

Abstract
Although the folding of α-helical repeat proteins has been well characterized, much less is known about the folding of repeat proteins containing β-sheets. Here we investigate the folding thermodynamics and kinetics of the leucine-rich repeat (LRR) domain of Internalin B (InlB), an extracellular virulence factor from the bacterium Lysteria monocytogenes. This domain contains seven tandem leucine-rich repeats, of which each contribute a single β-strand that forms a continuous β-sheet with neighboring repeats, and an N-terminal α-helical capping motif. Despite its modular structure, InlB folds in an equilibrium two-state manner, as reflected by the identical thermodynamic parameters obtained by monitoring its sigmoidal urea-induced unfolding transition by different spectroscopic probes. Although equilibrium two-state folding is common in α-helical repeat proteins, to date, InlB is the only β-sheet-containing repeat protein for which this behavior is observed. Surprisingly, unlike other repeat proteins exhibiting equilibrium two-state folding, InlB also folds by a simple two-state kinetic mechanism lacking intermediates, aside from the effects of prolyl isomerization on the denatured state. However, like other repeat proteins, InlB also folds significantly more slowly than expected from contact order. When plotted against urea, the rate constants for the fast refolding and single unfolding phases constitute a linear chevron that, when fitted with a kinetic two-state model, yields thermodynamic parameters matching those observed for equilibrium folding. Based on these kinetic parameters, the transition state is estimated to comprise 40% of the total surface area buried upon folding, indicating that a large fraction of the native contacts are formed in the rate-limiting step to folding.
Keywords: repeat protein, leucine-rich repeat, protein folding, kinetics
Repeat proteins, which are composed of linear tandem arrays of repeated secondary structural elements, make excellent model systems for folding studies. Repeat proteins have the same secondary structural elements and close packing interactions as globular proteins; however, the modular, linear structure of repeat proteins gives rise to a local topology and low contact order that permits the study of the limits of folding cooperativity. Moreover, the structural redundancy of repeat proteins facilitates a comparison of the energy distribution and kinetic pathway selection among similar structural elements (Kloss et al. 2007). Recent studies have elucidated the folding pathways of several naturally occurring α-helical repeat proteins (Tang et al. 1999; Zeeb et al. 2002; Bradley and Barrick 2006; Lowe and Itzhaki 2007b). Although most helical repeat proteins fold via an equilibrium, two-state mechanism, the kinetic pathways of these proteins are complex, often involving one or more on-pathway intermediates (Kloss et al. 2007).
Compared to α-helical repeat proteins, the folding of repeat proteins with β-sheet structure has not been studied extensively (Kamen et al. 2000; Kamen and Woody 2001; Junker et al. 2006). Here we investigate the equilibrium and kinetic folding mechanism of the leucine-rich repeat (LRR) domain of Internalin B (InlB), a virulence factor from Listeria monocytogenes. The LRR domain of InlB is composed of a linear array of seven tandemly repeated LRR motifs of 22 residues, each made up of a short β-strand, followed by a tight turn and a 310 helix (Fig. 1). Consecutive repeats are connected by a turn and stack linearly. The N terminus of the LRR array is capped by a 40-residue α-helical motif; the C terminus is capped by a single β-strand, which continues the LRR β-sheet (Marino et al. 1999).1 This modular architecture results in a local topology that is free of the long-range contacts that typify most globular proteins.
Figure 1.

Sequence and ribbon diagram of the seven leucine-rich repeats and α-helical capping motif (gray) of InlB. (A) Sequence of the LRR domain of InlB studied here. Secondary structure assignments (x = any residue, a = α-helix, b = β-strand, h = 310 helix, s = small residue) are taken from Marino et al. (1999). The single cysteine, located in the C-terminal β-strand, was replaced with a serine. Residues not visible in the crystal structure are shown in italics. (B) Ribbon diagram of the crystal structure of InlB (Marino et al. 1999). The six trans-proline residues are shown in CPK representation. The single tryptophan, located in the third leucine-rich repeat, is shown in black. Panel B was generated using PyMOL (DeLano Scientific).
Given its size, architecture, and spectroscopic properties, InlB is well suited for studies of the cooperativity and kinetics of folding of LRR domains. In a previous study, Freiberg et al. (2004) found a single coincident folding transition by circular dichroism- (CD) and fluorescence-monitored chemical denaturation. However, the unfolding transition was unusually steep and was found to become even steeper upon addition of Ca2+. In combination with crystallographic evidence for Ca2+-mediated interactions, the high and variable steepness of the equilibrium unfolding transition observed by Freiberg et al. (2004) suggests that folding may be coupled to native-state association.
To better characterize the equilibrium folding mechanism of monomeric InlB and to characterize the kinetic mechanism of folding, we measured folding and unfolding of InlB using multiple spectroscopic probes, both at equilibrium and in kinetic studies. We have measured equilibrium unfolding transitions by far-UV CD in the β-sheet region, tryptophan fluorescence, and near-UV CD using a multiwavelength analysis. We have also investigated the potential for oligomer formation using analytical ultracentrifugation. These equilibrium spectroscopic and hydrodynamic studies allow us to examine the cooperativitiy of folding of a monomeric species, to assess whether the modular structure of this domain results in modular thermodynamics. The kinetic studies permit us to analyze the mechanism of folding, test for kinetic intermediates, evaluate the influence of the local topology on folding rate, and begin to investigate the structural and energetic properties of the transition state ensemble. This study lays the foundation for residue-specific analysis of LRR folding.
Results
Urea-induced equilibrium denaturation of Internalin B
To determine the stability and the extent of folding cooperativity of InlB, we monitored urea-induced denaturation by CD at 217 nm, tryptophan fluorescence, and near-UV CD. The primary contribution of the CD signal at 217 nm is β-sheet structure, which is distributed across the entire LRR domain. The primary contribution of fluorescence change upon unfolding is a single tryptophan residue located in the third repeat of the LRR domain of InlB (Fig. 1). The primary contributions to CD in the near-UV region, which reflects the amount of rigid tertiary structure in the protein, are specific side-chain interactions involving the aromatic residues (1 trp, 3 tyr) (see Supplemental Fig. S1 for full far- and near-UV CD spectra). Taken together, these three spectroscopic probes allow a comparison of local and global unfolding of InlB.
