

Abstract
This study was undertaken to examine the mechanistic significance of two highly conserved residues positioned in the active site of pyruvate dehydrogenase kinase, Glu-243 and His-239. We used site-directed mutagenesis to convert Glu-243 to Ala, Asp, or Gln and His-239 to Ala. The resulting mutant kinases demonstrated a greatly reduced capacity for phosphorylation of pyruvate dehydrogenase. The Glu-243 to Asp mutant had ~2% residual activity, whereas the Glu-243 to Ala or Gln mutants exhibited less than 0.5 and 0.1% residual activity, respectively. Activity of the His-239 to Ala mutant was decreased by ~90%. Active-site titration with [α-32P]ATP revealed that neither Glu-243 nor His-239 mutations affected nucleotide binding. All mutant kinases showed similar or even somewhat greater affinity than the wild-type kinase toward the protein substrate, pyruvate dehydrogenase complex. Furthermore, neither of the mutations affected the inter-subunit interactions. Finally, pyruvate dehydrogenase kinase was found to possess a weak ATP hydrolytic activity, which required Glu-243 and His-239 similar to the kinase activity. Based on these observations, we propose a mechanism according to which the invariant glutamate residue (Glu-243) acts as a general base catalyst, which activates the hydroxyl group on a serine residue of the protein substrate for direct attack on the γ phosphate. The glutamate residue in turn might be further polarized through interaction with the neighboring histidine residue (His-239).
Mammalian pyruvate dehydrogenase complex (PDC)1 catalyzes oxidative decarboxylation of pyruvate with concomitant formation of acetyl-CoA and NADH. Under physiological conditions this reaction is irreversible and, therefore, largely defines the metabolic fate of pyruvic acid and carbohydrate fuels in general (1). In well oxygenated tissues such as brain, skeletal muscle, heart, and kidney, PDC commits pyruvate to further oxidation through the Krebs cycle, thus supplying the oxidative fuel for the generation of ATP. In lipogenic tissues such as liver, fat, and mammary gland, PDC provides acetyl-CoA primarily for biosynthesis of fatty acids, allowing the excessive carbohydrates obtained with diet to be spared. To fulfill these opposing functions, the activity of mammalian PDC is tightly regulated by reversible phosphorylation (2). Phosphorylation of the dehydrogenase component of the complex (E1 component) by dedicated pyruvate dehydrogenase kinase (PDK) renders the entire complex inactive (3). Phospho-PDC can be re-activated through the action of another dedicated enzyme, pyruvate dehydrogenase phosphatase (4). In mammals, both kinase and phosphatase components exist in several isozymic forms (four for PDK (5) and two for pyruvate dehydrogenase phosphatase (6)), which are likely to contribute to the tissue-specific regulation of PDC. The latter is evidenced by the tissue-specific distribution of different isozymes (5) as well as by their different responses to the naturally occurring inhibitors and activators of the kinase (7) and phosphatase reactions (6).
Despite the important role that phoshorylation of PDC plays in regulation of carbohydrate metabolism, little is known about molecular mechanisms underlying the phosphorylation reaction. Kinase-driven inactivation of PDC occurs as a result of phosphorylation of three serine residues usually referred to as phosphorylation sites 1, 2, and 3 (8). This makes PDK a strictly Ser-specific protein kinase. Surprisingly, PDK (5) along with the homologous mitochondrial protein kinase that phosphorylates the branched chain α-ketoacid dehydrogenase complex (BCKDC) and BCKDC kinase, respectively) (9), does not show any sequence similarity to the other Ser/Thr-specific protein kinases. Instead, it resembles histidine kinases (10), a diverse group of enzymes involved in regulation of various signal transduction pathways in bacteria. All structural elements characteristic of histidine kinases (so-called boxes H, N, G1, G2, and G3) can be readily identified in the amino acid sequence of PDK (11). Recently, the three-dimensional structures of two histidine kinases involved in the regulation of chemotaxis and osmolarity (CheA (12) and EnvZ (13), respectively) have been determined by x-ray crystallography and NMR. These structures reveal a very characteristic nucleotide binding domain that, in contrast to the catalytic domains of Ser/Thr-specific protein kinases, folds as an α/β sandwich consisting of five strands and three helices with unique left-handed connectivity. Furthermore, certain amino acids of boxes N, G1, G2, and G3 are intimately involved in the anchoring of the nucleotide substrate in the active site of the kinase molecule (13). When we probed the corresponding residues of PDK2 by site-directed mutagenesis, the resulting mutant kinases were catalytically defective due to the impaired ability to bind the nucleotide substrate, strongly suggesting that the nucleotide binding domain of PDK is folded similarly to the nucleotide binding domain of histidine kinases (14).
On the other hand, the differences between these two groups of enzymes are also quite apparent. Histidine kinases use ATP to phosphorylate their own histidine residue positioned within, external to the nucleotide-binding site histidine-bearing domain (12, 13). It is generally believed that the phospho-accepting histidine residue directly attacks the γ phosphate of ATP (12). PDK, in contrast, catalyzes the transfer of γ phosphate to the side chain of the serine residue of the exogenous substrate, E1. To date, there is no evidence for existence of a phospho-enzyme intermediate formed during catalysis by either PDK (15) or a related branched chain α-ketoacid dehydrogenase complex kinase (16). This prompted us to suggest that PDK, in contrast to histidine kinases, uses a general base catalysis to promote a direct nucleophilic attack on the γ phosphate by the hydroxyl group of a serine residue of the protein substrate (14). Here we report the first evidence that the invariant glutamic acid (Glu-243 in PDK2) serves as a general base catalyst of phosphorylation reaction. The results reported here are also consistent with the idea that, at least in mammalian kinases, the nucleophilicity of Glu-243 might be further increased through the interaction with the neighboring histidine residue (His-239 of PDK2).
