

Abstract
Human autoimmune diseases are thought to develop through a complex combination of genetic and environmental factors. Genome-wide linkage searches of autoimmune and inflammatory/immune disorders have identified a large number of non-major histocompatibility complex loci that collectively contribute to disease susceptibility. A comparison was made of the linkage results from 23 published autoimmune or immune-mediated disease genome-wide scans. Human diseases included multiple sclerosis, Crohn’s disease, familial psoriasis, asthma, and type-I diabetes (IDDM). Experimental animal disease studies included murine experimental autoimmune encephalomyelitis, rat inflammatory arthritis, rat and murine IDDM, histamine sensitization, immunity to exogenous antigens, and murine lupus (systemic lupus erythematosus; SLE). A majority (≈65%) of the human positive linkages map nonrandomly into 18 distinct clusters. Overlapping of susceptibility loci occurs between different human immune diseases and by comparing conserved regions with experimental autoimmune/immune disease models. This nonrandom clustering supports a hypothesis that, in some cases, clinically distinct autoimmune diseases may be controlled by a common set of susceptibility genes.
Autoimmune diseases are common chronic conditions that involve immune attack of one or more organ systems and affect approximately 5% of the population. The specific etiologies of virtually all human autoimmune diseases are unknown, although they are thought to arise through a complex combination of genetic and environmental factors. Several reviews of autoimmunity and autoimmune diseases have recently been published (1–3).
One of the central genetic factors recognized in autoimmune diseases is the major histocompatibility complex (MHC). Recently, MHC and non-MHC genetic loci have been identified through genome scanning methods in autoimmune or inflammatory animal models and in human diseases. These include multiple sclerosis (MS) (4–7), IDDM (8–10), Crohn’s disease (CD) (11, 12), familial psoriasis (PS) (13, 14), asthma (AS) (15), rat inflammatory arthritis (16), murine lupus (SLE) (17, 18), and experimental autoimmune encephalomyelitis (EAE) (19, 20). Linkage results from genome scans for autoimmune/immune disorders tend to have complex patterns as compared with traditional linkage studies of monogenic traits (21). Autoimmune genome-wide scans report a greater number of linked loci (≈5–20) of lower significance levels, suggesting a complex genetic etiology.
Guidelines for interpretation of genome scan results have been described to provide standard criteria for reporting linkage data (22). By these criteria, for monogenic Mendelian traits, P values of less than 0.05 in a complete genome scan are recognized as potentially significant with logarithm of odds (lod) scores >1.0 and χ2 values >4.0 commonly reported. In contrast, candidate loci from gene scans of complex diseases, including autoimmune disease, are more speculative, with frequent mapping of multiple loci, often covering wide physical distances (10–40 cM).
Here we have compared the map location of all non-MHC candidate loci from published studies of autoimmune diseases by using suggestive, significant, and highly significant (22) linkage estimations. These loci fall into 18 clusters, suggesting a possible shared genetic basis among different autoimmune diseases.
There are many common elements among clinically distinct autoimmune diseases, including population frequencies (2), geographical distributions (23), clinical features (24), and therapeutic strategies. Most autoimmune diseases are thought to involve altered functions of humoral or cellular immunity. A sex ratio other than 1 in autoimmune disease is common, with women representing ≈75% of autoimmune patients (24). Familial clustering of different autoimmune diseases (2, 25), and coassociation of multiple autoimmune diseases in individuals has been frequently reported. The occurrence of common features of autoimmune diseases and the coassociation of multiple autoimmune diseases in the same individual or family supports the notion that there may be common genetic factors that predispose to autoimmunity. Vyse and Todd (2) have shown significant overlapping of susceptibility loci for autoimmune diseases in mouse.
In this study, a comparison was made between 23 autoimmune or inflammatory genome-wide scans or linkage studies. Human autoimmune diseases included MS (4–7), CD (11, 12), PS (13, 14), AS (15), and human type-I diabetes (IDDM-H) (8, 10, 26, 27). Animal disease studies included EAE (19, 20), rat inflammatory arthritis (16), rat IDDM (IDDM-R) (9), murine IDDM (IDDM-M) (28, 29), Bordetella pertussis-induced histamine sensitization (30), immunity to exogenous antigens (31), and murine SLE (17, 18). As controls, seven nonautoimmune human disease genome scans performed in a similar manner were also included. These included human type II diabetes (NIDDM) (32), schizophrenia (SZ) (33, 34), bipolar (BP) disorder (35, 36), leptin-associated obesity (37), and hypertension (HT) (38).