The CD at 217 nm and fluorescence-monitored urea denaturation curves are sigmoidal, have well-defined baselines, and can each be described by an equilibrium two-state model in which unfolding free energy is linearly dependent on denaturant concentration (Fig. 2A). The fitted thermodynamic parameters from far-UV CD and fluorescence are the same, within error. The near-UV CD-monitored denaturation also captures a sharp unfolding transition. Because there is substantial noise at each wavelength in the near-UV region, we collected full near-UV CD spectra as a function of urea concentration and globally fitted these spectra from 260 to 287 nm using a two-state model. As an independent means of analyzing the data, we integrated the near-UV spectra over the same wavelength range and fitted the resulting transition to a two-state model (Fig. 2B). Thermodynamic unfolding parameters fitted from the near-UV CD spectra using both methods of analysis match those determined by CD at 217 nm and by fluorescence (Table 1). This close agreement supports an equilibrium two-state folding pathway.
Figure 2.

Urea-induced equilibrium unfolding of InlB. (A) Denaturation curves were monitored by CD at 217 nm (circles) and tryptophan fluorescence (squares), and fit with a two-state model using the linear extrapolation method. (B) Urea-induced unfolding of InlB monitored by near-UV CD. Near-UV spectra were integrated from 260 to 287 nm at each urea concentration (circles) and fit with a two-state model (solid line). Conditions: 3 μM protein, 150 mM NaCl, 25 mM Tris (pH 8.0), 25°C.
Table 1.
Thermodynamic parameters determined from equilibrium and kinetic studies

The steepness of the unfolding transition permits a quantitative measure, through the m-value, of the cooperativity of unfolding. An observed linear relationship between the steepness of two-state unfolding transitions and chain length predicts an m-value of 2.45 kcal.mol−1.M−1 for InlB, assuming the helical capping motif and LRR domain are thermodynamically coupled (Myers et al. 1995). The close agreement between this predicted m-value and the m-values observed by monitoring equilibrium unfolding transitions by fluorescence, CD (217 nm), and near-UV CD (Table 1) further suggests that the seven LRRs unfold in a concerted reaction that includes the α-helical capping motif.
The m-values and free energies of unfolding that we determined from our equilibrium denaturation experiments are ∼30% smaller than those previously published (Freiberg et al. 2004). One possible explanation for this discrepancy is that the previous measurements were made at lower ionic strength, temperature, and pH.2 To explore this possibility, we monitored equilibrium denaturation in solution conditions identical to those used by Freiberg et al. (2004). Regardless of conditions, we obtain the same fitted thermodynamic parameters (data not shown). Therefore, the difference between the thermodynamic parameters we measured and those previously published do not appear to result from a difference in ionic strength or pH.
Association state of Internalin B
A second possible explanation for the higher m-value and free energy of unfolding reported by Freiberg et al. (2004) is self-association of folded InlB. The crystal structure of InlB reveals a large interface for potential self-association; this interface includes binding sites for two Ca2+ ions (Marino et al. 1999). In support of native-state association via this interface and its contribution to the anomalous m-value, Freiberg et al. (2004) found that the m-value further increases in the presence of Ca2+.
To investigate the state of association of InlB under the conditions used here and to examine the effects of Ca2+ on potential association, we performed sedimentation equilibrium analytical ultracentrifugation experiments of InlB in various concentrations of Ca2+. We find that a model involving a single, monomeric species is adequate to describe the sedimentation equilibrium of InlB both in the absence and presence of 50 mM Ca2+ (Fig. 3). The residuals from these monomeric fits are small and are not significantly decreased by the inclusion of a dimeric species. The molecular weights of the single species in these fits match the calculated molecular weight of monomeric InlB, suggesting that the protein is monomeric under these conditions (Table 2). This finding simplifies the analysis of both equilibrium and kinetic folding data, although it does not explain the higher cooperativity seen by Freiberg et al. (2004).
Figure 3.

Equilibrium analytical ultracentrifugation of InlB. Absorbance at 280 nm versus radial position at three different protein concentrations and rotor speeds in the presence of (A) 0 mM and (B) 50 mM Ca2+. Loading concentrations of InlB were 44 (inner sector), 26 (middle sector), and 9 μM (outer sector). Symbols indicate speeds of 23 (circles), 26 (triangles), and 30 krpm (X's). Solid lines are the result of simultaneous fitting of an ideal single-species model to the data. Upper panels show combined residuals for all three speeds and concentrations.
Table 2.
Molecular weights of InlB in varying concentrations of Ca2+ determined by analytical ultracentrifugation

Recently, a second mode of self-association of the LRR domain of InlB has been identified (Banerjee et al. 2004). This mode of asocciation is mediated by disulfide bond formation involving the single cysteine located at the C terminus of the LRR domain. This cysteine is present in the construct studied by Freiberg et al. (2004); however, we have replaced it with a serine to promote long-term stability and avoid intermolecular association (see Materials and Methods). The elimination of this dimer-promoting residue may account for the differences between the thermodynamic parameters we report and those published by Freiberg et al. (2004).
Refolding and unfolding kinetics of Internalin B
The equilibrium two-state folding and monomeric association state make InlB an ideal candidate for kinetic studies. To gain insight into the kinetic folding pathway of InlB, we monitored refolding and unfolding in various concentrations of urea both by tryptophan fluorescence and by CD at 217 nm. We found fluorescence-monitored refolding curves to be multiphasic, as evidenced by the nonrandom residuals generated by fitting a single exponential (Equation 6, i = 1) to the data (black curve, Fig. 4A). In contrast, refolding curves are well-fitted by a double exponential (Equation 6, i = 2; red curve, Fig. 4A), resulting in much smaller and more randomly distributed residuals (top panel, Fig. 4A). Higher order exponential fits (Equation 6, i = 3 or more) do not significantly improve χr 2 (data not shown). At 0.18 M urea, the fast refolding phase is the dominant phase, with an amplitude of ∼70% of the total observed signal change (Table 3, see below).