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis
Point mutations within the amino acid sequence of rat PDK2 were introduced using oligonucleotide-directed mutagenesis (17). The sequences of mutagenic oligonucleotides were as follows: 5′-CCA CAT GCT CTT TGA TCT CTT CAA GAA TGC C-3′ for Glu-243 to Asp mutant; 5′-CCA CAT GCT CTT TCA ACT CTT CAA GAA TGC C-3′ for Glu-243 to Gln mutant; 5′-CCA CAT GCT CTT TGC ACT CTT CAA GAA TGC C-3′ for Glu-243 to Ala mutant; 5′-CCA CCT CTA CGC CAT GCT CTT TGA ACT C-3′ for His-239 to Ala mutant; and 5′-ATC AAA ATG AGT GAG CGA GGC GGG GGT-3′ for Asp-282 to Glu mutant (the amino acid residues are numbered according to the sequence of mature rat PDK2; the altered bases are underlined). Mutagenesis reactions were carried out on double-stranded DNA of rat PDK2 (5) subcloned into pUC 19 using the ExSite™ site-directed mutagenesis kit (Stratagene, La Jolla, CA). The reactions were set up essentially as recommended by the manufacturer. The mutations as well as the fidelity of the rest of DNAs were confirmed by direct sequencing (19).
Expression and Purification of the Mutant Kinases
PDK2 cDNAs (~1.2 kilobases) carrying the point mutations were cut out of the pUC 19 DNA with SacI and HindIII restriction enzymes and religated into the pET-28a expression vector (Novagen, Madison, WI) between SacI/HindIII sites of the vector (pPDK2 vector). Plasmids containing the inserts of the correct size (~1.2 kilobases) were identified by restriction analysis. Positive plasmids were cotransfected into BL21(DE3) cells (Novagen) along with pGroESL plasmid carrying the genes coding for molecular chaperones GroEL and GroES under the control of an iso-propyl-1-thio-β-D-galactopyranoside-inducible promoter (the respective plasmid was obtained as a generous gift from Dr. Anthony Gatenby at DuPont Central Research and Development, Wilmington, DE). Double-transformants were selected on yeast-tryptone agar containing kana-mycin and chloramphenicol (50 μg/ml each) (7). Several individual colonies from every transformation were tested for their ability to produce significant amounts of soluble, recombinant kinase. Clones expressing the greatest amount of the soluble kinase were used for further analysis.
The expression of the mutant kinases was performed essentially as described previously (14). Their purification was carried out using TALON™ (CLONTECH Laboratories, Inc., Palo Alto, CA) affinity resin as described elsewhere (7). The protein composition of each preparation was evaluated by SDS/PAGE analysis. Gels were stained with Coomassie R250. All enzyme preparations used in the present study were more than 90% pure.
Other Enzyme Preparations
Human recombinant PDC was expressed in Escherichia coli as described in Harris et al. (20). The complex was purified by polyethylene glycol 8000 (Sigma) precipitation, gel filtration on Sepharose 4B (Amersham Pharmacia Biotech), and high speed centrifugation (20). The E1 component of PDC was expressed and purified as described elsewhere (21). All enzyme preparations used in this study were more than 90% pure as judged by Coomassie-stained SDS/PAGE.
Kinase Pull-down Assay
To construct “bait” vectors for pull-down experiments, unique NdeI and XhoI restriction sites flanking the coding region of the kinase cDNA were introduced by site-directed mutagenesis. Respective cDNAs were subcloned between NdeI and XhoI sites of pET-28a vector (Novagen), producing inframe fusion with the vector sequence coding for a His6-Tag. “Catch” plasmids were constructed by subcloning kinase cDNAs flanked by SstI and XhoI restriction sites into pET-23a (Novagen) cut with SstI and XhoI, producing in-frame fusion with a T7-tag sequence coded by the vector. An expression cassette carrying the T7 promoter, the cDNA of interest, and a T7 terminator was cut out of catch vectors using BbsI and DraIII restriction enzymes. After blunt-ending, it was subcloned into an appropriate bait vector cut with DraIII, blunt-ended with T4 polymerase, and dephosphorylated with calf intestinal phosphatase. Thus, the resulting catch/bait plasmids were carrying two expression cassettes: one directing synthesis of His6-tagged kinase and the other directing synthesis of T7-tagged kinase. Expression of the respective proteins was performed in BL21(DE3) cells co-transformed with appropriate catch/bait plasmid and pGroESL vector, essentially as described above. Respective recombinant kinases were isolated using metal affinity chromatography on TALON™ resin (CLONTECH) (7). Isolated proteins were analyzed using SDS/PAGE and Western blotting with anti-His6-tag antibodies (CLONTECH) or anti-T7-tag antibodies (Novagen). Immunoreactive bands were visualized with 125I-protein A (ICN Biomedicals, Inc., Costa Mesa, CA) followed by autoradiography (22).
Standard PDK Activity Assay
Kinase activity was determined by following [32P]phosphate incorporation from [γ-32P]ATP into the E1α subunit of PDC, essentially as described previously (14). Phosphorylation reactions were incubated at 37 °C in a final volume of 100 μl containing 20 mM Tris-HCl, pH 7.8, 5 mM MgCl2, 50 mM KCl, 5 mM dithiothreitol, 1.0 mg/ml PDC, and nucleotide substrate (the specific activity of [γ-32P]ATP was ~200–500 cpm/pmol). For assaying recombinant kinases, the respective proteins were reconstituted with recombinant human PDC before the assay. Reconstituted preparations were kept on ice for 30 min. The final protein concentrations of the recombinant proteins in the assay mixture were as follows: the E1-E2·E3BP subcomplex at 1.0 mg/ml and the corresponding wild-type kinase at 5 μg/ml. When we tried to apply the above assay for the analysis of kinase mutants with minute activity, it was difficult to obtain reliable estimates even when reactions were conducted for 30 min. To improve the sensitivity of the assay, the amount of kinase protein added to the reaction was increased (see legend to Fig. 1). Under these conditions, there was a linear incorporation of [32P]phosphate into PDC with time for at least 30 min of the reaction for all mutants tested. The rates of phosphorylation reactions were proportional to the amount of added kinase, indicating that the protein substrate is not limiting under the conditions used. Protein-bound radioactivity was determined as described previously (14). The activity of wild-type kinase was determined in the standard assay and was calculated based on incorporation of [32P]phosphate during the first 30 s of the reaction. The activities of mutant kinases were calculated based on incorporation of [32P]phosphate during 30 min of the reaction. The concentrations of nucleotide substrate (ATP) and the concentrations of inhibitors used in particular experiments are given in the legends to figures. Radicicol was added from a solution made with dimethyl sulfoxide. The final concentration of dimethyl sulfoxide in the reaction mixture was 1% (v/v) at all concentrations of inhibitor tested. Under these conditions, dimethyl sulfoxide had no effect on the kinase activity as established in preliminary experiments. Phosphorylation reactions in this case were initiated by the addition of wild-type PDC or reconstituted PDC kinase after equilibration at 37 °C for 30 s. The volume of PDC added was 1/10 of the total reaction volume. All assays were conducted in duplicates.
