

Abstract
Ventral structures in the central nervous system are patterned by signals emanating from the underlying mesoderm as well as originating within the neuroectoderm. Mutations in the zebrafish, Danio rerio, are proving instrumental in dissecting these midline signals. The cyclops mutation leads to a loss of medial floor plate and to severe deficits in ventral forebrain development, leading to cyclopia. Here, we report that the cyclops locus encodes the nodal-related protein Ndr2, a member of the transforming growth factor type β superfamily of factors. The evidence includes identification of a missense mutation in the initiation codon and rescue of the cyclops phenotype by expression of ndr2 RNA, here renamed “cyclops.” Thus, in interaction with other molecules implicated in these processes such as sonic hedgehog and one-eyed-pinhead, cyclops is required for ventral midline patterning of the embryonic central nervous system.
In vertebrates, the neuroectoderm is induced in dorsal ectoderm by signals from dorsal mesoderm and endoderm (reviewed in refs. 1 and 2). Through a series of inductive interactions, the neural ectoderm is subsequently patterned along the anteroposterior and dorsoventral axes to eventually generate a differentiated nervous system. Signals from both the mesoderm and from within the neuroectoderm are thought to be involved in these patterning events that eventually are elaborated in the specification of many distinct neuronal cell types (reviewed in ref. 3). Thus, the nature and mechanism of action of these patterning signals is of great interest for the understanding of the development of the nervous system. Elegant experiments, mainly done in chickens, implicated the notochord in the induction of the floor plate and suggested a role for both notochord and floor plate in dorsoventral patterning of the spinal cord, e.g., the specification of motor neurons (3–7). The signaling molecule Sonic hedgehog (Shh) can mediate some of these signals when applied to explants of neural ectoderm in vitro (reviewed in ref. 8). Mice with a disrupted shh gene initiate but do not maintain the expression of ventral neural markers like HNF-3β and are defective in floor plate and ventral forebrain development, showing a condition similar to holoprosencephaly (9). Mutations at the shh locus also have been implicated as a cause of congenital holoprosencephaly in humans (10, 11).
In zebrafish, the floor plate can be distinguished into medial and lateral regions by marker expression and the existence of mutations that affect either subtype of floor plate cells (12), suggesting the involvement of multiple signals in the induction of ventral identity in the central nervous system (CNS). Among the factors involved, the shh gene is specifically required for the development of lateral floor plate cells, leaving medial floor plate cells unaffected (12); the lateral floor plate may correspond to the lateral HNF-3β-expressing domain in the ventral neural tube of the mouse (13). In contrast, mutations at the one-eyed-pinhead (oep) locus, which recently was shown to encode a membrane-bound ligand with epidermal growth factor-like repeats (14), affect the formation of medial floor plate and prechordal plate, resulting in a reduction of anterior structures and fusion of the eyes.
Zebrafish embryos with mutations in the cyclops (cyc) gene show severe defects in the development of medial floor plate and the ventral brain, leading to cyclopia caused by incomplete splitting of the eye field (15, 16). Abnormalities in prechordal mesoderm, notochord, axon guidance, general body shape (“curly tail down”), and randomization of left–right asymmetry are additional phenotypes of the mutation, which eventually leads to larval lethality (15–21). Although midline expression of shh is lost in the CNS of cyc mutants (22), the cyc phenotype itself is not rescued effectively by experimental activation of the hedgehog signal transduction pathway (23, 24), although some differences exist in the interpretation of the extent of rescue (25). On balance, these observations lend credence to the idea that cyc does not encode part of the hedgehog signaling pathway. In confirmation of this view, we show here that cyc encodes a new nodal-related factor within the transforming growth factor type β superfamily.
Members of the nodal subgroup of the transforming growth factor type β superfamily are involved in mesoderm formation, gastrulation, and establishment of laterality (26, 27). In zebrafish, we have studied two members of this family named “Ndr1” and “Ndr2” (28). Ndr1 is expressed in the blastoderm margin during blastula stages and then decays rapidly; it has mesoderm-inducing ability in the Xenopus animal cap assay. Ndr2, which we show here to be identical to cyc, is expressed in the hypoblast of the shield (organizer region) and subsequently in axial mesendoderm, especially the prechordal plate, and ventral neuroectoderm at the tailbud stage; some expression also is seen in the tailbud (ref. 28; see also Fig. 4). After disappearance from the embryo, ndr2 RNA is transiently re-expressed on the left side in the lateral plate mesoderm and in a small region of the posterior diencephalon. In this paper, we present evidence that ndr2 is identical to cyclops and consider the implications of the structure and expression pattern of this gene for our understanding of midline signaling in ventral CNS patterning.