Figure 4.

Fluorescence-detected refolding and unfolding of InlB. (A) Refolding was initiated by rapid dilution of urea from 3 M to 0.18 M. Refolding data (circles) were fitted with single- and double-exponential functions (black and red solid lines, respectively) according to Equation 6. (B) Unfolding was initiated by rapid addition of urea from 0 M to 2.95 M. Unfolding data (circles) were fitted with a single-exponential function (black line) according to Equation 6. Upper panels show residuals for each fit. For clarity, only every 10th data point is shown in each panel.
Table 3.
Fitted parameters for fluorescence- and CD-detected refolding and unfolding of InlB

The CD-detected refolding kinetics are noisier than those collected by fluorescence. As a result, the residuals from a single-exponential fit to stopped-flow CD data (black curve, Fig. 5A) are substantially larger than those from fluorescence, and deviation from single-exponential decay is less obvious than for fluorescence (Fig. 4A). However, at early refolding times, a clear, systematic deviation can be seen between the stopped-flow CD data and a fitted single-exponential decay. This deviation is eliminated when a second exponential phase is included (red curve, Fig. 5A), and χr 2 decreases significantly.
Figure 5.

CD-detected refolding and unfolding of InlB. (A) Refolding was initiated by rapid dilution of urea from 3 M to 0.2 M. Data (circles) were fitted with single- and double-exponential fits (black and red solid lines, respectively). For the double-exponential fit, the slow rate and the relative amplitudes of the fast and slow phases were fixed at the values determined by fitting the tryptophan fluorescence-monitored kinetics at the same denaturant concentration. (B) Unfolding was initiated by rapid addition of urea from 0 M to 3.0 M. Data (circles) were fitted with a single-exponential function (black line) according to Equation 6. Upper panels show residuals for each fit.
Although these results provide clear indication that, like fluorescence-monitored refolding, the CD-monitored refolding transitions are best described by a double exponential, the low signal-to-noise ratio prevents an accurate determination of rate constants and amplitudes of both phases. To test whether the rate constant of the major CD-detected refolding phase matches that from fluorescence, CD-detected refolding curves were fitted with double-exponential functions in which the rate constant for the slower phase and the relative amplitudes (k 2, A 1, and A 2, Equation 6) were held fixed at the values extracted from the fluorescence data at each urea concentration. With these constraints, the rate constant for the fast folding phase (k 1) of InlB determined by CD is within error of that determined by fluorescence (Table 3).
In contrast to refolding, the unfolding of InlB appears to be a single-exponential process both by fluorescence and CD. For both probes, unfolding curves are well described by a single exponential (black curves, Figs. 4B, 5B), resulting in randomly distributed residuals (top panels, Figs. 4B, 5B). Inclusion of a second exponential does not result in a significant improvement in χr 2 (data not shown).
The role of prolyl isomerization in the slow refolding phase
InlB contains six trans proline residues, two in the helical capping motif and four distributed among the LRR repeats (Marino et al. 1999; Fig. 1). Two observations suggest that these six proline residues give rise to the slow kinetic refolding phase. First, the rate constant for the slow refolding phase (0.09 s−1 in 0.18 M urea; Table 3) is within the range reported for prolyl isomerization in unstructured peptides (0.01–0.1 s−1) (Brandts et al. 1975). Second, the rate constant for the slow phase has a shallow urea dependence (Fig. 7A, below), as are prolyl isomerization-limited refolding phases in other proteins (Schmid 1992).
Figure 7.

Chevron plot of InlB. (A) Folding and unfolding monitored by tryptophan fluorescence (fast phase, circles; slow phase, squares) and by CD at 217 nm (fast phase, diamonds). The fast phases detected by fluorescence and CD data were fitted simultaneously with a kinetic two-state model (black line), according to Equation 7. Fitting the fluorescence data alone yields similar values of 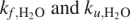 . Error bars are standard deviations on the mean of rate constants from five independent progress curves. (B) Relative amplitudes of the fast (circles) and slow (squares) refolding phases of InlB, determined by fitting fluorescence-monitored refolding data with a double-exponential function.
. Error bars are standard deviations on the mean of rate constants from five independent progress curves. (B) Relative amplitudes of the fast (circles) and slow (squares) refolding phases of InlB, determined by fitting fluorescence-monitored refolding data with a double-exponential function.
To further test whether these proline residues give rise to the slow refolding phase, we monitored the effects of cyclophilin, a peptidyl-prolyl isomerase, on the major and minor refolding rates of InlB by fluorescence. Cyclophilin increases the rate constant of the slow refolding phase by nearly fourfold, reaching a plateau at ∼2 μM cyclophilin (Fig. 6). Although the rate constant of the fast refolding phase appears to increase slightly in the presence of concentrations of cyclophilin above 2.5 μM, this may be a result of the convergence of the rates of the slow and fast phases at high concentrations of cyclophilin. Indeed, kinetic refolding curves are fitted equally well, based on χr 2 values, by a double exponential in which the rate constant of the fast phase is held fixed at the value determined in the absence of cyclophilin, whereupon the rate constant of the slow phase increases with cyclophilin in the same manner as when both rate constants are allowed to vary (not shown).
Figure 6.

The effect of cyclophilin on refolding rate constants of InlB. Denatured InlB was refolded by rapid dilution from 3 M into 0.27 M urea and varying concentrations of cyclophilin. Refolding rate constants (fast phase, circles; slow phase, squares) were determined by fitting with a double-exponential model.