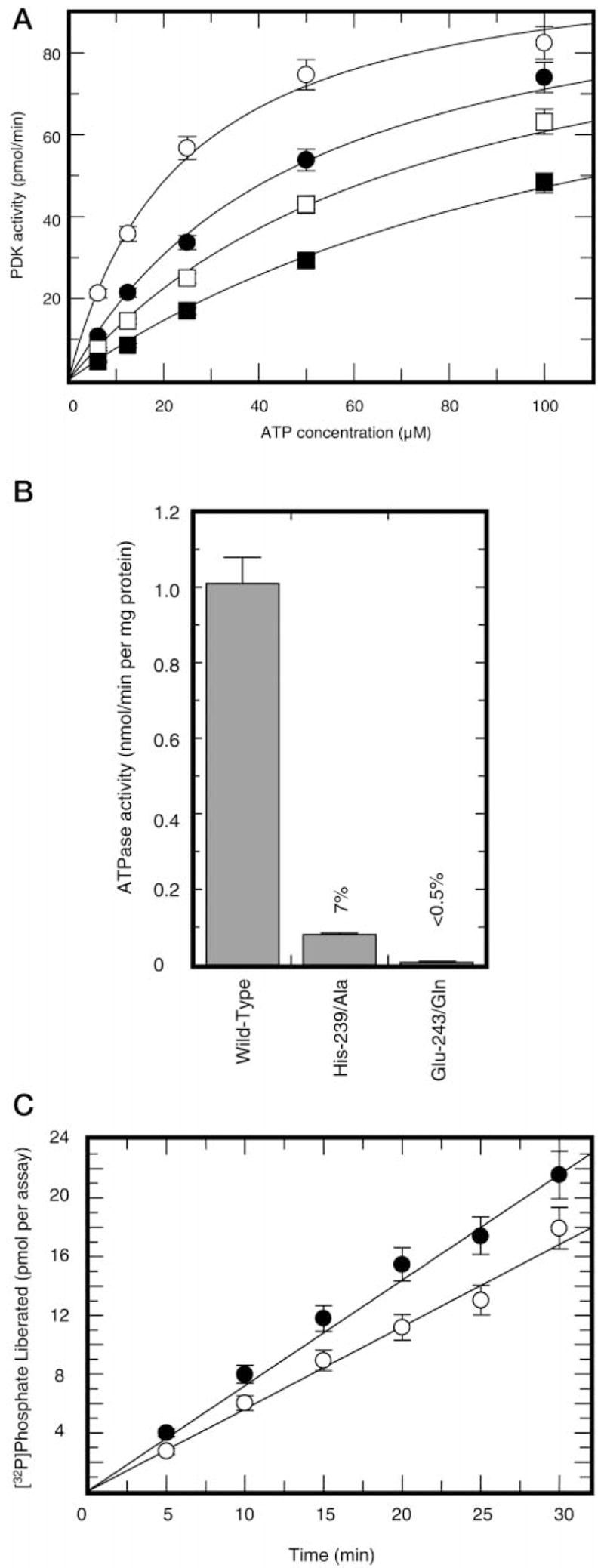
Fig. 1. Kinetics of PDC phosphorylation by wild-type and mutant PDKs.
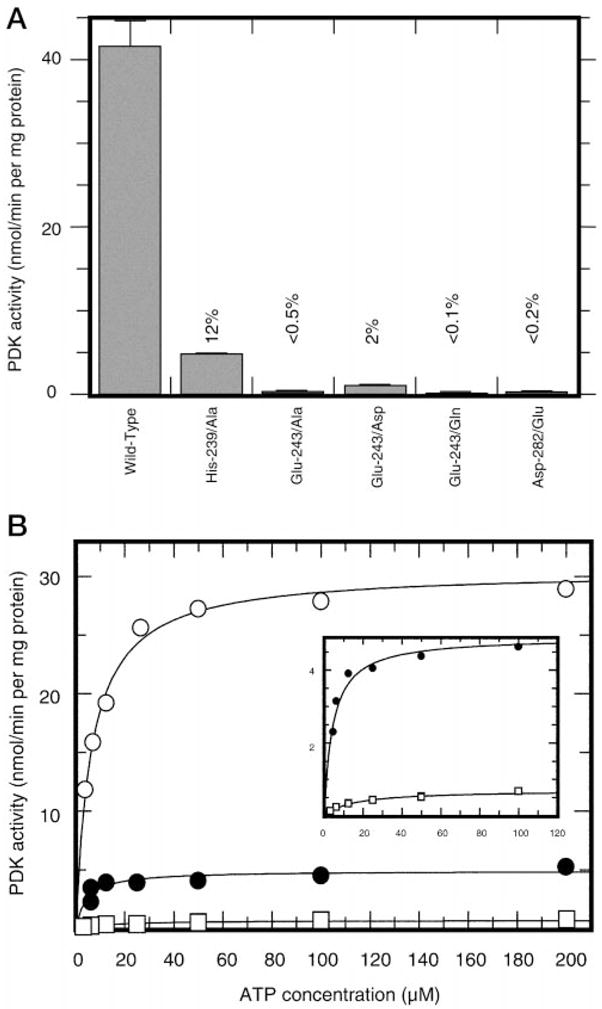
Panel A, average activities of various PDK preparations analyzed in this study. Experiments were conducted with 200 μM ATP and 1.0 mg/ml recombinant PDC as described under “Experimental Procedures” section. Respective enzymes, except for the wild-type PDK2, were used at final concentrations of 20 μg/ml. The activity of wild-type kinase was calculated based on incorporation of [32P]phosphate during the first 30 s of the reaction. Activities of mutant kinases were calculated based on the incorporation of [32P]phosphate during 30 min of the reaction. Panel B, representative results showing ATP dependence of PDC phosphorylation by wild-type PDK2 (○) and His-239 → Ala (●) and Glu-243 → Asp (□) mutants. The insert shows respective curves for His-239 → Ala (●) and Glu-243 → Asp (□) mutants. ATP concentration was varied from 3 to 200 μM.