There were marked differences between most of these studies in experimental design, patient populations, sample size, markers used, and calculations of results. However, despite the use of different analytic approaches, these studies come to similar conclusions; namely, that in almost all common autoimmune/inflammatory diseases there may be no single genes exerting a predominate effect. Nonetheless, there do appear to be multiple loci of less significance that are candidates in the complex genetic etiology of these diseases.
METHODS
This study uses MS as a reference disease, because a recent series of reports (4–7) demonstrated a large number of positive loci, including data on markers of lower significance. Studies were compared with MS studies in the following way:
(i) Significance minimums were established. Markers were defined as a positive marker (PM) if the highest lod score for that marker was >0.90, significance values (P values) were <0.05, and/or χ2 was >4.0.
(ii) All human markers were placed on the University of Southampton Location Database summary maps (39).
(iii) Contiguous intervals (cIs) for each locus in a study were defined to approximate unavailable locus confidence intervals. cIs are defined as contiguous PMs, from the same study, which were within approximately 12.75 cM of another PM from the same study. The average intermarker distance of all human autoimmune studies was 12.75 cM (range 7.5–20 cM).
(iv) An autoimmune cluster was defined as any two contiguous intervals from different human diseases that were within approximately 12.75 cM of each other. A PM with no flanking markers was considered an independent cI. One MS study (5) published data as a continuous confidence interval. These data were used as a cI. Individual clusters (Cl) are designated by their chromosomal locations (i.e., Cl-1p, Cl-3p, etc.). Overlapping cIs of the same disease or isolated individual PMs were not sufficient to define an autoimmune cluster (except where noted, Table 1). The remaining human markers (35%) that did not tend to cluster (as defined here) with markers from other autoimmune diseases are shown in Table 2. All data from the short arm of human chromosome 6, including the MHC, was ignored so as to minimize MHC effects.
Table 1.
Clustering of autoimmune loci

| Cl | Chrom/ Marker | Sp | C | Ex Dis | Ch.reg | P value | lod/χ2 | cMphym | Ref. | CI | Chrom/Marker | Sp | C | Ex Dis | Ch.reg | P value | lod/χ2 | cMphym | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Cl, Cluster; Chrom/Marker, human chromosome, marker or marker gene. Homologs of mouse or rat marker genes were used for cross-species comparisons. In some cases, the centiMorgan distance from the rodent centromere was used for homology comparisons to human gene maps. Rodent markers (in parentheses). Markers in bold are identical markers used in separate studies; Sp, Species; C, used for statistical significance; Ex, Exceptions to contiguous interval and cluster definition Dis, disease; MS, multiple sclerosis; CD, Crohn disease; CD/UC, Crohn disease/ulcerative colitis; EAE, experimental autoimmune encephalomyelitis; IA, inflammatory arthritis; PS, psoriasis; IDDM, insulin-dependent diabetes melitis; AS; asthma; HR, B. pertussis-induced histamine sensitization; HI, humoral immunity; SLE; systemic lupus erythematosus. Ch.reg., cytogenetic band; LDB–gmaps; na, not available; lod or χ2 lod scores or χ2; cM-phym, distance from the top of the human chromosome in centimorgans; LDB–gmaps, na, in cross-species comparisons.
Table 2.
Nonclustered autoimmune loci

| Chrom/Marker | C Dis | Ch.reg | P value | lod/ χ2 | cM-phym | Ref. | Chrom/Marker | C Dis | Ch.reg | P value | lod/ χ2 | cM-phym | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
See Table 1 legend for key to abbreviations.
Animal data were not used in defining a cluster or in the calculation of statistical significance, although these data were highlighted if it was found on conserved regions with a defined human cluster (Fig. 1 and Table 1). Cross-species comparisons were made in the following order: (i) USH–gmap locations of human homologs of marker genes, (ii) physical distance from the centromere of the marker reported by (a) the author or (b) found on the Whitehead mouse map: www-genome.wi.mit.edu/cgi-bin/mouse/index. These locations were then compared with the National Center for Biotechnology Information mouse–human homology map at: www3.ncbi.nlm.nih.gov/Homology/, or (iii) the human homolog of candidate genes identified by the author and found on USH–gmaps.
Figure 1.