Urea dependence of refolding and unfolding rates of Internalin B
For proteins that fold by simple two-state kinetic mechanisms, the rate constants for folding and unfolding show log-linear dependences on urea concentrations, and together constitute a simple V-shaped “chevron” plot. In contrast, proteins that fold via more complicated mechanisms often display curvature in the folding or unfolding limb of their chevron plots (Baldwin 1996). For InlB, the fast refolding phase and the unfolding phase constitute a linear chevron plot (Fig. 7A), suggesting a simple kinetic two-state model. Both the fast refolding phase and unfolding phase have a steep urea dependence. In contrast, the rate constant for the slow refolding phase is much less sensitive to urea concentration. A kinetic two-state model (Equation 7) fits the fluorescence rate constants well (solid line, Fig. 7A) and yields rates of folding and unfolding in water (kf and ku, respectively) of 1.2 s−1 and 1.3 × 10−3 s−1, respectively. Although the deviations between the rates determined by the two probes is greater than their associated errors at each urea concentration, similar rate constants are obtained when the fluorescence and CD rate constants are globally fitted to a kinetic two-state model (1.1 s−1 and 1.7 × 10−3 s−1, respectively; Table 3), suggesting that the fluorescence and CD describe the same folding and unfolding transitions. The amplitudes of the major and minor refolding phases are also sensitive to urea concentration (Fig. 7B). At low urea concentrations, the fast refolding phase is dominant; however, its amplitude decreases, at the expense of the slow refolding phase, as urea concentration increases. The two amplitudes cross at ∼1 M, above which the slow refolding phase becomes dominant.
A test for burst-phase intermediates
The linear chevron described by the major refolding and unfolding rate constants of InlB is suggestive of two-state kinetics. However, it is possible for a rapidly forming refolding intermediate to be populated within the dead time of the stopped flow (Kuwajima 1989; Kuwajima et al. 1991; Mann and Matthews 1993; Shastry and Roder 1998; Myers and Oas 2002). To test for such an intermediate, we compared initial values observed in stopped-flow fluorescence refolding experiments with values expected for the denatured and native states of InlB (YD and YN, respectively). At each urea concentration, YD values were estimated from a linear extrapolation of fluorescence signals arising from denatured protein in high concentrations of urea. YN values were measured by directly equilibrating native protein at each urea concentration. Fractional burst-phase changes (fB) were calculated as
 |
where Y 0 is the starting fluorescence signal in a stopped-flow refolding trace.
At all denaturant concentrations, the burst-phase amplitudes detected by fluorescence are small (Fig. 8). At the lowest urea concentration we examined (0.27 M), the burst-phase amplitude was 14% of the expected signal (that is, 86% of the expected total amplitude is observed). By 1 M urea, the burst-phase amplitude decreases to 5%. The linear decrease in burst-phase signal for InlB differs from the sigmoidal burst phases of several proteins with rapid refolding intermediates (Kuwajima 1989; Kuwajima et al. 1991; Mann and Matthews 1993; Shastry and Roder 1998; Myers and Oas 2002). Taken together, the small burst-phase signal and linear chevron support direct refolding from the denatured state.
Figure 8.

Fractional burst-phase amplitudes (circles) for refolding as a function of urea concentration in fluorescence-monitored experiments. Burst-phase amplitudes are calculated as the fractional difference between the observed and expected spectroscopic signal changes as determined by Equation 1. Total observed amplitudes (1 − fB, squares) are the fraction of the expected signal change that is captured by the refolding progress curve.
Discussion
Leucine-rich repeat-containing proteins such as InlB share several characteristics that make them interesting targets of folding studies. Free of the three-dimensional packing constraints that typify globular proteins, these elongated proteins can be quite large and thus may be expected to fold via complicated pathways involving both equilibrium and kinetic intermediates. Most LRR domains are also flanked by helical capping motifs at either or both termini (Ceulemans et al. 1999; Marino et al. 1999; Wu et al. 2000; Huyton and Wolberger 2007; Kim et al. 2007); in addition to enhancing the solubility of LRR domains, these caps may also contribute to the folding and possible self-association. The goal of this study is to determine and quantify the equilibrium and kinetic folding mechanisms of InlB, to gain insight into cooperativity, to test for potential folding intermediates, and to gain insight into the rate-limiting steps and transition state structure that control the folding of these unique LRR architectures.
Two-state equilibrium folding of InlB
The LRR domain of InlB folds in an equilibrium two-state manner, as evidenced by the coincident all-or-none transitions and the agreement between the thermodynamic parameters extracted from our equilibrium denaturation experiments monitored by different spectroscopic probes (far-UV CD, near-UV CD, and tryptophan fluorescence). The finding here that InlB folds by an equilibrium two-state mechanism is consistent with the finding of Freiberg et al. (2004), who reported identical far-UV CD- and tryptophan fluorescence-detected unfolding curves.
The fitted thermodynamic parameters reported by Freiberg et al. (2004) and the m-value in particular are significantly larger than those extracted from our equilibrium folding studies. This discrepancy does not result from a difference in solution conditions. Whereas the increased m-value observed by Freiberg et al. (2004) is suggestive of noncovalent self-association, we have found that InlB is monomeric in solution and that Ca2+ does not mediate association. It is possible that the discrepancy results from the single cysteine residue in the construct of Freiberg et al. (2004) (which we substituted with a serine residue in our construct), which has recently been implicated in self-association (Banerjee et al. 2004). Regardless of the source of discrepancy, confirmation that the native state of the InlB construct used in the present study is monomeric simplifies studies of both the thermodynamics and kinetics of folding.
The contribution of proline residues to refolding heterogeneity
InlB shows two well-resolved kinetic refolding phases. Several characteristics of the slow refolding phase are consistent with prolyl isomerization. First, the rate associated with the slow phase falls within the expected range for prolyl isomerization (0.01–0.1 s−1). Second, the rate of this phase is insensitive to urea concentration, while its associated amplitude increases as a function of urea concentration (Fig. 7A,B), as is often seen when folding is coupled to prolyl isomerization (Kiefhaber et al. 1992; Bradley and Barrick 2005; Mello et al. 2005). In addition, peptidyl-prolyl isomerase increases the rate of the minor phase while the rate of the major phase remains constant (Fig. 6). The amplitude associated with the slow refolding phase matches the amplitude of the slow phase of the Notch ankyrin repeat domain, which contains approximately the same number of proline residues (Bradley and Barrick 2005). Taken together, these observations suggest that the refolding heterogeneity arises from the cis–trans isomerization reactions of proline residues in the denatured state of InlB. Thus, the fast refolding phase can be considered to reflect the barrier (or barriers) to folding, independent of prolyl isomerization.