ATP Binding Assay
Nucleotide binding studies were conducted using a modified vacuum filtration assay developed by Pratt and Roche (23). Briefly, the recombinant kinases were used in a final concentration of 0.1 mg/ml in binding buffer (20 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 50 mM KCl, 5 mM dithiothreitol). Binding reactions (total volume of 100 μl) were initiated by the addition of [α-32P]ATP (with a specific radioactivity of 200–500 cpm/pmol). The final concentration of ATP in the binding buffer was varied from 2.5 to 50 μM. The reactions were incubated at room temperature for 2 min. Protein-bound radioactivity was determined essentially as described previously (14). All binding experiments were conducted in triplicate.
ATPase Activity Assay
ATPase activity was assayed essentially as described by Singh and Cerione (24). Briefly, reactions were set up in a final volume of 100 μl containing 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol, 5% (v/v) glycerol, and 10 μM [γ-32P]ATP (specific radioactivity of 200–400 cpm/pmol). Reactions were initiated by adding the respective preparation of recombinant kinase to a final protein concentration of 0.1 mg/ml. Assays were conducted at room temperature. At the indicated times, reactions were terminated by the addition of 1 ml of ice-chilled 5% (w/v) Norit A (Sigma) in 50 mM NaH2PO4. The mixture was centrifuged, and 100 μl of supernatant was mixed with 5 ml of scintillation fluid. The release of [32P]phosphate as the outcome of ATP hydrolysis was measured in a scintillation counter. Radicicol (final concentration 200 μM) was added from the solution made with dimethyl sulfoxide. The final concentration of dimethyl sulfoxide in the reaction mixture was less than 1% (v/v). Reactions made without radicicol received an appropriate amount of dimethyl sulfoxide for control. When E1 component was used as phosphate acceptor, its final concentration was 1.0 mg/ml.
PDC Binding Assay
PDC binding reactions were set up in 100-μl volumes containing 20 mM Tris-HCl, pH 7.8, 5 mM MgCl2, 50 mM KCl, 1 mM dithiothreitol, 1.0 mg/ml recombinant PDC, and various amounts of PDK2. Kinase was allowed to bind to the complex for 15 min at room temperature. By the end of the incubation, reactions were loaded on the columns containing 1 ml of Sephacel S300 (Amersham Pharmacia Biotech) equilibrated with 20 mM Tris-HCl, pH 7.8, 5 mM MgCl2, 50 mM KCl, 1 mM dithiothreitol, and 0.1% (w/v) Tween 20. Before the experiment, columns were centrifuged at 2,000 rpm for 1 min in a IEC Centra CL2 centrifuge (International Equipment Co., Needham Heights, MA) equipped with a bucket rotor. Immediately after loading, columns were centrifuged again at 2,000 rpm for 1 min. The flow-through containing the PDC-kinase complex was collected in Eppendorf tubes. The resulting preparations of PDC-bound kinase were analyzed further by Western blotting with anti-PDK2 antibodies essentially as described (22).
Analysis of Kinetic and Binding Data
Raw kinetic and binding data were analyzed using GraFit version 3 software (Erithacus Software Ltd, Middlesex, UK). The apparent inhibition constants were determined by measuring the initial rates of the phosphorylation reaction at various concentrations of nucleotide substrate and inhibitor (matrix of 24 different conditions). The resulting matrixes were analyzed as a set to determine the respective kinetic parameters. Shown are representative results obtained with one out of three preparations of each enzyme analyzed for this study.
Other Procedures
SDS/PAGE was carried out according to Laemmli (25). Protein concentrations were determined according to Lowry et al. (26) with bovine serum albumin as standard.
RESULTS AND DISCUSSION
Kinase Activity of Wild-type and Mutant Proteins
In this study, we used site-directed mutagenesis to test a hypothesis that the invariant glutamic acid residue of mammalian PDK (Glu-243 in rat PDK2) serves as a catalyst in the phospho-transfer reaction. We also analyzed the functional significance of two additional residues: His-239, which may be involved in polarization of Glu-243, and Asp-282, which contributes to the binding of ATP in the active site (14). The latter residue has been chosen as a control because its substitution produces a kinase with impaired catalytic ability. However, this effect, as previously established (14), is achieved by different means, through the knockout of nucleotide binding. The invariant Glu-243 of rat PDK2 was altered to Ala, Asp, and Gln and His-239 was altered to Ala. The invariant Asp-282 was changed to Glu. Wild-type PDK2 and respective mutant kinases were expressed in E. coli cells and purified to near homogeneity as discribed under “Experimental Procedures.” When the activities of wild-type and mutant enzymes were assayed in the standard phosphorylation assay (see “Experimental Procedures”), all mutant kinases showed a markedly decreased ability to phosphorylate PDC (Fig. 1A). Substitution of His-239 to Ala resulted in reduced kinase activity (~12% of that of wild-type enzyme). Kinases with mutations of Glu-243 to Asp, Ala, or Gln had ~2%, < 0.5%, and < 0.1% residual activity, respectively. PDK2 carrying the substitution of Asp-282 to Glu showed less than 0.2% activity of wild-type enzyme (Fig. 1A). This outcome strongly suggests that all residues targeted for mutagenesis in this study are important for kinase function.