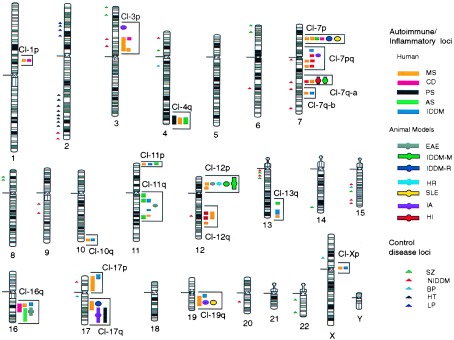
Clustering of autoimmune candidate loci. Positions of PMs for autoimmune diseases that fall into clusters are to the right of each chromosome relative to their cytogenetic location found in Table 1. Solid bar, human autoimmune disease PMs; circle or circle/bar, animal disease models; Human nonautoimmune diseases are to the left of each chromosome and are denoted by triangles. MS, multiple sclerosis; CD, Crohn disease; EAE, experimental autoimmune encephalomyelitis; IA, rat inflammatory arthritis; PS, familial psoriasis; AS, asthma; IDDM, insulin-dependent diabetes (type I) (IDDM-H, human; IDDM-M, mouse; IDDM-R, rat); HR, B. pertussis-induced histamine sensitization; SLE, murine lupus; HI, humoral immunity; SZ, schizophrenia; NIDDM, non-insulin-dependent diabetes (type II); BP, bipolar disorder; LP, leptin-associated obesity; and HT, hypertension.
Significance of the clustering of human PMs was evaluated by using two randomization tests. Method I: cI locations, from human studies, were randomly positioned on either a 12.75, 10, 5, or 1 cM grid covering the human genome, minus chromosome 6p (3,163 cM). After each repositioning, the number of independent cIs was determined. A cI was independent if it was not within the designated distance (12.75, 10, 5, 1 cM) of a cI for a different autoimmune disease. MS markers were fixed in the randomization test because the test was based on independently repositioning cIs and the large number of PMs made it difficult to determine which markers identified independent regions. Holding markers for one disease fixed did not interfere with the validity of the randomization test. This test was repeated 10,000 times. The statistical significance level was the number of replications resulting in a number of independent cIs no greater than that observed in the true data. The randomization probability (adjusted P value) that the smallest of the four significance levels was no greater than that in the data (0.043) was computed based on 10,000 replications. This value (0.08) represents the overall significance level adjusted for the four grid sizes used.
Method II: We repeated these analyses after merging cIs from different studies of the same disease that were within 12.75 cM. We also performed the randomization test including the MS loci in the repositioning procedure. Results of these sensitivity analyses gave smaller statistical significance levels for autoimmune diseases than those cited above (Table 3).
Table 3.
Statistical significance of Al cl clustering
| Method | 12.75 cM | 10 cM | 5 cM | 1 cM | Adjusted P value |
|---|---|---|---|---|---|
| I Autoimmune cl | 0.21 | 0.049 | 0.048 | 0.043 | 0.08 |
| Control cl | 0.23 | 0.46 | 0.76 | 0.76 | 0.30 |
| II Autoimmune cl | 0.074 | 0.034 | 0.014 | 0.0086 | 0.021 |
A few PMs are included in clusters (Table 1) that fail to strictly satisfy the 12.75 cM cluster definition; these are noted and not used for statistical significance.
RESULTS
Fig. 1 shows 18 clusters of autoimmune loci. Clusters defined within a 12.75-cM interval included those on chromosomes 1p, 7p, and two regions on 7q, 10q, 11p, 12p, and Xp. Other clusters of PMs occur over broader intervals and contain multiple markers for individual diseases on chromosomes 3p, 4q, 7pq, 11q, 12q, 16q, 17p, 17q, and 19q. Analysis of PMs for five nonautoimmune human diseases did not significantly overlap within the clusters defined for autoimmune disease (Fig. 1 and Tables 3 and 4). These clusters were also compared with cDNAs mapped on the human transcript map (40) and did not tend to be associated with regions of high gene density (not shown).
Table 4.
Percentage of human positive markers in autoimmune clusters
| Disease | Positive markers in clusters/total positive markers | % in clusters | Ref. |
|---|---|---|---|
| Autoimmune | |||
| MS | 18/24 | 75.0 | 5 |
| MS | 3/8 | 37.5 | 4 |
| MS | 9/21 | 43.0 | 6 |
| CD | 6/6 | 100 | 11 |
| CD | 10/13 | 76.9 | 12 |
| PS | 9/9 | 100 | 13 |
| PS | 4/4 | 100 | 14 |
| AS | 16/19 | 84.2 | 15 |
| IDDM | 15/32 | 46.8 | 8 |
| Control | |||
| NIDDM | 3/22 | 13.6 | 32 |
| BP | 0/3 | 0 | 35 |
| SZ | 0/5 | 0 | 33 |
| SZ | 0/5 | 0 | 34 |
| HT | 0/20 | 0 | 38 |
| BP | 0/1 | 0 | 36 |
| LP | 0/4 | 0 | 37 |
As shown in Table 1, MS and CD linkages were both reported at Cl-1p using the same marker (D1S236). This chromosomal region is conserved with the mouse locus eae3 (20), as well as Idd10 (41). Similarly, the cluster on chromosome 3p (Cl-3p) (Table 1) contains a region with an MS reported lod score of approximately 1.0 between markers D3S1289 and D3S1261. This interval of 23 cM also contains two markers linked to CD/UC, D3S1573 and D3S1076, and the human homolog of a marker gene for rat arthritis, ACAA. The autoimmune cluster on chromosome 7p (Cl-7p) has PMs from MS, AS, and CD found within an 11-cM interval. Finally, the cluster on chromosome 4q (CL-4q) contains PMs for PS, AS, and MS within an interval of 19 cM. Identical markers were found in all three diseases.