Figure 4.

The expression pattern of cyc at the bud stage is modified in cyctf219 mutant embryos. (A) The wild-type pattern (28). Cyc RNA is seen in the tailbud, and the anterior notochord and prechordal plate including the polster (hatching gland precursor). (B, C, and F) Wild-type sibs. (D, E, and G) cyctf219 mutant embryos. Anterior views (B and D) and dorsal views (C–G) are shown. At the tailbud stage, expression in tailbud and polster is essentially unaffected in cyctf219, but expression in the prechordal plate, ventral neuroectoderm, and anterior notochord is largely lost. At the 20-somite stage, asymmetry of staining in the lateral plate and diencephalon is lost in mutant embryos.
MATERIALS AND METHODS
Genetic Linkage.
Linkage was assessed by mapping an Aci I restriction polymorphism between cyctf219 carriers and the WIK strain. The following cyc mapping primer pair was used: NDR2MF1: 5′-GAAAGGAGATCCACAGTGGGCCTAG-3′; NDR2MR1: 5′-TGGTCTCTGAGGCCCACTCTGGA-3′.
PCR conditions were: 40 μCi 32P-dCTP, 100 ng of genomic DNA, 50 ng of each primer; hot start, 94°, 1 min; 65°, 1 min; 72°, 45 s, 45 cycles. For sequencing of cyctf219 DNA, high fidelity PCR (Klentaq, CLONTECH, or TaqPlus Precision, Stratagene) was used with primers based on the wild-type sequence (28) to generate fragments spanning the ndr2 gene in mutant DNA.
Expression Constructs.
The wild-type construct in the CS2+ vector (29) contained the entire cyc ORF (28). The mutant construct, designed to initiate translation at the second methionine (position 38), was constructed by annealing the 5′ phosphorylated oligos MALTF and MALTR and ligating the annealed product into HindIII/SphI cut pCS2+Cyc. Oligo MALTR is the complement of MALTF, designed to give an annealed product with cohesive SphI and HindIII ends. Oligo MALTF: 5′-AGCTTGATTTAGGTGACACTATAGAATACAAGCTACTTGTTCTTTTTGCAGGATCCCACCATGTCACACCGCATG-3′. Injections and assays in Xenopus animal caps were done as described (28).
Additional PCR Primers.
For detecting ndr2 and ndr1 sequences in cycb16 mutant embryos, we used cyc primer pair one (amino acids 23–76): NDR2F2: 5′-AAAGCACACGCGTTATC-3′; NDR2R8: 5′-TCCTGATTGTGTCTGCGT-3′; cyc primer pair two (amino acids 473–501): NDR2FA: 5′-CAGCGCTCTCAGTATGCTGTAC-3′; NDR2RB: 5′-TCACAGGCATCCGCACTCCT-3′; and ndr1 primer pair: NDR1F15: 5′-TGAACTTAGATGACCGGCCAG-3′ NDR1R10L: 5′-GGCCAAAACTACGCTCAGGAG-3′. The following primers were used to genotype embryos: NDR2R20: 5′-GGTGTCTCAGAATCATC-3′ NDR2FB: 5′-AAGAGCACCACCTGCAGGAG-3′. PCR conditions were: hot start, 94°, 1 min; 55°, 1 min; 72°, 1 min, for 35 to 47 cycles, 0.5 embryo equivalent of template, 50 ng of each primer.
In Vitro-Coupled Transcription–Translation.
Reactions with or without dog pancreatic microsomes were carried out with the TNT kit (Promega), and endoglycosidase H treatment was done according to the manufacturer’s instructions (New England Biolabs). Proteins were electrophoresed on 4–20% polyacrylamide gels.
RESULTS
cyclops Encodes the Nodal-Related Factor Ndr2.
In previous work, we have characterized two nodal-related factors in zebrafish, named “Ndr1” and “Ndr2” (28). By using a “candidate gene approach,” we identified Ndr2 as the product of the cyclops gene. The γ ray-induced cycb16 allele is thought to be a deletion covering the cyc locus (15, 30). Approximately one-quarter of the progeny of an intercross of cycb16 heterozygous carriers did not show any ndr2 expression at the gastrula stage when examined by in situ hybridization (Fig. 1A). PCR templated by DNA from homozygous cycb16 mutant embryos, collected at a stage when the phenotype is visible, failed to give an amplification product with ndr2-specific primers, demonstrating the absence of the ndr2-coding region in the cycb16 genome (Fig. 1B).
Figure 1.