Two-state kinetic folding of InlB
The urea dependences of the rates of the fast refolding and unfolding phases give rise to a linear chevron (Fig. 7A). This chevron is consistent with a simple kinetic model where only two states, the native and denatured states, are populated upon folding:
 |
Here, D represents a denatured state ensemble composed of denatured proteins with all kinetically limiting proline residues in the trans configuration. However, a linear chevron may also result in a three-state mechanism in which the denatured state immediately and quantitatively converts to a kinetic intermediate during the dead time of the reaction at all denaturant concentrations where refolding is observed:
 |
In such cases the observed refolding kinetics monitors the formation of N from I, rather than D (Baldwin 1996). Such rapid refolding intermediates may be expected to show a large burst-phase refolding amplitude with a sigmoidal denaturant dependence. In contrast, InlB has a small burst-phase amplitude with a shallow linear urea dependence (Fig. 8). These observations do not support the formation of refolding intermediates, but instead support the two-state kinetic model.
A simple test for two-state kinetics is to compare equilibrium and two-state kinetic folding parameters. If the folding kinetics are two state, the thermodynamic parameters (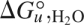 and m-value) determined from the ratio of folding and unfolding rate constants should match those determined from equilibrium measurements.3 For InlB, and m-values are within 0.1–0.2 kcal.mol−1 and 0.2–0.4 kcal.mol−1.M−1 of equilibrium values, respectively (Table 1). The agreement between these kinetic and equilibrium parameters strongly supports a kinetic two-state mechanism for InlB folding.
and m-value) determined from the ratio of folding and unfolding rate constants should match those determined from equilibrium measurements.3 For InlB, and m-values are within 0.1–0.2 kcal.mol−1 and 0.2–0.4 kcal.mol−1.M−1 of equilibrium values, respectively (Table 1). The agreement between these kinetic and equilibrium parameters strongly supports a kinetic two-state mechanism for InlB folding.
Size of the folding transition state
Because of the simple two-state kinetic folding mechanism, the rate constant of folding of InlB can be used to directly infer structural features of the transition state ensemble. The Tanford beta value (βT), which relates the urea dependence of the rate of folding to the equilibrium dependence (see Materials and Methods), provides an estimate of the degree of solvent accessibility of the transition state relative to the native state (Tanford 1968, 1970). The value of βT for InlB is 0.40 (Table 1), suggesting that its folding transition state ensemble involves a little less than half of the protein. This value may reflect partial structure formation throughout the protein or complete structure formation in one region of the protein.
Comparison with other repeat proteins
Although exceptions exist, most helical repeat proteins have been found to fold in a thermodynamic, two-state transition, in which intermediate states are not populated at equilibrium (Tang et al. 1999; Zweifel and Barrick 2001; Mosavi et al. 2002; Lowe and Itzhaki 2007a). This result is surprising given the local topology and absence of long-range interactions characteristic of repeat proteins. It has been suggested that this high level of cooperativity arises from stabilizing interactions at the interfaces between energetically unstable repeats (Mello and Barrick 2004; Kajander et al. 2005). In contrast, two β-sheet-containing repeat proteins for which equilibrium folding studies have been reported, pertactin and pelC, show multistate equilibrium unfolding (Kamen et al. 2000; Junker et al. 2006).
The two-state equilibrium unfolding of InlB demonstrates that such behavior is not exclusive to helical repeat proteins, but can be maintained in β-sheet-containing LRRs. This behavior may reflect the fact that the elongated β-sheet structure of InlB involves interrepeat hydrogen bonding spanning all seven repeats, although such a structure is apparently not sufficient to impart two-state unfolding on pertactin and pelC. It is also quite likely that, like ankyrin and tetratricopeptide repeats, individual folded LRRs are very energetically unstable and are unlikely to populate folded conformations without docking to one another. This intrinsic instability decreases the likelihood of populating intermediates composed of only a few folded repeats at equilibrium, resulting in an all-or-none transition.
In contrast to the high degree of cooperativity revealed by equilibrium experiments, the kinetics of naturally occurring repeat proteins are much more complex, as evidenced by their multiple kinetic phases and nonlinear chevron plots (Tang et al. 1999; Zeeb et al. 2002; Mello et al. 2005). This complexity is consistent with on-pathway kinetic refolding and unfolding intermediates. Surprisingly, the folding kinetics of InlB conforms to a simple two-state mechanism. To our knowledge, InlB is the first naturally occurring repeat protein to be shown to possess such a simple kinetic mechanism.4 It remains to be seen whether two-state kinetics is a general feature of LRR domains or whether it is specific to InlB. Irrespective of generality, InlB is an ideal candidate for further residue-specific analysis of its folding pathway via Φ-value analysis.
Another common feature of repeat-protein folding is slow kinetics (Tang et al. 1999; Zeeb et al. 2002; Mello et al. 2005; Lowe and Itzhaki 2007a). Slow folding of repeat proteins is surprising because an empirical inverse relationship between relative contact order and folding rate predicts that repeat proteins should have very fast folding rates (Plaxco et al. 1998). InlB is no exception to this trend. With an extrapolated rate constant of 1.2 s−1 and a relative contact order of 4%, InlB folds approximately six orders of magnitude slower than predicted. It has been suggested that repeat proteins may fold slowly because two or more intrinsically unstable repeats must fold and subsequently dock together in the formation of the folding transition state (Mello and Barrick 2004; Kloss et al. 2007). A similar mechanism may limit the rate of folding of InlB, whose transition state buries approximately half its solvent-accessible surface area, based on the βT-value determined here.