Overall, comparative analysis identified 18 clusters of candidate loci. In 14 cases (Table 1, boldface type), identical markers were assigned for autoimmune diseases by independent groups in the following clusters: Cl-1p, Cl-3p, Cl-4q, Cl-7p, Cl-7q-a, Cl-11p, Cl-12p, Cl-17p, Cl-17q, and Xp. There does not appear to be a coassociation of markers from any particular disease with any other (i.e., IDDM with MS).
Clusters were analyzed for statistical significance on a chromosomal grid by using two different randomization tests. Clustering was shown to be statistically significant by using both tests with either 10, 5, or 1 cM grids (Table 3). Clustering of nonautoimmune control markers did not reach statistical significance.
Once clusters were defined, nine human autoimmune studies and seven nonautoimmune studies were compared for the percentage of total PMs, from a given study, that are contained within the defined autoimmune clusters. Table 4 shows that a large majority of PMs from complete human autoimmune studies are clustered with other autoimmune diseases, whereas only 5.0% of the markers from nonautoimmune studies fall within the autoimmune clusters.
The 18 clusters defined here contain a large number of genes of known and unknown function. The interval defining Cl-19q (D19S49–D19S246), covering approximately 24 cM, contains at least 75 known genes (39) and 155 unknown transcripts (40). Genes in defined clusters that have previously been linked to autoimmune disease include: COL7A1 (42), IGER (43), SSA1 (44), C1NH (45), CD4 (46), C1R (47), ITGB3 (48), BAX (49), SNRP70 (50), TIMP2 (51), LIG1 (52), and CLC1 (53). CD4, found in Cl-12p, encodes the type I membrane glycoprotein that defines the helper/inducer subset of T cells. The CD4 gene has been associated with SLE (54), as well as type I diabetes (55).
Of interest, five clusters of autoimmune loci contain linkages or associations to susceptibility/resistance to infectious diseases. Cl-3p contains the candidate gene CMKBR5. A 32 bp deletion in this chemokine receptor has been associated with resistance to HIV (56). The chromosomal region of Cl-19q13 has been associated with resistance/susceptibility to picornaviruses including polio (57), ECHO-11 (58), and Coxsackie-B3 (59). This region, 19q13, is also conserved in mouse with the Hsv2 locus on mouse chromosome 7, which is associated with mouse hepatitis virus (60). Clusters 7pq, 12p, 11q, and 19q, as well as the nonclustered MS locus 5p15, are conserved in rodents to regions linked to resistance to Leishmania major (61).
DISCUSSION
A genetic component to autoimmune susceptibility has been clearly shown by twin and adoption studies, and by increased risk to siblings (2). A number of non-MHC genetic loci in autoimmune diseases have recently been mapped. As described here, a majority (65%) of these PMs appear to cluster nonrandomly into 18 groups. This clustering occurs within different human autoimmune diseases, as well as across species with experimental autoimmune/inflammatory disease models. In some cases, this coincidence of disease markers occurs at the same marker, or within 12.75 cM, whereas in others it is over a broader interval with multiple significant markers for the same disease.
Based on our results, the observed clustering of autoimmune loci occurring by random chance is statistically unlikely. Similar statistical findings have been reported in autoimmune animal models (2, 31). The clustering of loci in both experimental animal models and human disease may suggest a common gene, or multiple genes within an interval, influencing autoimmune disease susceptibility. Furthermore, we recognize that multiple genes in an interval may (i.e., the MTT gene cluster -16q) or may not be functionally related.
The overlapping of autoimmune loci was recently described in animal model systems (2, 16, 31, 61). Although mouse/human chromosome homologies and genetic maps are not uniformly comparable, at least nine human clusters seem to share conserved genetic locations with animal autoimmune loci: Cl-3p, Cl-7p, Cl-7qa, Cl-11q, Cl-12p, Cl-16q, Cl-17q, and Cl-19q.