(A) ndr2 RNA is present in wild-type (WT) but not homozygous cycb16 embryos, as seen by in situ hybridization at the shield stage (28). (B) ndr2 is deleted in cycb16 DNA, visualized by PCR. Lanes: 1, ndr2 primer pair 1 + ndr2 plasmid; 2, ndr2 primer pair 1 + wild-type genomic DNA; 3, ndr2 primer pair 1 + cycb16 homozygous genomic DNA; 4, ndr2 primer pair 2 + ndr2 plasmid; 5, ndr1 primers + ndr2 primer pair 2 + cycb16 genomic DNA; 6, ndr1 primers + ndr1 plasmid.
Because cycb16 is a deletion of unknown extent (30), we turned to an ethyl nitroso urea-induced allele, cyctf219 (31), for further linkage analysis, using a restriction fragment length polymorphism in the ndr2 gene between the cyctf219 carrier strain and the WIK reference strain. The segregation of this restriction fragment length polymorphism was followed into phenotypically mutant and wild-type sibling pools of 50 F2 embryos derived from a cross between a cyctf219 heterozygous carrier and the WIK line. The restriction fragment length polymorphism segregated with cyc in two independent experiments, demonstrating that ndr2 and cyc are linked (Fig. 2A).
Figure 2.
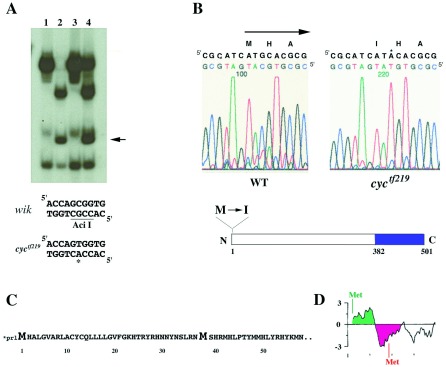
(A) ndr2 is linked to cyctf219, as shown by segregation of an Aci I restriction polymorphism in ndr2 sequences (16) amplified from genomic DNAs of a mapping cross. Lanes: 1, heterozygous cyctf219 G0 fish; 2, homozygous wild-type G0 fish (WIK strain); 3, F2 mutant sibs (homozygous cyctf219); 4, F2 phenotypically wild-type (heterozygous and homozygous wild-type) sibs. Arrow denotes fragment generated by Aci I site present in the WIK but not the cyctf219 strain; this restriction fragment length polymorphism segregates with the cyc mutation, demonstrating linkage. (B) The first methionine of the ndr2 ORF is changed to isoleucine in cyctf219; the M→I mutation was verified in two independent amplification products. In the schematic drawing, shading denotes region of mature Ndr2 polypeptide (382–501). (C) Translation of ndr2 sequence showing the first 61 aa of the ORF (upper case). Asterisk denotes the first in-frame stop codon upstream of the initiating methionine. (D) Hydrophobicity plot of the N-terminal region of Ndr2, generated with the pepplot program of the GCG package; green and red denote hydrophobic and hydrophilic regions, respectively.
To determine the molecular nature of the cyctf219 mutation, we sequenced the ndr2 protein-coding region including the exon/intron boundaries in DNA from cyctf219 homozygous mutant embryos. Several sequence alterations in the cyctf219 allele were observed by comparison to the originally published sequence of ndr2 (Table 1). We further determined the ndr2 coding sequence in the ethyl nitroso urea-mutagenized founder fish from which the cyctf219 mutation was derived. Only a single nucleotide difference, changing the methionine initiation codon to an isoleucine, was identified between the cyctf219 allele and the founder fish (Fig. 2B), whereas the other changes constitute polymorphisms. Two independent PCR amplification and sequencing experiments were carried out to confirm the methionine-to-isoleucine mutation in the initiation codon.
Table 1.
Polymorphisms in the ndr2 gene
| Position | WT | Founder | Change |
|---|---|---|---|
| 34 | T | C | – |
| 87 | G | A | R → H |
| 577 | A | T | – |
| 673 | G | A | – |
| 720 | C | T | A → V |
| 725 | G | A | A → T |
| 728 | C | T | P → S |
| 839 | G | C | V → L |
| 928 | A | T | – |
| 1057 | G | A | – |
| 1063 | T | C | – |
| 1360 | T | C | – |
Position refers to the sequence of the cloned ndr2 cDNA reported previously (28), which is listed as WT (wild type); only protein-coding sequence is considered. Sequence changes in the founder fish in which the cyctf219 mutation was induced are listed, and resulting amino acid changes are given. The founder fish was phenotypically wild type, so these changes are considered polymorphisms. The changes at positions 720, 725, and 728 also were identified as polymorphisms within the cDNA library from which the original ndr2 clone was isolated.
