have general relevance to GPCR biology because they suggest that individual agonists can induce distinct activated conformations of a GPCR that differ significantly in their physiological regulation. Our observations suggest that it is possible, in principle, to design agonists for GPCRs that strongly activate receptor signaling through their cognate heterotrimeric G proteins without promoting GRK and/or arrestin-mediated desensitization of receptors, thereby circumventing a major limitation in the therapeutic usefulness of a wide variety of agonist drugs used currently in clinical medicine.
Acknowledgments
We thank Jeff Benovic for supplying cDNA and antibody for β-arrestin. We thank Richard Vallee for HA-tagged Dyn and K44E cDNA. We thank Uwe Klein for constructing the EE-tagged version of β-arrestin and providing advice on nucleotide exchange assays. We thank Peter Chu for technical assistance. We thank Taroh Iiri and Sankuratri Suryanarayana for advice with adenylyl cyclase assays. We thank David Julius, Robert Malenka, Robert Edwards, and Roger Nicoll for critical reading of the manuscript and thank the other members of the laboratory for many good suggestions and discussions. J.L.W. thanks Stig Hansen for his support. This work was supported by National Institutes of Health Grant DA10711 (to M.v.Z.) and National Institutes of Health Training Grant MH19552 and National Research Service Award Grant DA05844 (to J.L.W.).
ABBREVIATIONS
- GPCR
G protein-coupled receptors
- GRK
GPCR kinase
- DAMGO
[d-Ala2, N-McPhe4, Gly5-ol]enkephalin
- HEK
Human embryonic kidney
- Dyn
dynamin
- FLAG
epitope (DYKDDDDK)
- EE
epitope (EEEEYMPME)
Note Added in Proof
While this manuscript was under review, complementary studies of a rat μ opioid receptor were reported by Zhang et al. (28).
References
- 1.Evans C J. In: Diversity Among the Opioid Receptors. Korenman S G, Barchas J D, editors. New York: Oxford University Press; 1993. pp. 31–48. [Google Scholar]
- 2.Grudt T J, Williams J T. Rev Neurosci. 1995;6:279–286. doi: 10.1515/revneuro.1995.6.3.279. [DOI] [PubMed] [Google Scholar]
- 3.Matthes H W, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le M M, Dolle P, et al. Nature (London) 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 4.Sora I, Takahashi N, Funada M, Ujike H, Revay R S, Donovan D M, Miner L L, Uhl G R. Proc Natl Acad Sci USA. 1997;94:1544–1549. doi: 10.1073/pnas.94.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Y, Zhang L, Yin X, Sun H, Uhl G R, Wang J B. J Biol Chem. 1997;272:28869–28874. doi: 10.1074/jbc.272.46.28869. [DOI] [PubMed] [Google Scholar]
- 6.Pei G, Kieffer B L, Lefkowitz R J, Freedman N J. Mol Pharmacol. 1995;48:173–177. [PubMed] [Google Scholar]
- 7.Keith D E, Murray S R, Zaki P A, Chu P C, Lissin D V, Kang L, Evans C J, von Zastrow M. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- 8.Arden J R, Segredo V, Wang Z, Lameh J, Sadee W. J Neurochem. 1995;65:1636–1645. doi: 10.1046/j.1471-4159.1995.65041636.x. [DOI] [PubMed] [Google Scholar]
- 9.Tao P L, Chang L R, Law P Y, Loh H H. Brain Res. 1988;462:313–320. doi: 10.1016/0006-8993(88)90559-8. [DOI] [PubMed] [Google Scholar]
- 10.Law P Y, Hom D S, Loh H H. J Biol Chem. 1984;259:4096–4104. [PubMed] [Google Scholar]
- 11.Cvejic S, Trapaidze N, Cyr C, Devi L A. J Biol Chem. 1996;271:4073–4076. doi: 10.1074/jbc.271.8.4073. [DOI] [PubMed] [Google Scholar]
- 12.von Zastrow M, Keith D E, Jr, Evans C J. Mol Pharmacol. 1993;44:166–172. [PubMed] [Google Scholar]
- 13.von Zastrow M, Keith D E, Zaki P, Evans C J. Regul Pept. 1994;54:315–316. [Google Scholar]
- 14.Sternini C, Spann M, Anton B, Keith D J, Bunnett N W, von Z M, Evans C, Brecha N C. Proc Natl Acad Sci USA. 1996;93:9241–9246. doi: 10.1073/pnas.93.17.9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keith D E, Anton B, Murray S R, Zaki P A, Chu P C, Lissin D V, Monteillet-Agius G, Stewart P L, Evans C J, von Zastrow M. Mol Pharmacol. 1998;53:377–384. [PubMed] [Google Scholar]
- 16.Duttaroy A, Yoburn B C. Anesthesiology. 1995;82:1226–1236. doi: 10.1097/00000542-199505000-00018. [DOI] [PubMed] [Google Scholar]
- 17.Chu P, Murray S, Lissin D, von Z M. J Biol Chem. 1997;272:27124–27130. doi: 10.1074/jbc.272.43.27124. [DOI] [PubMed] [Google Scholar]
- 18.Damke H, Baba T, Warnock D E, Schmid S L. J Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herskovits J S, Burgess C C, Obar R A, Vallee R B. J Cell Biol. 1993;122:565–578. doi: 10.1083/jcb.122.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodman O J, Krupnick J G, Santini F, Gurevich V V, Penn R B, Gagnon A W, Keen J H, Benovic J L. Nature (London) 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 21.Barak L S, Ferguson S S, Zhang J, Caron M G. J Biol Chem. 1997;272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- 22.Klein, U. & von Zastrow, M. (1998) Methods in Receptor Biology, in press.
- 23.Lohse M J, Benovic J L, Codina J, Caron M G, Lefkowitz R J. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 24.Gurevich V V, Dion S B, Onorato J J, Ptasienski J, Kim C M, Sterne M R, Hosey M M, Benovic J L. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 25.Gurevich V V, Benovic J L. J Biol Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- 26.Simantov R, Lotem J, Levy R. Neuropeptides. 1984;5:197–200. doi: 10.1016/0143-4179(84)90061-1. [DOI] [PubMed] [Google Scholar]
- 27.Lenoir D, Barg J, Simantov R. Brain Res. 1984;304:285–290. doi: 10.1016/0006-8993(84)90332-9. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Ferguson S S, Barak L S, Bodduluri S R, Laporte S A, Law P Y, Caron M G. Proc Natl Acad Sci USA. 1988;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]