The Cia3 locus in rat inflammatory arthritis (16) contains a number of candidate genes whose human homologs map to Cl-12p, including CD4, CD27, and CD69. This cluster is linked to human MS and in rodent systems for EAE, B. pertussis-induced histamine sensitization, and IDDM. Cia3 also appears to overlap with the rat locus IDDM1/Lyp as defined in BB rats (9, 16). This locus, which contains the marker gene NPY, has been associated with lymphopenia, diabetes (9), and autoimmune thyroiditis (62). The human equivalent of NPY maps to Cl-7p, which contains markers linked to MS, CD, and AS (Table 1).
A characteristic of autoimmune loci from model systems is that specific autoimmune traits are linked to different loci and may be additive, resulting in a compound phenotype in a given autoimmune disease. In mouse lupus, the loci Lbw 1,7,8 are associated with antichromatin antibody production; Lbw 3,4,5 with mortality; and Sbw 1,2 with splenomegaly (17). Animal autoimmune loci clustering with human disease loci suggests that this pattern could occur in human disease as well.
Particularly intriguing are reports of resistance/susceptibility to infectious diseases that map within autoimmune clusters: Leishmania resistance (61) (Cl-7pq, 12p, 11q, 19q); susceptibility to picornaviruses, including polio and coxsackie virus (57, 59) (Cl-19q), and HIV resistance (56) (Cl-3p). An association of picornavirus has been described in EAE (63), as well as coxsackie virus for IDDM (64) and autoimmune myocarditis (65).
A number of autoimmune candidate loci from different diseases did not fall into identifiable clusters (Table 2). These singleton loci may be independent loci; may contribute to disease specific susceptibility, tissue or organ tropism; or may be false positives. They may also group with other loci under different definitions of a cluster.
This clustering of autoimmune candidate loci into 18 groups from multiple autoimmune diseases and model systems suggests: (i) that there may be a related genetic background contributing to the susceptibility of clinically distinct autoimmune/inflammatory diseases; (ii) that it may be likely that the genes found at these clusters are involved in primary or secondary regulation of the immune system; (iii) that other diseases thought to have an autoimmune component (i.e., narcolepsy) may be compared with the genetic loci of more defined autoimmune diseases; and (iv) that shared genes among distinct diseases may lead to common early diagnostic criteria and therapeutic strategies.
Acknowledgments
We thank Drs. W. Bias, E. Blankenhorn, P. Meltzer, G. Papanicolaou, N. Rose, C. Teuscher, and J. Todd for reading the manuscript. We thank D. Leja for graphic assistance.
ABBREVIATIONS
- MHC
major histocompatibility complex
- MS
multiple sclerosis
- IDDM
type-I diabetes
- CD
Crohn’s disease
- PS
familial psoriasis
- AS
asthma
- EAE
experimental autoimmune encephalomyelitis
- lod
logarithm of odds
- PM
positive marker
- cI
contiguous interval
- SZ
schizophrenia
- BP
bipolar
- HT
hypertension
- LP
leptin-associated obesity
- systemic lupus erythematosus.
References
- 1.Theofilopoulos A N. Immunol Today. 1995;16:150–159. doi: 10.1016/0167-5699(95)80133-2. [DOI] [PubMed] [Google Scholar]
- 2.Vyse T J, Todd J A. Cell. 1996;85:311–318. doi: 10.1016/s0092-8674(00)81110-1. [DOI] [PubMed] [Google Scholar]
- 3.Todd J A. Proc Natl Acad Sci USA. 1995;92:8560–8565. doi: 10.1073/pnas.92.19.8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ebers G C, Kukay K, Bulman D E, Sadovnick A D, Rice G, Anderson C, Armstrong H, Cousin K, Bell R B, Hader W, et al. Nat Genet. 1996;13:472–476. doi: 10.1038/ng0896-472. [DOI] [PubMed] [Google Scholar]