Materials and Methods
Protein expression and purification
The leucine-rich repeat domain and N-terminal capping motif of InlB were expressed from the plasmid pET28a (Novagen), a gift from the laboratories of Partho Ghosh and Pascale Cossart. The single cysteine in the leucine-rich repeat domain was replaced with a serine using the QuikChange Mutagenesis Kit (Stratagene) to avoid dimerization and promote long-term stability and reversible denaturation. InlB was expressed in Escherichia coli BL21 (DE3) cells by addition of 1 mM IPTG at OD600 = 0.6–0.8 for 4 h. Bacteria were lysed in 600 mM NaCl, 15 mM Tris (pH 8.0), 15 mM Imidazole and InlB was purified by Ni2+ chromatography, followed by a Sephacryl S-100 gel filtration column. Purified protein was dialyzed into 150 mM NaCl, 25 mM Tris (pH 8.0) and frozen at −80°C. Human cyclophilin was expressed in E. coli strain XA90 from the plasmid pHNJ, a gift from the laboratory of Dr. Christopher Walsh. Cyclophilin was expressed and purified as previously described (Liu et al. 1990; Mello et al. 2005).
Equilibrium folding studies
Ultrapure urea was purchased from Amresco. Urea was dissolved in water and treated with a mixed bed resin at a concentration of 5 g per 100 mL. Protein was denatured in 6–8 M urea, 25 mM Tris (pH 8.0), 150 mM NaCl, and titrated into a square 1-cm fluorescence cell containing native protein in 25 mM Tris (pH 8.0), 150 mM NaCl, using a Hamilton Microlab 500 titrator. Samples were equilibrated for 300 s at each urea concentration prior to signal acquisition. Urea-induced unfolding transitions were performed at 25°C and tracked by CD at 217 nm and by tryptophan fluorescence emission using a perpendicular 320-nm cutoff filter following excitation at 280 nm in an Aviv 62A DS Spectropolarimeter (Aviv Associates). Near-UV CD-monitored unfolding transitions were performed in the same way as described above in a J-810 spectropolarimeter (JASCO, Inc.). All equilibrium folding experiments were performed at 3 μM protein concentration.
Data were analyzed using the linear extrapolation method (Pace 1986; Bolen and Santoro 1988; Santoro and Bolen 1988), which assumes a linear relationship between the free energy of unfolding and urea concentration:
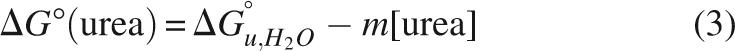 |
Applying the standard relationship between reaction free energy and equilibrium constant yields the following equation:
A two-state model relates the observed signal (Yobs), which is a population-weighted average of signals arising from the native and denatured states, to the unfolding equilibrium (Ku = [D]/[N]):
where fN and fD are the fraction of native and denatured protein, respectively. YN and YD depend linearly on urea. For far-UV CD- and fluorescence-monitored transitions, Equation 5 was fitted to urea-induced denaturation curves to obtain 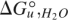 and m-values using the nonlinear least-squares analysis tool of Kaleidagraph 3.0 (Synergy Software). For near-UV CD spectra, global fitting was performed using the program Profit 6.0.0 (Quantum Soft) In the global fitting, a single and m-value were assumed to describe unfolding at all wavelengths, whereas the baseline parameters were treated as wavelength-specific local parameters.
and m-values using the nonlinear least-squares analysis tool of Kaleidagraph 3.0 (Synergy Software). For near-UV CD spectra, global fitting was performed using the program Profit 6.0.0 (Quantum Soft) In the global fitting, a single and m-value were assumed to describe unfolding at all wavelengths, whereas the baseline parameters were treated as wavelength-specific local parameters.
Analytical ultracentrifugation studies
Equilibrium analytical ultracentrifugation data were collected using a Beckman XL-A/XL-I ultracentrifuge (Beckman Coulter). Protein in 25 mM Tris (pH 8.0), 150 mM NaCl was dialyzed extensively at 4°C into the same buffer containing 0 mM, 10 mM, and 50 mM CaCl2 prior to data collection. Protein concentrations ranged from ∼8–45 μM. Samples were centrifuged at 25°C at 23,000, 26,000, and 30,000 rpm. Sedimentation equilibrium was established when concentration distributions, determined using absorbance optics, measured 2 h apart were coincident. Data were analyzed using the program SEDANAL (Stafford and Sherwood 2004).
Kinetic unfolding and refolding studies
Fluorescence-detected kinetic measurements of unfolding and refolding were made on an Applied Photophysics SX.18MV-R stopped-flow fluorometer. Fluorescence changes were detected perpendicular to excitation at 280 nm using a 320-nm cutoff filter to monitor the changes in the environment surrounding the single tryptophan located in the third repeat of the LRR domain. Circular dichroism-detected kinetic measurements were made on a J-810 spectropolarimeter with a Bio-Logic SFM-20 rapid mixing stopped-flow attachment (JASCO, Inc.; Bio-Logic). CD changes were monitored at 217 nm. Final protein concentrations were typically 2–4 μM and 4–10 μM for stopped-flow fluorescence and CD, respectively. Samples contained 150 mM NaCl, 25 mM Tris (pH 8.0) and were at 25°C.
Amplitudes and rate constants for unfolding and refolding were determined using nonlinear least-squares analysis to fit the following equation to individual progress curves:
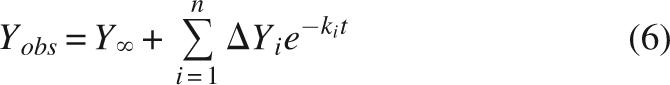 |
Y ∞ represents the fluorescence or CD signal at equilibrium, ΔYi represents the spectroscopic change contributed by the ith phase, and ki represents the rate constant for the ith phase. Progress curves were considered to be best described by the minimum number of phases (i = 1, 2, or 3) that produced a satisfactory fit, based on distribution residuals and reduced χ2 values.