Kinetics of Phosphorylation Reaction for Wild-type and Mutant Kinases
If Glu-243 and His-239 contribute to the catalysis of phosphotransfer reaction, substitution of these residues would be expected to affect the catalytic efficiency and should be manifested as a decrease in the apparent Vmax, with little if any effect on the apparent Km value for the nucleotide substrate. To test this hypothesis, we determined the kinetics of the phosphorylation reaction using kinase mutants showing substantial residual activity: Glu-243 to Asp and His-239 to Ala. Determinations were made at a single fixed concentration of PDC of 1.0 mg/ml and several concentrations of ATP varied from 3 to 200 μM (Fig. 1B). As expected, substitution of either His-239 to Ala or Glu-243 to Asp resulted in a decrease in apparent Vmax values, 7- and 50-fold, respectively (Fig. 1B, insert). On the other hand, the apparent Km value for ATP of the His-239 to Ala mutant was equal to the Km value of the wild-type enzyme within experimental error (4.4 ± 1.3 versus 6.4 ± 1.2 μM). The Glu-243 to Asp mutant showed a small increase in the apparent Km value for ATP (~14.7 ± 2.1 μM).
Active-site Titration with [α-32P]ATP
If His-239 and Glu-243 contribute to the catalysis of phosphotransfer reaction, it would be expected that the respective mutant proteins would still be capable of binding ATP at levels similar to the wild-type enzyme. The results of kinetic experiments described above are consistent with this hypothesis. However, due to the limits of detection of the phosphorylation assay, we could not apply this approach to the mutant proteins having very low kinase activity. Furthermore, a drastic reduction in the activity of the mutant proteins could be attributed to the fact that only a small portion of the protein in each preparation binds ATP and phosphorylates PDC, whereas the majority cannot bind the nucleotide substrate. Thus, to determine whether all kinase molecules were capable of binding ATP, we performed an active-site titration with [α-32P]ATP as a ligand. The binding experiments showed that both Glu-243 and His-239 mutants bind [α-32P]ATP at levels comparable with the wild-type enzyme with the Kd value of ~4 μM (Fig. 2). The stoichiometry of binding for the wild-type enzyme and for the His-239 to Ala mutant was close to the unity. For kinases carrying the substitutions at Glu-243, the stoichiometry of nucleotide binding was ~0.8–0.9 mol of [α-32P]ATP per mol of kinase, suggesting that neither Glu-243 nor His-239 contribute to the binding of nucleotide substrate to a great extent.
Fig. 2. [α-32P]ATP binding by the wild-type PDK2 and mutant enzymes.
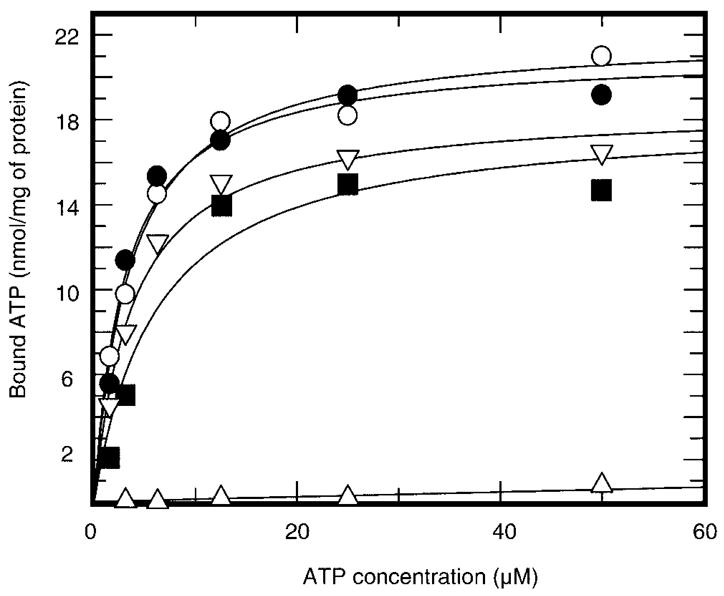
Recombinant PDK2, wild-type (○), His-239 → Ala (●) Glu-243 → Ala (▽), Glu-243 → Gln (■), or Asp-282 → Glu (Δ) mutants were incubated with the indicated concentrations of [α-32P]ATP (specific radioactivity ~200 cpm/pmol) for 2 min. Free and protein-bound nucleotides were separated using a vacuum-filtration assay as described under “Experimental Procedures.” The specific binding was determined after subtraction of nonspecific binding from the total binding. The nonspecific binding was determined in the presence of 1,000-fold excess of cold ATP.
In contrast, the substitution of Asp-282 severely affected nucleotide binding. The Asp-282 to Glu mutant did not exhibit any binding of [α-32P]ATP in excess of the background values, in agreement with its role in providing the major specific interaction between the adenine base and the protein (14). Interestingly, Asp-282 to Glu is a conserved substitution that does not change the charge of the side chain. Nevertheless, this mutation has a great impact on the nucleotide binding. The latter is entirely consistent with the available structural information (12, 13). The nucleotide-binding sites of histidine kinases appear to be very tightly packed and the additional methylene group of glutamic acid should put severe conformational constraints on the positioning of ATP within the active site.
PDC Binding
It is generally believed that the kinase component is an integral part of PDC, and every complex contains 2–3 tightly bound molecules of kinase (27). Association with the complex alone accounts for more than a 10-fold increase in the kinase activity (28). Therefore, an essential decrease in activity would be expected if the mutations affected the interaction between kinase and PDC directly or indirectly. To explore this possibility, we measured the PDC-kinase interaction using gel filtration through Sephacel S-300 columns (see “Experimental Procedures”). This procedure allows for fast separation of free and complex-bound kinase, thus decreasing the possibility of kinase dissociation during the procedure. As shown in Fig. 3A and B, under these conditions binding of wild-type PDK2 to the complex was readily detectable. To evaluate the ability of PDK2 active-site mutants to interact with the protein substrate, we conducted a series of experiments to determine their relative affinities for PDC (Fig. 3C). It appeared that all mutant kinases tested bind PDC at levels similar to the wild-type kinase. This suggests that the ability to catalyze the phosphotransfer reaction or the ability to bind the nucleotide substrate has no profound effect on the protein-protein interactions involved in kinase binding. On the other hand, we have noticed that all mutant proteins had a somewhat higher affinity for the protein substrate. The latter was especially apparent for the Asp-282 to Glu mutant, which consistently showed 2–3-fold higher affinity for PDC. The rationale for this phenomenon is currently unknown.
Fig. 3. Binding of wild-type PDK2 and respective mutant kinases to recombinant PDC.