- 5.Sawcer S, Jones H B, Feakes R, Gray J, Smaldon N, Chataway J, Robertson N, Clayton D, Goodfellow P N, Compston A. Nat Genet. 1996;13:464–468. doi: 10.1038/ng0896-464. [DOI] [PubMed] [Google Scholar]
- 6.Haines J L, Ter-Minassian M, Bazyk A, Gusella J F, Kim D J, Terwedow H, Pericak-Vance M A, Rimmler J B, Haynes C S, Roses A D, et al. Nat Genet. 1996;13:469–471. doi: 10.1038/ng0896-469. [DOI] [PubMed] [Google Scholar]
- 7.Kuokkanen S, Sundvall M, Terwiliger J D, Tienari P J, Wikstrom J, Holmdahl R, Pettersson U, Peltonen L. Nat Genet. 1996;13:477–480. doi: 10.1038/ng0896-477. [DOI] [PubMed] [Google Scholar]
- 8.Davies J L, Kawaguchi Y, Bennett S T, Copeman J B, Cordell H J, Pritchard L E, Reed P W, Gough S C, Jenkins S C, Palmer S M, et al. Nature (London) 1994;371:130–136. doi: 10.1038/371130a0. [DOI] [PubMed] [Google Scholar]
- 9.Jacob H J, Pettersson A, Wilson D, Mao Y, Lernmark A, Lander E. Nat Genet. 1992;2:56–60. doi: 10.1038/ng0992-56. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto L, Habita C, Beressi J P, Delepine M, Besse C, Cambon-Thomsen A, Deschamps I, Rotter J I, Djoulah S, James M R, et al. Nature (London) 1994;371:161–164. doi: 10.1038/371161a0. [DOI] [PubMed] [Google Scholar]
- 11.Hugot J-P, Laurent-Puig P, Gower-Rousseau C, Olson J M, Lee J C, Beaugerie L, Naom I, Dupas J L, Van Gossum A, Orholm M, et al. Nature (London) 1996;379:821–823. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 12.Satsangi J, Parker M, Louis E, Hashimoto L, Kato N, Welsh K, Terwilliger J D, Lathrop G M, Bell J I, Jewell D P. Nat Genet. 1996;14:199–202. doi: 10.1038/ng1096-199. [DOI] [PubMed] [Google Scholar]
- 13.Matthews D, Fry L, Powels A, Weber J, McCarthy M, Fisher E, Davies K, Williamson R. Nat Genet. 1996;14:231–233. doi: 10.1038/ng1096-231. [DOI] [PubMed] [Google Scholar]
- 14.Tomfohrde J, Silverman A, Barnes R, Fernandez-Vina M A, Young M, Lory D, Morris L, Wuepper K D, Stastny P, Menter A, et al. Science. 1994;264:1141–1145. doi: 10.1126/science.8178173. [DOI] [PubMed] [Google Scholar]
- 15.Daniels S E, Bhattacharrya S, James A, Leaves N I, Young A, Hill M R, Faux J A, Ryan G F, le Souef P N, Lathrop G M, et al. Nature (London) 1996;383:247–250. doi: 10.1038/383247a0. [DOI] [PubMed] [Google Scholar]
- 16.Remmers E F, Longman R E, Du Y, O’Hare A, Cannon G W, Griffiths M M, Wilder R L. Nat Genet. 1996;14:82–85. doi: 10.1038/ng0996-82. [DOI] [PubMed] [Google Scholar]
- 17.Kono D H, Burlingame R W, Owens D G, Kuramochi A, Balderas R S, Balomenos D, Theofilopoulos A N. Proc Natl Acad Sci USA. 1994;91:10168–10172. doi: 10.1073/pnas.91.21.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morel L, Rudofsky U H, Longmate J A, Schiffenbauer J, Wakeland E K. Immunity. 1995;1:219–229. doi: 10.1016/1074-7613(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 19.Baker D, Rosenwasser O A, O’Neill J K, Turk J. J Immunol. 1995;155:4046–4051. [PubMed] [Google Scholar]
- 20.Sundvall M, Jirholt J, Yang H-T, Jansson L, Engstrom A, Pettersson U, Holmdahl R. Nat Genet. 1995;10:313–317. doi: 10.1038/ng0795-313. [DOI] [PubMed] [Google Scholar]
- 21.Goodfellow P N, Schmidtt K. Nature (London) 1994;371:104–105. doi: 10.1038/371104a0. [DOI] [PubMed] [Google Scholar]
- 22.Lander E, Kruglyak L. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 23.Patrick S L, May C S, LaPorte R E. Diabetes Metab Rev. 1989;5:571–578. doi: 10.1002/dmr.5610050704. [DOI] [PubMed] [Google Scholar]