The cyctf219 Allele Encodes an Inactive Protein.
The next AUG codon downstream of the initiation codon in ndr2 occurs in frame at position 38. Initiation at this codon is expected to lead to a protein lacking an effective signal sequence, as judged by hydrophobicity plots (Fig. 2 C and D). To test for possible residual activity of such a truncated protein, we prepared a construct in which the N-terminal 37 codons were deleted and an optimized Kozak sequence was generated at the second methionine; this construct parallels the wild-type ndr2 expression construct used for comparison (28). Coupled in vitro transcription–translation showed that the wild-type and truncated proteins were made with equal efficiency (Fig. 3A). When dog pancreas microsomes were added to the translation reaction, approximately one-third of the wild-type protein product was converted to a slower migrating form that was sensitive to endoglycosidase H; in contrast, no endoglycosidase-sensitive form was detected in the translation products of the truncated protein (Fig. 3A). These results indicate that truncated ndr2 RNA can be translated but not processed.
Figure 3.
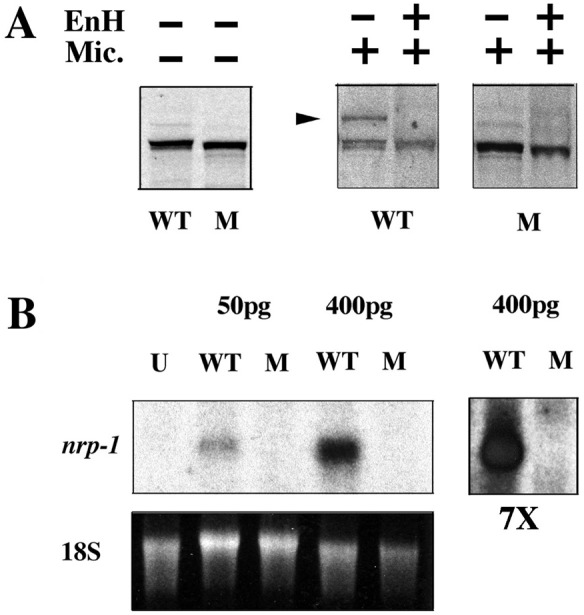
(A) The Ndr2 protein encoded by the cyctf219 allele lacks a functional signal sequence, as judged by the failure to be glycosylated. Proteins from coupled transcription–translation reactions with expression plasmids that initiate at the wild-type met (WT) or the second met at position 38 (corresponding to cyctf219, denoted M) were analyzed under denaturing and reducing conditions. Aliquots were treated with or without dog pancreatic microsomes (Mic.) and±endoglycosidase H. The arrow indicates the glycosylated form of the polypeptide. (B) Northern blot analysis of the in vivo activity of wild-type (WT) and mutant (M) RNA in the Xenopus animal cap assay, using nrp-1 as a probe for neural induction (28). U, uninjected; pg, amount of RNA injected per embryo; 18S rRNA was used as a loading control; 7×, sevenfold longer exposure of the two lanes at the right to illustrate the inactivity of the mutant RNA.
A bioassay for the function of the mutant protein was based on previous work showing that Ndr2 can induce neural markers in Xenopus animal caps (28). Whereas the product encoded by wild-type ndr2 RNA was effective in inducing the neural marker nrp-1 as shown previously (28), the truncated form encoded by the mutant RNA was inactive (Fig. 3B). These results indicate that the cyctf219 locus is likely to produce a protein, but this protein has little or no biological activity because of deficient secretion and/or processing. Our results agree with the fact that cyctf219 is a strong allele, with a phenotype that is almost as severe as the deletion allele cycb16 (15, 31). Taken together, the linkage data, sequence alteration, and functional analysis of mutant RNA provide strong evidence that the Ndr2 protein is encoded by the cyc locus; thus, we rename ndr2 as “cyclops.”
Autoregulation of cyclops Expression in Late Gastrulation.
