

Abstract
Wallerian degeneration is the degeneration of the distal stump of an injured axon. It normally occurs over a time course of around 24 hr but it is delayed in the slow Wallerian degeneration mutant mouse (C57BL/Wlds) for up to 3 weeks. The gene, which protects from rapid Wallerian degeneration, Wld, previously has been mapped to distal chromosome 4. This paper reports the fine genetic mapping of the Wld locus, the generation of a 1.4-Mb bacterial artificial chromosome and P1 artificial chromosome contig, and the identification of an 85-kb tandem triplication mapping within the candidate region. The mutation is unique to C57BL/Wlds among 36 strains tested and therefore is a strong candidate for the mutation that leads to delayed Wallerian degeneration. There are very few reports of tandem triplications in a vertebrate and no evidence for a mutation mechanism so this unusual mutation was characterized in more detail. Sequence analysis of the boundaries of the repeat unit revealed a minisatellite array at the distal boundary and a matching 8-bp sequence at the proximal boundary. This finding suggests that recombination between short homologous sequences (“illegitimate” or “nonhomologous” recombination) was involved in the rearrangement. In addition, a duplication allele was identified in two Wlds mice, indicating some instability in the repeat copy number and suggesting that the triplication arose from a duplication by unequal crossing over.
Wallerian degeneration is the common endpoint of many forms of axonal pathology. It occurs in the peripheral and central nervous system and can be studied experimentally in the distal stump of an axon that has been cut or crushed (1). Early events in the Wallerian degeneration pathway, usually occurring within 24 hr of a lesion, include disintegration of the axonal cytoskeleton and breakdown of the axonal membrane. This degeneration is followed by secondary changes in other cell types, such as breakdown of the myelin sheath and macrophage infiltration (2). Hypotheses about what initiates Wallerian degeneration include loss of trophic support from the cell body and activation of calpain by calcium influx (3, 4). In contrast, studies on the C57BL/slow Wallerian degeneration mutant mouse (C57BL/Wlds suggest that Wallerian degeneration could be an active process akin to apoptosis (5).
The C57BL/Wlds mouse has an autosomal dominant mutation that protects its neurons from undergoing rapid Wallerian degeneration (6, 7), and a stimulated sciatic nerve from C57BL/Wlds consequently will transmit a compound action potential for up to 3 weeks after the nerve has been severed. The mutation occurred spontaneously at Harlan-Olac (Bicester, U.K.) and is not harmful. In fact, the C57BL/Wlds mouse is indistinguishable from C57BL/6J in appearance, behavior, histocompatibility, and the genotype of more than 50 microsatellites and restriction fragment length polymorphisms. The C57BL/Wlds mouse previously has been distinguished only by the remarkable ability of its axons to survive without a nucleus after separation from the cell body.
The Wlds mutation originally was thought to act by slowing macrophage infiltration into a damaged nerve, but a series of transplantation experiments has shown since that the phenotype is inherent in the axon (8, 9). It also can be reproduced in vitro by using primary explants of dorsal root ganglion or superior cervical ganglion (5). Neurites grow from the explant in the presence of nerve growth factor and, when severed from the cell body by using a scalpel, survive for up to 10 times as long if the tissue has been taken from a C57BL/Wlds mouse. This phenomenon is seen even if neurons are plated as single cells and in the absence of any Schwann cells.
Attempts to intervene in some neurological disorders will have to address degeneration both of the axon and the cell body. For example, the introduction of a bcl-2 transgene or treatment with glial cell line-derived neurotrophic factor (GDNF) protects cell bodies in the motor neuron disease mouse pmn but, by themselves, these treatments do not prevent axonal loss nor do they alleviate the symptoms of the disease (10, 11). Furthermore, studies using C57BL/Wlds have shown that rapid Wallerian degeneration is required for the subsequent regeneration of sensory nerves to proceed efficiently (12, 13). Unfortunately, it is not clear how to manipulate Wallerian degeneration because little is known about its regulation. The identification of the Wld gene should give an insight into this problem.
Wld has been mapped to distal mouse chromosome 4 close to Nppa (14), and further studies have excluded some potential candidate genes in this region (15). Interestingly, an axonal form of Charcot-Marie-Tooth (CMT2A) characterized by axonal loss, has been mapped to the homologous location in humans, 1p36 (16). This paper reports the identification and characterization of a tandem triplication within the Wlds candidate region. In addition to being an important step toward the identification of the Wld gene, this paper characterizes a vertebrate tandem triplication in a way that allows a mutation mechanism to be proposed. Clues are provided by the identification of a short homologous sequence at each boundary of the repeat unit and by the identification of a duplication allele.
MATERIALS AND METHODS
Genetic Mapping. The mapping followed the method of Lyon et al. (14), with one additional step. A distal segment of sciatic nerve was fixed in 2% glutaraldehyde, 2% paraformaldehyde, 0.1 M sodium phosphate (pH 7.2) at the time of the electrophysiological assay, and critical recombinants were rescored by electron microscopy. There were no discrepancies between the two assays.
Physical Mapping.