- 24.Rose N R. Hospital Practice. 1997;32:147–154. doi: 10.1080/21548331.1997.11443469. [DOI] [PubMed] [Google Scholar]
- 25.Bias W B, Reveille J D, Beaty T H, Meyers D A, Arnett F C. Am J Hum Genet. 1986;39:584–602. [PMC free article] [PubMed] [Google Scholar]
- 26.Rowe R R, Wapelhorst B, Bell G I, Risch N, Spielman R S, Concannon P. Nat Genet. 1995;10:240–242. doi: 10.1038/ng0695-240. [DOI] [PubMed] [Google Scholar]
- 27.Field L L, Tobias R, Magnus T. Nat Genet. 1994;8:189–194. doi: 10.1038/ng1094-189. [DOI] [PubMed] [Google Scholar]
- 28.Gosh S, Palmer S M, Rodrigues N R, Cordell H J, Hearne C M, Cornall R J, Prins J B, McShane P, Lathrop G M, Peterson L B, et al. Nat Genet. 1993;4:404–409. doi: 10.1038/ng0893-404. [DOI] [PubMed] [Google Scholar]
- 29.Dallas-Pedretti A, McDuffie M, Haskins K. Proc Natl Acad Sci USA. 1995;92:1386–1390. doi: 10.1073/pnas.92.5.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sudweeks J D, Todd J A, Blankenhorn E P, Wardell B B, Woodward S R, Meeker N D, Estes S S, Teuscher C. Proc Natl Acad Sci USA. 1993;90:3700–3704. doi: 10.1073/pnas.90.8.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu J, Longmate J A, Adamus G, Hargrave P A, Wakeland E K. J Immunol. 1996;157:2498–2505. [PubMed] [Google Scholar]
- 32.Hanis C L, Boerwinkle E, Chakraborty R, Ellsworth D L, Concannon P, Stirling B, Morrison V A, Wapelhorst B, Spielman R S, Gogolin-Ewens K J, et al. Nat Genet. 1996;13:161–166. doi: 10.1038/ng0696-161. [DOI] [PubMed] [Google Scholar]
- 33.Coon H, Jensen S, Holik J, Hoff M, Myles-Worsley M, Reimherr F, Wender P, Waldo M, Freedman R, Leppert M, et al. Am J Med Genet. 1994;54:59–71. doi: 10.1002/ajmg.1320540111. [DOI] [PubMed] [Google Scholar]
- 34.Pulver A E, Lasseter V K, Kasch L, Wolyniec P, Nestadt G, Blouin J L, Kimberland M, Babb R, Vourlis S, Chen H, et al. Am J Med Genet. 1995;60:252–260. doi: 10.1002/ajmg.1320600316. [DOI] [PubMed] [Google Scholar]
- 35.Ginns E I, Ott J, Egeland J A, Allen C R, Fann C S J, Pauls D L, Weissenbachoff J, Carulli J P, Falls K M, Keith T P, Paul S M. Nat Genet. 1996;12:431–435. doi: 10.1038/ng0496-431. [DOI] [PubMed] [Google Scholar]
- 36.LaBuda M C, Maldonado M, Marshall D, Otten K, Gerhard D S. Am J Hum Genet. 1996;59:1343–1362. [PMC free article] [PubMed] [Google Scholar]
- 37.Comuzzie A G, Hixson J E, Almasy L, Mitchell B D, Mahaney M C, Dyer T D, Stern M P, MacCluer J W, Blangero J. Nat Genet. 1997;15:273–276. doi: 10.1038/ng0397-273. [DOI] [PubMed] [Google Scholar]
- 38.Nichols W C, Koller D L, Slovis B, Foroud T, Terry V H, Arnold N D, Siemieniak D R, Wheeler L, Phillips J A, Newman J H, et al. Nat Genet. 1997;15:277–280. doi: 10.1038/ng0397-277. [DOI] [PubMed] [Google Scholar]
- 39.Collins A, Frezal J, Teague J, Morton N E. Proc Natl Acad Sci USA. 1996;93:14771–14775. doi: 10.1073/pnas.93.25.14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuler G D, Boguski M S, Stewart E A, Stein L D, Gyapay G, Rice K, White R E, Rodriguez-Tome P, Aggarwal A, Bajorek E, et al. Science. 1996;274:540–546. [PubMed] [Google Scholar]
- 41.Podolin P L, Denny P, Lord C J, Hill N J, Todd J A, Peterson L B, Wicker L S, Lyons P A. J Immunol. 1997;159:1835–1843. [PubMed] [Google Scholar]
- 42.Lapiere J-C, Woodley D T, Parente M G, Iwasaki T, Wynn K C, Christiano A M, Uitto J. J Clin Invest. 1993;92:1831–1839. doi: 10.1172/JCI116774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cookson W O C M, Sharp P A, Faux J A, Hopkin J M. Lancet. 1989;i:1292–1295. doi: 10.1016/s0140-6736(89)92687-1. [DOI] [PubMed] [Google Scholar]