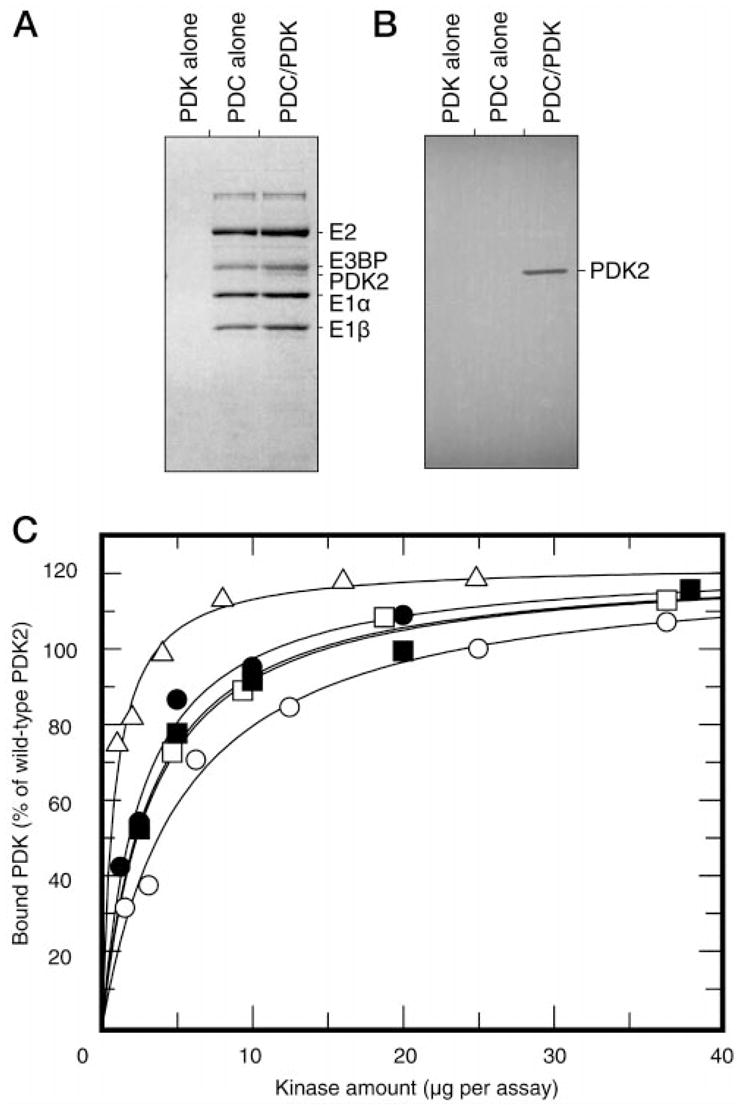
Panel A, SDS/PAGE analysis of wild-type PDK2, recombinant PDC, and PDC-kinase complex centrifuged through Sephacel S300 columns. Panel B, Western blot analysis of corresponding preparations with monoclonal antibodies against His6-tag. Immunoreactive bands were visualized by [125I]protein A staining followed by autoradiography. Panel C, concentration-dependence curves for binding of wild-type PDK2 (○), His-239 → Ala (□), Glu-239 → Asp (■), Glu-243 → Gln (●), and Asp-282 → Glu (△) mutants to recombinant PDC. Binding curves were constructed based on the results of scanning densitometry of the respective Western blots stained with 125I-protein A. Data was analyzed using UN-SCAN-IT gel™ software (Silk Scientific, Inc., Orem, UT). Results are expressed as percent of wild-type PDK2 bound to PDC.
Inter-subunit Interaction
In solution, PDK exists as a dimer (3). Dimerization was suggested to be very important for kinase function, allowing the kinase to move around PDC, phosphorylating multiple copies of the E1 component without dissociating from the complex (30). Thus, if the mutations somehow compromise the inter-subunit interactions within the dimer, this might have a deleterious effect on kinase activity. To explore this possibility, we employed a genetic approach, co-expressing His6-tagged and T7-tagged kinase molecules in E. coli. This allowed the use of the His6-tagged kinase as a bait to pull down T7-tagged kinase on an affinity resin. The resulting preparations can be characterized for the presence of T7-tagged species by Western blot analysis with anti-T7-tag anti-bodies. This way, it is possible to establish the formation of mixed species and draw conclusions about the strength of inter-subunit interaction for different mutant kinases. When the respective constructs were expressed in E. coli, His6-tagged species were purified on TALON™ resin and probed with anti-T7-tag antibodies using Western blot analysis; it was found that all preparations tested contained T7-tagged species, which cannot directly bind TALON™ resin (Fig. 4, panels A and B). The latter strongly suggests that the wild-type PDK2 as well as kinases carrying amino acid substitutions within the kinase domain exist as dimers. Furthermore, the ratios between T7-tagged and His6-tagged species in each preparation were very similar, indicating that the ability to catalyze phosphotransfer reaction or the ability to bind adenyl nucleotides is not required for dimer formation.
Fig. 4. Western blot analysis of differentially tagged preparations of PDK purified by metal affinity chromatography.
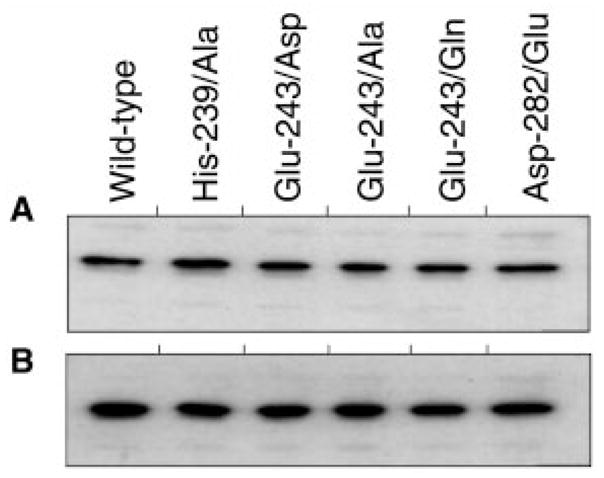
Panel A, affinity-purified kinase preparations probed with antibodies raised against T7-tag. Panel B, corresponding preparations probed with anti-H6-tag antibodies. Immunoreactive bands were visualized by 125I-protein A staining followed by autoradiography.