P1 artificial chromosomes (P1) were isolated from the library of the Resource Centre Primary Database (Berlin) (17) by PCR and hybridization and bacterial artificial chromosomes (BAC) from the Research Genetics (Huntsville, AL) library by hybridization. End clones were obtained by inverse PCR using vector-derived primers or by shotgun subcloning followed by hybridization with a vector-specific probe lying close to the cloning junction. Many end clones were placed on the genetic map by restriction fragment length polymorphism mapping to detect any chimerism (although in fact none was encountered in these libraries) and to locate recombination breakpoints.
Extraction of Genomic DNA.
Solution DNA was extracted from mouse spleen by using the Nucleon II kit (Scotlab, Shelton, CT) or by phenol extraction. High molecular weight DNA for pulsed-field gel electrophoresis also was extracted from spleen as follows. Half a spleen was dissociated in 1 ml of PBS by multiple passage through increasingly narrower needles (down to 25 gauge) and strained through a Becton Dickinson cell strainer (70 μm). The cells were mixed with an equal volume of 1% low-melting point agarose (Flowgen, Lichfield, U.K.) in distilled water and poured into a Bio-Rad plug mold on ice. The embedded cells then were lysed by three overnight incubations in 0.5 M EDTA, 1% sodium-N-laurylsarcosine, and 25 μg/ml of proteinase K and stored long term in the same solution.
DNA Hybridization.
DNA was Southern-blotted overnight onto Hybond N+ (Amersham) in 0.4 M NaOH. Probes were radiolabeled by using the Amersham Megaprime kit and [α-32P]dCTP (DuPont/NEN). Posthybridization washes were normally done at a stringency of 0.5× standard saline citrate (SSC), 0.1% SDS, unless the probe was less than 200 bp (1× SSC, 0.1% SDS) or repetitive (0.1× SSC, 0.1% SDS). Radioactive filters were exposed to x-ray film (X-Ograph, Malmesbury, U.K.) for between 1 hr and 4 days at −70°C.
PCR.
The method of Saiki et al. (18) was followed by using Taq DNA polymerase (Promega) or, for long-range PCR or products for sequencing, Pfu polymerase (Stratagene). Custom oligonucleotides were obtained from GIBCO/BRL and D4Mit map pairs from Research Genetics.
Automated DNA Sequencing.
P1 and BAC subclones in pGEM3zf(−) (Promega) were sequenced by dye primer technology by using the Amersham Thermosequenase Dye Primer kit and Applied Biosystems fluorescently labeled universal primers. PCR products were sequenced with the same reagents after blunt-end cloning of Pfu polymerase products into pCR-Script Amp SK(+) (Stratagene). Sequencing reactions were analyzed at Alta Bioscience (University of Birmingham) by using an Applied Biosystems 373a or 377 prism sequencer and the corresponding software.
Pulsed-Field Gel Electrophoresis.
Before digestion, agarose plugs were equilibrated with five changes of 5 ml of 10 mM Tris⋅HCl (pH 7.5), 1 mM EDTA, and then with 0.5 ml of the appropriate manufacturer’s restriction digest buffer. Thirty units of enzyme in a volume of 100 μl was used to digest 1/2 fragments of plugs for 3 hr at 37°C. Pulsed-field gel electrophoresis was performed in 0.5× 90 mM Tris/64.6 mM boric acid/2.5 mM EDTA, pH 8.3 by using a Bio-Rad Chef-DR electrophoresis cell and running conditions of 6 V/cm, 10°C, switch times ramped from 15 to 45 sec, and a run length of 21–24 hr.
Quantification of Hybridization Signals.
Hybridization signals were analyzed by using a Storm 840 PhosphorImager and Molecular Dynamics version 4.1 imagequant software.
Subcloning BAC into Phage.
BAC 269-F8 was partially digested (to a 10- to 30-kb size range) with MboI, ligated to LambdaGEM12 XhoI half-site arms (Promega) and packaged into Gigapack IIXL packaging extract (Stratagene). Packaged phage were used to infect LE392 cells. The DNA was transferred by the standard phage lift protocol to Hybond N (Amersham) and analyzed by hybridization.
RESULTS
Genetic and Physical Mapping of the Wld Gene.
Wld previously has been mapped to distal mouse chromosome 4 (14) by using a 282-animal cross of (C57BL/Wlds × Mus spretus) × C57BL/6J. We have extended this cross to 1,262 animals and made a second 227-animal cross in which M. spretus was replaced by DBA, bringing the total to 1,489. Both crosses indicated the same location for Wld between Nppa and D4Mit33 and, in addition to this, the DBA cross placed Wld proximal to D4Mit127. The composite locus order is centromere - (Tnfr2, Nppa) - (0.40 ± 0.16) - (Wld, D4Mit49, D4Mit225, D4Mit310) - (0.07 ± 0.07) - D4Mit127 - (0.07 ± 0.07) - D4Mit33 - (0.64 ± 0.28) - Ly63 - telomere.