The availability of the cyctf219 missense mutation allowed us to test whether the Cyc protein regulates the expression of its own gene. The expression pattern of cyc RNA in the cyctf219 background was unaffected in the axial hypoblast of the shield (not shown; see ref. 28). At the bud stage, the cyc RNA has a major anterior and a smaller posterior expression domain (Fig. 4A). Among these regions, expression in the polster, the precursor to the hatching gland, was unaffected, whereas expression in the rest of the prechordal plate, anterior notochord, and ventral neuroectoderm was greatly reduced in homozygous cyctf219 mutant embryos (Fig. 4 B–E). The expression in the tail bud was largely unaffected by the mutation (Fig. 4 C and E). In a weaker allele, cyctu43x, cyc expression is altered in a similar but less severe way (not shown). Cyc RNA disappears from the embryo shortly after tail bud and reappears transiently at the 20-somite stage in a left–right asymmetric manner in the lateral plate and a restricted region of the diencephalon (28). When phenotypically mutant cyctf219 embryos were examined by in situ hybridization at the 20-somite stage, staining was detected bilaterally in the brain and lateral plate (Fig. 4 F and G). Thus, Cyclops activity appears to be required to maintain the expression of the cyc gene in the anterior axial mesoderm and ventral neuroectoderm. It is also possible that the differentiation of cells that express cyc is impaired in the mutant, preventing normal expression of the gene. These experiments further show that cyc activity is required for the maintenance of its asymmetric expression during later development. Cyclops gene expression also is affected by other mutations that act in midline differentiation. For example, as we reported previously, the anterior domain of cyc RNA expression is widened in no tail mutant embryos (28), in which medial floor plate also is expanded (32).
Rescue of cyc Mutants by Injection of Expression Constructs.
Cell transplantation experiments suggested that expression of the wild-type cyc gene product in mutant embryos could locally rescue some aspects of the phenotype (15, 16). To test for possible rescue with the cloned gene product, we injected DNA constructs containing the cyc gene (see Materials and Methods) into the progeny of an intercross of cycb16 individuals. The resulting embryos were examined by morphology and by in situ hybridization with marker genes and then genotyped by PCR with primers spanning an intron (see Fig. 5J). As summarized in Table 2, 74% of injected mutant embryos showed rescue, in some cases to essentially wild-type appearance. The twhh gene is expressed widely in the ventral brain and is a useful marker because most of this expression is lost in cyc mutants (ref. 33; Fig. 5 A and B). In some injected embryos (Fig. 5C), much of the normal expression pattern of twhh has been restored; this embryo is genotypically mutant (Fig. 5J). Likewise, expression of twhh in the medial floor plate was restored in either a patchy or continuous manner by DNA injection (Fig. 5 D–F and K), and floor plate rescue also could be seen by inspection under DIC. Partial rescue of both head structure and floor plate is illustrated by staining with different markers, shh and pax2, in Fig. 5 G–I. At the dose used in these experiments, little if any ectopic marker expression was seen, emphasizing the specificity of the phenotypic rescue.
Figure 5.
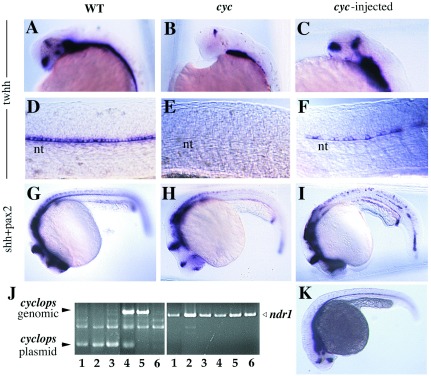
Rescue of the cycb16 phenotype by injection of a cyc expression construct. Wild-type (A, D, and G), cyc (B, E, and H), and cyc rescued by injection of 0.5 pg DNA per embryo (C, F, I, and K) are shown, as hybridized with twhh (A–F and K) or shh and pax2 (G–I). (J) After staining, the embryos were genotyped by PCR with primers that span an intron in the cyc gene; injected embryos also showed amplification product from the intronless expression plasmid. Aliquots of the same DNA samples also were amplified with primers for the unlinked ndr1 gene as a positive control. Lanes: 1, amplification of DNA from embryo in C; 2, F; 3, I; 4, injected wild type; 5, uninjected wild type; 6, uninjected cyc.
Table 2.
Phenotypic rescue of cycb16 mutant embryos by injection of a cyc expression construct
| Expt. | n | cyc−/− | Rescue
|
No rescue | |||
|---|---|---|---|---|---|---|---|
| Extensive | H and FP | H | FP | ||||
| 1 | 46 | 8 | 3 | 1 | 1 | 1 | 2 |
| 2 | 55 | 11 | 2 | 2 | 1 | 3 | 3 |
Mutant embryos were identified by PCR with genomic DNA (Fig. 5J). Among mutant embryos, the extent of rescue is classified as extensive if they were not distinguishable from wild type by visual inspection, and twhh expression in the floor plate was continuous. Some embryos showed partial rescue in head (H), floor plate (FP), or both. See Fig. 5 for examples. In sum, 14 of 19 (74%) of injected mutant embryos showed some level of rescue.