The rate constants for refolding and unfolding were analyzed as a function of urea using a linear, kinetic two-state model, with the equation:
where mf and mu impart linear urea dependences on logkf and logku, respectively.
The Tanford beta value was calculated according to the following equation:
 |
where meq is the slope of the dependence of the rate constant of the fast folding phase on urea concentration and mkin is the kinetic m-value determined by fitting the chevron plot with a simple two-state model (Tanford 1968, 1970).
Acknowledgments
We thank Dr. Partho Ghosh and Dr. Pascale Cossart for providing us with the InlB clone, Dr. Christopher A. Walsh and C. Gary Marshall for providing a human cyclophilin expression construct, and Devon Sheppard for making the cysteine to serine substitution in the LRR domain of InlB. This work was supported by NIH grant GM68462, awarded to D.B.
Footnotes
In addition, in the full-length protein, the LLR domain is stabilized by a C-terminal immunoglobulin-like domain (Freiberg et al. 2004).
Unfolding transitions reported by Freiberg et al. (2004) were performed in 40 mM Tris-HCl, 1 mM EDTA (pH 7.0), 20°C; here we use 150 mM NaCl, 25 mM Tris-HCl (pH 8.0), 25°C.
In this comparison, the kinetically determined value must be corrected for the population of denatured state polypeptides with nonnative prolyl isomers. Here we used the relative amplitudes of the slow and fast refolding phases to estimate and correct for this population.
Myotrophin, a four ankyrin repeat-containing protein, has been suggested to fold by a kinetic two-state mechanism with a moving transition state. However, a separate report from the same group invokes an on-pathway intermediate. As a result, the kinetic mechanism of this protein remains unclear (Lowe and Itzhaki 2007a, b).
Supplemental material: see www.proteinscience.org
Reprint requests to: Doug Barrick, T.C. Jenkins Department of Biophysics, The Johns Hopkins University, 3400 North Charles Street, Baltimore, MD 21218, USA; e-mail: barrick@jhu.edu, fax: (410) 516-4118.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.073166608.
References
- Baldwin, R.L. On-pathway versus off-pathway folding intermediates. Fold. Des. 1996;1:R1–R8. doi: 10.1016/S1359-0278(96)00003-X. [DOI] [PubMed] [Google Scholar]
- Banerjee, M., Copp, J., Vuga, D., Marino, M., Chapman, T., van der Geer, P., Ghosh, P. GW domains of the Listeria monocytogenes invasion protein InlB are required for potentiation of Met activation. Mol. Microbiol. 2004;52:257–271. doi: 10.1111/j.1365-2958.2003.03968.x. [DOI] [PubMed] [Google Scholar]
- Bolen, D.W., Santoro, M.M. Unfolding free energy changes determined by the linear extrapolation method. 2. Incorporation of ΔG degrees N-U values in a thermodynamic cycle. Biochemistry. 1988;27:8069–8074. doi: 10.1021/bi00421a015. [DOI] [PubMed] [Google Scholar]
- Bradley, C.M., Barrick, D. Effect of multiple prolyl isomerization reactions on the stability and folding kinetics of the Notch ankyrin domain: Experiment and theory. J. Mol. Biol. 2005;352:253–265. doi: 10.1016/j.jmb.2005.06.041. [DOI] [PubMed] [Google Scholar]
- Bradley, C.M., Barrick, D. The Notch ankyrin domain folds via a discrete, centralized pathway. Structure. 2006;14:1303–1312. doi: 10.1016/j.str.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Brandts, J.F., Halvorson, H.R., Brennan, M. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis–trans isomerism of proline residues. Biochemistry. 1975;14:4953–4963. doi: 10.1021/bi00693a026. [DOI] [PubMed] [Google Scholar]
- Ceulemans, H., De Maeyer, M., Stalmans, W., Bollen, M. A capping domain for LRR protein interaction modules. FEBS Lett. 1999;456:349–351. doi: 10.1016/s0014-5793(99)00965-5. [DOI] [PubMed] [Google Scholar]
- Freiberg, A., Machner, M.P., Pfeil, W., Schubert, W.D., Heinz, D.W., Seckler, R. Folding and stability of the leucine-rich repeat domain of internalin B from Listeria monocytogenes . J. Mol. Biol. 2004;337:453–461. doi: 10.1016/j.jmb.2004.01.044. [DOI] [PubMed] [Google Scholar]
- Huyton, T., Wolberger, C. The crystal structure of the tumor suppressor protein pp32 (Anp32a): Structural insights into Anp32 family of proteins. Protein Sci. 2007;16:1308–1315. doi: 10.1110/ps.072803507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker, M., Schuster, C.C., McDonnell, A.V., Sorg, K.A., Finn, M.C., Berger, B., Clark, P.L. Pertactin β-helix folding mechanism suggests common themes for the secretion and folding of autotransporter proteins. Proc. Natl. Acad. Sci. 2006;103:4918–4923. doi: 10.1073/pnas.0507923103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajander, T., Cortajarena, A.L., Main, E.R., Mochrie, S.G., Regan, L. A new folding paradigm for repeat proteins. J. Am. Chem. Soc. 2005;127:10188–10190. doi: 10.1021/ja0524494. [DOI] [PubMed] [Google Scholar]
- Kamen, D.E., Woody, R.W. A partially folded intermediate conformation is induced in pectate lyase C by the addition of 8-anilino-1-naphthalenesulfonate (ANS) Protein Sci. 2001;10:2123–2130. doi: 10.1110/ps.19801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamen, D.E., Griko, Y., Woody, R.W. The stability, structural organization, and denaturation of pectate lyase C, a parallel β-helix protein. Biochemistry. 2000;39:15932–15943. doi: 10.1021/bi001900v. [DOI] [PubMed] [Google Scholar]