ATPase Activity in Preparations of PDK
Many phosphokinases (31), including some protein kinases (32, 33), have been reported to possess an intrinsic hydrolytic activity toward ATP. If PDK uses general base catalysis to activate the hydroxyl group of the serine residue for the direct attack on the ATP γ phosphate, it also must be able to utilize the hydroxyl of water as a phosphoryl acceptor instead of an amino acid hydroxyl group, although at a slower rate. To investigate this possibility, we determined ATPase activity in various preparations of PDK. When wild-type enzyme was incubated in a reaction mixture containing the standard components of the ATPase assay, and aliquots were removed and assayed for the release of [32P]Pi, there was a linear formation of Pi with time. Further-more, when Mg2+ was omitted from the standard ATPase assay, no activity was observed, suggesting that, indeed, preparations of PDK contain some ATP hydrolytic activity (data not shown). However, these results do not exclude the possibility that the source of the observed ATPase activity is a contaminating ATP hydrolase.
In several studies, the specificity of ATP hydrolytic reaction was demonstrated using compounds acting as potent inhibitors of ATP binding (34, 35). Unfortunately, to date, there are no PDK inhibitors available that are specific for the nucleotide binding domain. However, we reasoned that some of ATP-binding site inhibitors designed for enzymes with nucleotide binding domains arranged similarly to the nucleotide binding domain of histidine kinases and, therefore, to the nucleotide binding domain of PDK might inhibit PDK activity (18, 36). Indeed, one of these compounds, radicicol (monorden) (36), potently inhibited phosphorylation of PDC when tested in standard kinase assay (Fig. 5, panel A). Inhibition was competitive with respect to ATP, with the apparent Ki value of 23.3 ± 1.8 μM, making radicicol an invaluable tool as an inhibitor of the nucleotide binding domain of PDK.
Fig. 5. ATP hydrolytic activity in preparations of PDK2.
Panel A, inhibition of pyruvate dehydrogenase kinase activity by radicicol. Kinase activity was determined without radicicol (○) or in the presence of radicicol at concentrations of 25 μM (●), 50 μM (■), or 100 μM (□). Panel B, radicicol-sensitive ATP hydrolytic activity in preparations of wild-type, Glu-243 → Gln, and His-239 → Ala PDK2. Total ATP hydrolysis was measured in the absence of radicicol with 10 μM [γ-32P]ATP as a substrate. Nonspecific hydrolysis was determined in the presence of 200 μM radicicol. Shown is radicicol-sensitive ATPase activity (difference between total and nonspecific activity) intrinsic to PDK2. Panel C, radicicol-sensitive (○) versus E1-sensitive (●) ATPase activity in preparations of wild-type PDK2. Radicicol-sensitive ATPase activity was determined essentially as described in the legend to panel B. E1-sensitive ATPase activity was determined as a difference between total ATPase activity and ATPase activity measured in the presence of E1 component (final concentration 1.0 mg/ml).
The addition of radicicol to the standard ATPase assay inhibited ~25–30% of ATP hydrolytic activity in preparations of wild-type PDK2. The specific activity of radicicol-sensitive ATPase was ~1.0 nmol of Pi released/min/mg (Fig. 5, panel B), suggesting that PDK2 possesses intrinsic ATPase activity that comprises ~2.5% of kinase activity. Having analyzed the ATPase activity in preparations of wild-type PDK2, experiments were performed to establish whether the mutations within the active site of kinase affect the ATP hydrolytic activity. When kinases carrying the substitutions of Glu-243 and His-239 were assayed similarly to the wild-type PDK2, there was little if any radicicol-sensitive ATP hydrolysis that could be assigned to PDK2, indicating that these residues are essential for ATPase activity. To further explore the possible mechanistic significance of Glu-243 and His-239 in an ATP hydrolytic reaction, we prepared several highly purified preparations of PDK2 carrying substitutions of Glu-243 to Gln and His-239 to Ala. These preparations had a drastically reduced radicicol-insensitive ATPase activity. Subsequent analysis of these preparations revealed that His-239 to Ala enzyme had ~7% of the activity of the wild-type PDK2, whereas the activity of Glu-243 to Gln enzyme was very close to the background values, less than 0.5% of the wild type (Fig. 5, panel B). These results strongly suggest that the same residues of PDK2 are involved in catalysis of both hydrolytic and phosphotransferase reactions. To further analyze the relationship between kinase and ATPase activities, we characterized the effect of the substrate of kinase reaction (E1 component of PDC) on the rate of ATPase reaction. As shown in Fig. 5, panel C, inhibition of the total ATPase activity caused by the addition of E1 component was comparable with that caused by the addition of radicicol. This further confirms that PDK2 possesses an intrinsic ATPase activity and also shows that the kinase preferentially catalyzes the phosphorylation reaction under conditions when the native phosphoacceptor is provided.
Thus, it appears that both Glu-243 and His-239 are crucial for kinase activity. In conjunction with the results showing that Glu-243 and His-239 do not contribute to ATP binding, inter-subunit interaction, and binding to PDC, these data strongly suggest that Glu-243 and His-239 are required for catalysis of the phosphotransfer reaction. In accord with this idea is the observation that PDK possesses a weak ATP hydrolytic activity, which can be detected only in the absence of physiological substrate, the E1 component of PDC. Furthermore, the ATPase activity appears to depend on the presence of both Glu-243 and His-239. Substitution of either Glu-243 or His-239 resulted in a decrease in ATPase activity that closely corresponded to the decrease in kinase activity.
In conclusion, it is interesting to note that bacteria contain another protein of the same lineage, the so-called anti-sigma factor SpoIIAB (29). Its sequence is very similar to the bacterial histidine kinases but, like PDK, contains a properly spaced glutamic acid in the N box. Accordingly, SpoIIAB phosphorylates its substrate SpoIIAA on Ser-58 (29). This suggests that the ability of this type of catalytic domain to phosphorylate exogenous protein substrates on serine residues might be largely defined by the presence of glutamic acid in the N box.
Footnotes
This work was supported by United States Public Health Services Grants GM 51262 and DK 56898.