- 44.Schoenlebe J, Buyon J P, Zitelli B J, Friedman D, Greco M A, Knisely A S. Am J Dis Child. 1993;147:1072–1075. doi: 10.1001/archpedi.1993.02160340058014. [DOI] [PubMed] [Google Scholar]
- 45.Muhlemann M F, Macrae K D, Smith A M, Beck P, Hine I, Hegde U, Milford-Ward A, Carter G D, Wise P H, Cream J J. J Clin Pathol. 1987;40:518–523. doi: 10.1136/jcp.40.5.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukuda T, Matsunaga M, Kurata A, Mine M, Ikari N, Katamine S, Kanazawa H, Eguchi K, Nagataki S. Immunology. 1984;53:643–649. [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S L, Wallace S L, Barone R, Blum L, Chase P H. Arthritis Rheum. 1978;21:958–967. doi: 10.1002/art.1780210813. [DOI] [PubMed] [Google Scholar]
- 48.Kekomaki R, Jouhikainen T, Ollikainen J, Westman P, Laes M. Br J Haematol. 1993;83:306–310. doi: 10.1111/j.1365-2141.1993.tb08286.x. [DOI] [PubMed] [Google Scholar]
- 49.Knudson C M, Tung K S K, Tourtellotte W G, Brown G A J, Korsmeyer S J. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 50.Spritz R A, Strunk K, Surowy C S, Hoch S O, Barton D E, Francke U. Nucleic Acids Res. 1987;15:10373–10391. doi: 10.1093/nar/15.24.10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Becker K G, Mattson D H, Powers J M, Gado A M, Biddison W E. J Neuroimmunol. 1997;77:27–38. doi: 10.1016/s0165-5728(97)00045-3. [DOI] [PubMed] [Google Scholar]
- 52.Webster A D B, Barnes D E, Arlett C F, Lehmann A R, Lindahl T. Lancet. 1992;339:1508–1509. doi: 10.1016/0140-6736(92)91266-b. [DOI] [PubMed] [Google Scholar]
- 53.Ackerman S J, Corrette S E, Rosenberg H F, Bennett J C, Mastrianni D M, Nicholson-Weller A, Weller P F, Chin D T, Tenen D G. Immunity. 1993;150:456–468. [PubMed] [Google Scholar]
- 54.Stohl W, Crow M K, Kunkel H G. N Engl J Med. 1985;312:1671–1678. doi: 10.1056/NEJM198506273122604. [DOI] [PubMed] [Google Scholar]
- 55.Ghabanbasani M Z, Buyse I, Legius E, Decorte R, Marynen P, Bouillon R, Cassiman J J. Clin Exp Immunol. 1994;97:517–521. doi: 10.1111/j.1365-2249.1994.tb06119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu R, Paxton W A, Choe S, Ceradini D, Martin S R, Horuk R, MacDonald M E, Stuhlmann H, Koup R A, Landau N R. Nature (London) 1996;382:668–669. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 57.Siddique T, McKinney R, Hung W-Y, Bartlett R J, Bruns G, Mohandas T K, Ropers H H, Wilfert C, Roses A D. Genomics. 1988;3:156–160. doi: 10.1016/0888-7543(88)90147-4. [DOI] [PubMed] [Google Scholar]
- 58.Kaneda Y, Hayes H, Uchida T, Yoshida M C, Okada Y. Chromosoma. 1987;95:8–12. doi: 10.1007/BF00293835. [DOI] [PubMed] [Google Scholar]
- 59.Gerald P S, Bruns G A. Birth Defects Orig Art Ser XIV. 1978;6A:1–7. [PubMed] [Google Scholar]
- 60.Smith M S, Click R E, Plagemann P G. J Immunol. 1984;133:428–432. [PubMed] [Google Scholar]
- 61.Beebe A M, Mauze S, Schork N J, Coffman R L. Immunity. 1997;6:551–557. doi: 10.1016/s1074-7613(00)80343-x. [DOI] [PubMed] [Google Scholar]
- 62.Meeker N D, Hickey W F, Korngold R, Hansen W K, Sudweeks J D, Wardell B B, Griffith J S, Teuscher C. Proc Natl Acad Sci USA. 1995;92:5684–5688. doi: 10.1073/pnas.92.12.5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller S D, Vanderlugt C L, Begolka W S, Pao W, Yauch R L, Neville K L, Katz-Levy Y, Carrizosa A, Kim B S. Nat Med. 1997;10:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- 64.Fohlman J, Friman G. Ann Med. 1993;6:569–574. [PubMed] [Google Scholar]
- 65.Rose N R, Hill S L. Clin Immunol Immunopathol. 1996;80:S92–S99. doi: 10.1006/clin.1996.0146. [DOI] [PubMed] [Google Scholar]