DISCUSSION
The expression pattern and properties of the cyc gene product (28) are consistent with the phenotypes of the mutation. Although cyc RNA is not detected in the notochord, head mesenchyme, differentiated floor plate, or ventral diencephalon at a time when the mutant phenotype becomes apparent, the cyc RNA is detected during earlier stages of development in tissues that contain progenitor cells for the structures that are affected by the mutation (34, 35). Since cyc encodes a signaling molecule, this suggests that the inductive interactions underlying the specification of midline derivatives, especially the medial floor plate cells and ventral midline brain structures, occur earlier than might have been expected from the phenotype, i.e., during gastrulation when the cyc gene is expressed. This idea is supported by the observation that the expression of goosecoid within prechordal plate and prospective anterior neuroectoderm is affected as early as the midgastrula stage in cyclops mutants (20). This view is also consistent with the conclusion that notochord and floor plate have a common pool of precursor cells (36, 37), implying that midline signaling in ventral CNS patterning is initiated during the formation of both mesodermal and neural midline structures rather than after the notochord and neural tube already are formed. Nevertheless, it is also conceivable that the Cyc protein might perdure beyond the persistence of the RNA and could be required in the embryo during the time of differentiation of ventral structures in the CNS.
The results reported in this paper focus on Cyclops, a member of the nodal-related subgroup of the transforming growth factor type β superfamily, as an essential signaling factor in the specification of the medial floor plate, the ventral diencephalon, and other ventral fates within the zebrafish CNS. C. Wright and colleagues (K. Sampath, A. L. Rubinstein, A. M. S. Cheng, J. Liang, K. Fekany, L. Solnica-Krezel, V. Korzh, M. E. Halpern, and C. V. E. Wright, personal communication) have identified independently the same nodal-related factor as the product of the cyc locus. Nodal, the prototype and only known member in this group in the mouse, is required for gastrulation, mesoderm formation, and head development in that animal (38–41). In zebrafish, a genome duplication event apparently has partitioned these functions among multiple nodal-related factors, as Talbot and colleagues (B. Feldman, M. Gates, E. Egan, G. Rennebeck, A. Schier, and W. Talbot, personal communication) have shown that the squint gene, which interacts with cyc in gastrulation, in germ layer induction, and in the patterning of the CNS, encodes a distinct nodal-related factor (which we separately isolated and named Ndr1; ref. 28). In addition to their role in midline signaling, some nodal-related factors including Cyclops are implicated in the establishment of left–right asymmetry of the heart and other organs in different vertebrate animals (reviewed in ref. 27).
Multiple factors appear to be responsible for the midline signals required for patterning of the ventral CNS (3–8), both during and after gastrulation (37, 42–44). In addition to the nodal-related factors encoded by cyclops and squint, Shh (8, 9, 12), BMP-7 (42), and Oep (14) are implicated in patterning of the ventral CNS. Oep is required early during gastrulation, and its expression pattern partially overlaps that of cyc. It is conceivable that cyc and oep interact in common or parallel pathways in the induction of medial floor plate cells, and beyond this it is possible that some effects of the cyc mutation are caused by the loss of Shh and Twhh expression in many midline cells (22, 33). Because neither Shh nor Twhh suffices to completely rescue the cyclops phenotype (23, 24), the relation between Cyclops and the Hedgehogs is not likely to be in a linear pathway. One possibility is that Cyclops activity may be required in addition to Hedgehog signaling to antagonize dorsalizing factors (e.g., BMPs and Dorsalin) expressed within the roof plate and overlying ectoderm. This view is consistent with the failure to maintain expression of Shh-dependent genes in the ventral neural tube in mouse noggin null mutants in which the balance between BMP signaling and its antagonistic regulators is disrupted (45). Such a hypothesis also is consistent with the ability of Cyclops to act as a strong neuralizing inducer in the Xenopus animal cap assay (28), an activity that is likely to depend on antagonizing a BMP signal (1, 2). The identification of the product of the cyclops gene as a new nodal-related factor should aid in unraveling the relationships between the factors involved in midline signaling and CNS patterning.
Acknowledgments
We thank H.-G. Fronhöfer for mutant strains of zebrafish, C. Fricke, S. Geiger-Rudolph, and E. Laver for assistance, and C. Wright and W. Talbot for sharing unpublished information, and Ajay Chitnis for comments on the manuscript.