The abbreviations used are: PDC, pyruvate dehydrogenase complex; PDK, pyruvate dehydrogenase kinase; PDK1, PDK2, PDK3, and PDK4, isozymes 1, 2, 3, and 4 of pyruvate dehydrogenase kinase; E1, pyruvate dehydrogenase component of PDC; E2, dihydrolipoyl acetyltransferase component of PDC; E3, dihydrolipoamide dehydrogenase component of PDC; E3BP, E3-binding protein component of PDC; PAGE, polyacrylamide gel electrophoresis.
References
- 1.Randle PJ. Proc Nutr Soc. 1995;54:317–327. doi: 10.1079/pns19950057. [DOI] [PubMed] [Google Scholar]
- 2.Linn TC, Pettit FH, Reed LJ. Biochemistry. 1968;62:234–241. [Google Scholar]
- 3.Stepp LR, Pettit FH, Yeaman SJ, Reed LJ. J Biol Chem. 1983;258:9454–9458. [PubMed] [Google Scholar]
- 4.Teague WM, Pettit FH, Wu TL, Silberman SR, Reed LJ. Biochemistry. 1982;21:5585–5592. doi: 10.1021/bi00265a031. [DOI] [PubMed] [Google Scholar]
- 5.Popov KM, Kedishvili NY, Zhao Y, Gudi R, Harris RA. J Biol Chem. 1994;269:29720–29724. [PubMed] [Google Scholar]
- 6.Huang B, Gudi R, Wu P, Harris RA, Hamilton J, Popov KM. J Biol Chem. 1998;273:17680–17688. doi: 10.1074/jbc.273.28.17680. [DOI] [PubMed] [Google Scholar]
- 7.Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Biochem J. 1998;329:191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sale GJ, Randle PJ. Biochem J. 1982;203:99–108. doi: 10.1042/bj2030099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popov KM, Zhao Y, Shimomura Y, Kuntz MJ, Harris RA. J Biol Chem. 1992;267:13127–13130. [PubMed] [Google Scholar]
- 10.Robinson VL, Buckler DR, Stock AM. Nat Struct Biol. 2000;7:626–633. doi: 10.1038/77915. [DOI] [PubMed] [Google Scholar]
- 11.Popov KM, Kedishvili NY, Zhao Y, Shimomura Y, Crabb DW, Harris RA. J Biol Chem. 1993;268:26602–26606. [PubMed] [Google Scholar]
- 12.Bilwes AM, Alex LA, Crane BR, Simon MI. Cell. 1999;96:131–141. doi: 10.1016/s0092-8674(00)80966-6. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka T, Saha SK, Tomomori C, Ishima R, Liu D, Tong KI, Park H, Dutta R, Qin L, Swindells MB, Yamazaki T, Ono AM, Kainosho M, Inouye M, Ikura M. Nature. 1998;396:88–92. doi: 10.1038/23968. [DOI] [PubMed] [Google Scholar]
- 14.Bowker-Kinley M, Popov KM. Biochem J. 1999;344:47–53. [PMC free article] [PubMed] [Google Scholar]
- 15.Thelen JJ, Miernyk JA, Randall DD. Biochem J. 2000;349:195–201. doi: 10.1042/0264-6021:3490195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davie JR, Wynn RM, Meng M, Huang YS, Aalund G, Chuang DT, Lau KS. J Biol Chem. 1995;270:19861–19867. doi: 10.1074/jbc.270.34.19861. [DOI] [PubMed] [Google Scholar]
- 17.Kunkel TA. Proc Natl Acad Sci U S A. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis RJ, Singh OMP, Smith CV, Skarzynski T, Maxell A, Wonacott AJ, Wigley DB. EMBO J. 1996;15:1412–1420. [PMC free article] [PubMed] [Google Scholar]
- 19.Sanger F, Nicklen S, Coulson AR. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris RA, Bowker-Kinley MM, Wu P, Jeng J, Popov KM. J Biol Chem. 1997;272:19746–19751. doi: 10.1074/jbc.272.32.19746. [DOI] [PubMed] [Google Scholar]
- 21.Jeng J, Kallarakal AT, Kim SF, Popov KM, Song BJ. Comp Biochem Physiol B Biochem Mol Biol. 1998;120:205–216. doi: 10.1016/s0305-0491(98)10010-x. [DOI] [PubMed] [Google Scholar]
- 22.Wu P, Sato J, Zhao Y, Jaskiewicz J, Popov KM, Harris RA. Biochem J. 1998;329:197–201. doi: 10.1042/bj3290197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pratt ML, Roche TE. J Biol Chem. 1979;254:7191–7196. [PubMed] [Google Scholar]
- 24.Singh US, Cerione RA. J Biol Chem. 1996;271:27292–27298. doi: 10.1074/jbc.271.44.27292. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 27.Patel MS, Roche TE. FASEB J. 1990;4:3224–3233. doi: 10.1096/fasebj.4.14.2227213. [DOI] [PubMed] [Google Scholar]
- 28.Popov KM. FEBS Lett. 1997;419:197–200. doi: 10.1016/s0014-5793(97)01453-1. [DOI] [PubMed] [Google Scholar]
- 29.Najafi SMA, Willis AC, Yudkin MD. J Bacteriol. 1995;177:2912–2913. doi: 10.1128/jb.177.10.2912-2913.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S, Baker JC, Roche TE. J Biol Chem. 1995;270:793–800. doi: 10.1074/jbc.270.2.793. [DOI] [PubMed] [Google Scholar]
- 31.Knowles JR. Annu Rev Biochem. 1980;49:877–919. doi: 10.1146/annurev.bi.49.070180.004305. [DOI] [PubMed] [Google Scholar]
- 32.Moll GW, Jr, Kaiser ET. J Biol Chem. 1976;251:3993–4000. [PubMed] [Google Scholar]
- 33.Paudel HK, Carlson GM. J Biol Chem. 1991;266:16524–16529. [PubMed] [Google Scholar]
- 34.Panaretou B, Prodromou C, Roe MS, O’Brien R, Ladbury JE, Piper PW, Pearl LH. EMBO J. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Obermann WMJ, Sondermann H, Russo AA, Pavletich NP, Hartl FU. J Cell Biol. 1998;143:901–910. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roe SM, Prodromou C, O’Bien R, Ladbury JE, Piper PW, Pearl LH. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]