- Kiefhaber, T., Kohler, H.H., Schmid, F.X. Kinetic coupling between protein folding and prolyl isomerization. I. Theoretical models. J. Mol. Biol. 1992;224:217–229. doi: 10.1016/0022-2836(92)90585-8. [DOI] [PubMed] [Google Scholar]
- Kim, H.M., Oh, S.C., Lim, K.J., Kasamatsu, J., Heo, J.Y., Park, B.S., Lee, H., Yoo, O.J., Kasahara, M., Lee, J.O. Structural diversity of the hagfish variable lymphocyte receptors. J. Biol. Chem. 2007;282:6726–6732. doi: 10.1074/jbc.M608471200. [DOI] [PubMed] [Google Scholar]
- Kloss, E., Courtemanche, N., Barrick, D. Repeat protein folding: New insights into origins of cooperativity, stability, and topology. Arch. Biochem. Biophys. 2007 doi: 10.1016/j.abb.2007.08.03.4. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima, K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins. 1989;6:87–103. doi: 10.1002/prot.340060202. [DOI] [PubMed] [Google Scholar]
- Kuwajima, K., Garvey, E.P., Finn, B.E., Matthews, C.R., Sugai, S. Transient intermediates in the folding of dihydrofolate reductase as detected by far-ultraviolet circular dichroism spectroscopy. Biochemistry. 1991;30:7693–7703. doi: 10.1021/bi00245a005. [DOI] [PubMed] [Google Scholar]
- Liu, J., Albers, M.W., Chen, C.M., Schreiber, S.L., Walsh, C.T. Cloning, expression, and purification of human cyclophilin in Escherichia coli and assessment of the catalytic role of cysteines by site-directed mutagenesis. Proc. Natl. Acad. Sci. 1990;87:2304–2308. doi: 10.1073/pnas.87.6.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, A.R., Itzhaki, L.S. Biophysical characterisation of the small ankyrin repeat protein myotrophin. J. Mol. Biol. 2007a;365:1245–1255. doi: 10.1016/j.jmb.2006.10.060. [DOI] [PubMed] [Google Scholar]
- Lowe, A.R., Itzhaki, L.S. Rational redesign of the folding pathway of a modular protein. Proc. Natl. Acad. Sci. 2007b;104:2679–2684. doi: 10.1073/pnas.0604653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, C.J., Matthews, C.R. Structure and stability of an early folding intermediate of Escherichia coli trp aporepressor measured by far-UV stopped-flow circular dichroism and 8-anilino-1-naphthalene sulfonate binding. Biochemistry. 1993;32:5282–5290. doi: 10.1021/bi00071a002. [DOI] [PubMed] [Google Scholar]
- Marino, M., Braun, L., Cossart, P., Ghosh, P. Structure of the lnlB leucine-rich repeats, a domain that triggers host cell invasion by the bacterial pathogen L. monocytogenes . Mol. Cell. 1999;4:1063–1072. doi: 10.1016/s1097-2765(00)80234-8. [DOI] [PubMed] [Google Scholar]
- Mello, C.C., Barrick, D. An experimentally determined protein folding energy landscape. Proc. Natl. Acad. Sci. 2004;101:14102–14107. doi: 10.1073/pnas.0403386101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello, C.C., Bradley, C.M., Tripp, K.W., Barrick, D. Experimental characterization of the folding kinetics of the Notch ankyrin domain. J. Mol. Biol. 2005;352:266–281. doi: 10.1016/j.jmb.2005.07.026. [DOI] [PubMed] [Google Scholar]
- Mosavi, L.K., Williams, S., Peng, Z.Y. Equilibrium folding and stability of myotrophin: A model ankyrin repeat protein. J. Mol. Biol. 2002;320:165–170. doi: 10.1016/S0022-2836(02)00441-2. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Oas, T.G. Mechanism of fast protein folding. Annu. Rev. Biochem. 2002;71:783–815. doi: 10.1146/annurev.biochem.71.110601.135346. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., Scholtz, J.M. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C.N. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., Baker, D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- Santoro, M.M., Bolen, D.W. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- Schmid, F.X. Kinetics of unfolding and refolding of single-domain proteins. In: Creighton T.E., editor. Protein folding. W.H. Freeman; New York: 1992. pp. 197–241. [Google Scholar]
- Shastry, M.C., Roder, H. Evidence for barrier-limited protein folding kinetics on the microsecond timescale. Nat. Struct. Biol. 1998;5:385–392. doi: 10.1038/nsb0598-385. [DOI] [PubMed] [Google Scholar]
- Stafford, W.F., Sherwood, P.J. Analysis of heterologous interacting systems by sedimentation velocity: Curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys. Chem. 2004;108:231–243. doi: 10.1016/j.bpc.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Tanford, C. Protein denaturation. Adv. Protein Chem. 1968;23:121–282. doi: 10.1016/s0065-3233(08)60401-5. [DOI] [PubMed] [Google Scholar]
- Tanford, C. Protein denaturation. C. Theoretical models for the mechanism of denaturation. Adv. Protein Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- Tang, K.S., Guralnick, B.J., Wang, W.K., Fersht, A.R., Itzhaki, L.S. Stability and folding of the tumour suppressor protein p16. J. Mol. Biol. 1999;285:1869–1886. doi: 10.1006/jmbi.1998.2420. [DOI] [PubMed] [Google Scholar]
- Wu, H., Maciejewski, M.W., Marintchev, A., Benashski, S.E., Mullen, G.P., King, S.M. Solution structure of a dynein motor domain associated light chain. Nat. Struct. Biol. 2000;7:575–579. doi: 10.1038/76804. [DOI] [PubMed] [Google Scholar]
- Zeeb, M., Rosner, H., Zeslawski, W., Canet, D., Holak, T.A., Balbach, J. Protein folding and stability of human CDK inhibitor p19(INK4d) J. Mol. Biol. 2002;315:447–457. doi: 10.1006/jmbi.2001.5242. [DOI] [PubMed] [Google Scholar]
- Zweifel, M.E., Barrick, D. Studies of the ankyrin repeats of the Drosophila melanogaster Notch receptor. 2. Solution stability and cooperativity of unfolding. Biochemistry. 2001;40:14357–14367. doi: 10.1021/bi011436+. [DOI] [PubMed] [Google Scholar]