ABBREVIATIONS
- BMP
bone morphogenetic protein
- CNS
central nervous system
References
- 1.Sasai Y, De Robertis E M. Dev Biol. 1997;182:5–20. doi: 10.1006/dbio.1996.8445. [DOI] [PubMed] [Google Scholar]
- 2.Hemmati-Brivanlou A, Melton D. Annu Rev Neurosci. 1997;20:43–60. doi: 10.1146/annurev.neuro.20.1.43. [DOI] [PubMed] [Google Scholar]
- 3.Placzek M, Furley A. Curr Biol. 1996;6:526–529. doi: 10.1016/s0960-9822(02)00533-x. [DOI] [PubMed] [Google Scholar]
- 4.Placzek M, Tessier-Lavigne M, Yamada T, Jessell T, Dodd J. Science. 1990;250:985–988. doi: 10.1126/science.2237443. [DOI] [PubMed] [Google Scholar]
- 5.Clarke J D, Holder N, Soffe S R, Storm-Mathisen J. Development. 1991;112:499–516. doi: 10.1242/dev.112.2.499. [DOI] [PubMed] [Google Scholar]
- 6.Yamada T, Pfaff S L, Edlund T, Jessell T M. Cell. 1993;73:673–686. doi: 10.1016/0092-8674(93)90248-o. [DOI] [PubMed] [Google Scholar]
- 7.Tanabe Y, Jessell T M. Science. 1996;274:1115–1123. doi: 10.1126/science.274.5290.1115. [DOI] [PubMed] [Google Scholar]
- 8.Hammerschmidt M, Brook A, McMahon A P. Trends Genet. 1997;13:14–21. doi: 10.1016/s0168-9525(96)10051-2. [DOI] [PubMed] [Google Scholar]
- 9.Chiang C, Litingtung Y, Lee E, Young K E, Corden J L, Westphal H, Beachy P A. Nature (London) 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 10.Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell H F, Donis-Keller H, Helms C, Hing A V, et al. Nat Genet. 1996;14:353–356. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- 11.Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer S W, Tsui L C, Muenke M. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- 12.Schauerte H E, van Eeden F J M, Fricke C, Odenthal J, Strähle U, Haffter P. Development. 1998;125:2983–2993. doi: 10.1242/dev.125.15.2983. [DOI] [PubMed] [Google Scholar]
- 13.Monaghan A P, Kaestner K H, Grau E, Schütz G. Development. 1993;119:567–578. doi: 10.1242/dev.119.3.567. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Talbot W S, Schier A F. Cell. 1998;92:241–251. doi: 10.1016/s0092-8674(00)80918-6. [DOI] [PubMed] [Google Scholar]
- 15.Hatta K, Kimmel C B, Ho R K, Walker C. Nature. 1991;350:339–341. doi: 10.1038/350339a0. [DOI] [PubMed] [Google Scholar]
- 16.Hatta K, Püschel A W, Kimmel C B. Proc Natl Acad Sci USA. 1994;91:2061–2065. doi: 10.1073/pnas.91.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatta K. Neuron. 1992;9:629–642. doi: 10.1016/0896-6273(92)90027-b. [DOI] [PubMed] [Google Scholar]
- 18.Chen J N, van Eeden F J, Warren K S, Chin A, Nüsslein-Volhard C, Haffter P, Fishman M C. Development. 1997;124:4373–4382. doi: 10.1242/dev.124.21.4373. [DOI] [PubMed] [Google Scholar]
- 19.Chandrasekhar A, Moens C B, Warren J T, Kimmel C B, Kuwada J Y. Development. 1997;124:2633–2644. doi: 10.1242/dev.124.13.2633. [DOI] [PubMed] [Google Scholar]
- 20.Thisse C, Thisse B, Halpern M E, Postlethwait J H. Dev Biol. 1994;164:420–429. doi: 10.1006/dbio.1994.1212. [DOI] [PubMed] [Google Scholar]
- 21.Yan Y L, Hatta K, Riggleman B, Postlethwait J H. Dev Dyn. 1995;203:363–376. doi: 10.1002/aja.1002030308. [DOI] [PubMed] [Google Scholar]
- 22.Krauss S, Concordet J P, Ingham P W. Cell. 1993;75:1431–1444. doi: 10.1016/0092-8674(93)90628-4. [DOI] [PubMed] [Google Scholar]
- 23.Ungar A R, Moon R T. Dev Biol. 1996;178:186–191. doi: 10.1006/dbio.1996.0209. [DOI] [PubMed] [Google Scholar]
- 24.Strähle U, Fischer N, Blader P. Mech Dev. 1997;62:147–160. doi: 10.1016/s0925-4773(97)00657-6. [DOI] [PubMed] [Google Scholar]
- 25.Hammerschmidt M, Bitgood M J, McMahon A P. Genes Dev. 1996;10:647–658. doi: 10.1101/gad.10.6.647. [DOI] [PubMed] [Google Scholar]
- 26.Smith J C. Curr Opin Cell Biol. 1995;7:856–861. doi: 10.1016/0955-0674(95)80070-0. [DOI] [PubMed] [Google Scholar]
- 27.Levin M. Bioessay. 1997;19:287–296. doi: 10.1002/bies.950190406. [DOI] [PubMed] [Google Scholar]
- 28.Rebagliati, M. R., Toyama, R., Fricke, C., Haffter, P. & Dawid, I. B. (1998) Dev. Biol., in press. [DOI] [PubMed]
- 29.Turner D L, Weintraub H. Genes Dev. 1994;8:1434–1447. doi: 10.1101/gad.8.12.1434. [DOI] [PubMed] [Google Scholar]
- 30.Talbot W S, Egan E S, Gates M A, Walker C, Ullmann B, Neuhauss S C, Kimmel C B, Postlethwait J H. Genetics. 1998;148:373–380. doi: 10.1093/genetics/148.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brand M, Heisenberg C P, Warga R M, Pelegri F, Karlstrom R O, Beuchle D, Picker A, Jiang Y J, Furutani-Seiki M, van Eeden F J, et al. Development. 1996;123:129–142. doi: 10.1242/dev.123.1.129. [DOI] [PubMed] [Google Scholar]
- 32.Odenthal J, Haffter P, Vogelsang E, Brand M, van Eeden F J, Furutani-Seiki M, Granato M, Hammerschmidt M, Heisenberg C P, Jiang Y J, et al. Development. 1996;123:103–115. doi: 10.1242/dev.123.1.103. [DOI] [PubMed] [Google Scholar]
- 33.Ekker S C, Ungar A R, Greenstein P, von Kessler D P, Porter J A, Moon R T, Beachy P A. Curr Biol. 1995;5:944–955. doi: 10.1016/s0960-9822(95)00185-0. [DOI] [PubMed] [Google Scholar]
- 34.Kimmel C B, Warga R M, Schilling T F. Development. 1990;108:581–594. doi: 10.1242/dev.108.4.581. [DOI] [PubMed] [Google Scholar]
- 35.Woo K, Fraser S E. Development. 1995;121:2595–2609. doi: 10.1242/dev.121.8.2595. [DOI] [PubMed] [Google Scholar]
- 36.Catala M, Teillet M A, De Robertis E M, Le Douarin M L. Development. 1996;122:2599–2610. doi: 10.1242/dev.122.9.2599. [DOI] [PubMed] [Google Scholar]
- 37.Halpern M E, Hatta K, Amacher S L, Talbot W S, Yan Y L, Thisse B, Thisse C, Postlethwait J H, Kimmel C B. Dev Biol. 1997;187:154–170. doi: 10.1006/dbio.1997.8605. [DOI] [PubMed] [Google Scholar]
- 38.Zhou X, Sasaki H, Lowe L, Hogan B L, Kuehn M R. Nature (London) 1993;361:543–547. doi: 10.1038/361543a0. [DOI] [PubMed] [Google Scholar]
- 39.Conlon F L, Lyons K M, Takaesu N, Barth K S, Kispert A, Herrmann B, Robertson E J. Development. 1994;120:1919–1928. doi: 10.1242/dev.120.7.1919. [DOI] [PubMed] [Google Scholar]
- 40.Varlet I, Collignon J, Robertson E J. Development. 1997;124:1033–1044. doi: 10.1242/dev.124.5.1033. [DOI] [PubMed] [Google Scholar]
- 41.Iannaccone P M, Zhou X, Khokha M, Boucher D, Kuehn M R. Dev Dyn. 1992;194:198–208. doi: 10.1002/aja.1001940305. [DOI] [PubMed] [Google Scholar]
- 42.Dale J K, Vesque C, Lints T J, Sampath T K, Furley A, Dodd J, Placzek M. Cell. 1997;90:257–269. doi: 10.1016/s0092-8674(00)80334-7. [DOI] [PubMed] [Google Scholar]
- 43.Shimamura K, Rubenstein J L. Development. 1997;124:2709–2718. doi: 10.1242/dev.124.14.2709. [DOI] [PubMed] [Google Scholar]
- 44.Houart C, Westerfield M, Wilson S W. Nature (London) 1998;391:788–792. doi: 10.1038/35853. [DOI] [PubMed] [Google Scholar]
- 45.McMahon J A, Takada S, Zimmerman L B, Fan C M, Harland R M, McMahon A P. Genes Dev. 1998;12:1438–1452. doi: 10.1101/gad.12.10.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamada T, Pfaff S L, Edlund T, Jessell T M. Cell. 1993;73:673–686. doi: 10.1016/0092-8674(93)90248-o. [DOI] [PubMed] [Google Scholar]