The three nonrecombinant markers, D4Mit49, D4Mit225, and D4Mit310, were used to nucleate a physical map. The Wld region was poorly represented in yeast artificial chromosome (YAC) libraries and highly unstable in the few YACs that were found, so P1s and BACs were used to generate the contig (Fig. 1). The contig is estimated to be 1.4 Mb long with approximately 1.2 Mb lying within the candidate region. In a backcross of this size the most probable distance between adjacent recombination breakpoints is less than 150 kb so there has been a lower than average rate of recombination across the Wld region. Interestingly, five recombinations all map within a single P1 clone (236-D23) at the proximal end of the candidate region, indicating a hotspot of recombination.
Figure 1.

Physical and genetic map of the Wld region. Wld maps between Nppa and D4Mit127 in distal mouse chromosome 4. Five recombinations lying within the span of a single P1 (236-D23) define the proximal end of the Wld region, whereas a single recombination defines the distal end. A BAC and P1 contig estimated at 1.4 Mb was generated by using as a starting point three nonrecombinant markers, D4Mit49, D4Mit225, and D4Mit310. BACs (Research Genetics) are labeled “B” on their left side on this diagram and the other clones are P1s (Resource Centre Primary Database, RZPD). The coordinates of each clone from the respective libraries are shown. Five P1 and BAC end clones are shown to lie within the repeated region. L23/R and 75/R are the closest flanking end clones that are not repeated, and the location of 5P1 (used in Fig. 2a) is also shown.
A Tandem Repeat within the Wlds Candidate Region.
Several probes located approximately 100–200 kb from the proximal end of the Wld candidate region hybridize more strongly to genomic DNA from C57BL/Wlds than to C57BL/6J (Fig. 2a). This finding was consistent across 15 C57BL/Wlds mice from three different breeding colonies, and the most likely explanation is an increase in the copy number of the target sequence in C57BL/Wlds. The region whose copy number increased is substantially larger than 30 kb, because probes located across a 30-kb region all gave a greater signal intensity but no difference in fragment size with a range of frequent-cutter restriction enzymes (Fig. 2b).
Figure 2.

Identification by genomic Southern blotting of a tandemly repeated region on Wlds chromosome 4. (a) Three probes (56/L, 320/L, and 269/L) lying within 30 kb of one another show an amplified signal when hybridized to a Southern blot of Wld genomic DNA. Markers on either side (L23/R and 5P1) show an approximately equal signal in C57BL/Wlds and C57BL/6J when hybridized to the same filter. (b) Probes located centrally within the repeat unit (320/L is shown here) consistently show an increase in signal intensity but no difference in restriction fragment size with a range of frequent-cutter restriction enzymes. (c) Probe 59 (i) and (iii) located within the repeat unit immediately adjacent to the proximal boundary, hybridizes to a fragment of identical size in C57BL/Wlds and in C57BL/6J, but also to a Wld-specific fragment of altered size and with an increased intensity. A probe located 0.8–1.0 kb from the distal end of the repeat unit (ii and iv) detects the same Wld-specific fragment (indicated by arrows) as probe 59, together with a different fragment that is common to both C57BL/6J and C57BL/Wlds. (d) Tandem repeat model to account for the above observations. Probe 1 represents probes used in a and b. It lies centrally in the repeated region and detects a restriction fragment of increased dosage but unaltered size in C57BL/Wlds. Probe 2 represents probe 59 in c. It detects one restriction fragment that is present as a single copy in both C57BL/6J and C57BL/Wlds. However, when the target sequence of this probe is repeated in C57BL/Wlds, a shifted band appears because of the formation of a junction fragment. This band may be shifted up or down depending on the location of the restriction site shown on the left. Probe 3 is located at the other end of the repeat unit and detects the same shifted fragment as probe 2, but a different fragment that is common to both C57BL/6J and C57BL/Wlds.
There are several types of genomic rearrangement that could give rise to an increase in copy number, e.g., tandem repeats, inverted repeats, or dispersed repeats. To distinguish between them, abnormal junction fragments occurring at the end of the repeat unit were investigated. Probes were generated by walking outward from the above markers in a phage library of BAC 269-F8 and by inverse PCR (see below). Their hybridization patterns indicate the presence of a tandem repeat in C57BL/Wlds (Fig. 2c). First, probes from each end of the repeat unit detect Wlds-specific fragments of an identical size, despite being located at distant sites on the physical map. This finding suggests that these probes have been juxtaposed in C57BL/Wlds by the formation of a junction between adjacent tandem repeat units (Fig. 2d). Second, the absence of any further C57BL/Wlds-specific fragments is evidence against the existence of any inverted repeat. Finally, the presence of fragments common to both C57BL/Wlds and C57BL/6J indicates that there are no local rearrangements in C57BL/Wlds at either end of the repeat array. An analogous restriction pattern is seen in other tandem repeats, such as the human chromosome 17 duplication in Charcot-Marie-Tooth type 1A (CMT1A) (19).
The Rearrangement Is Unique to C57BL/Wlds.
To test the likelihood that this genomic rearrangement is the Wlds mutation, genomic DNAs from 36 other mouse strains were obtained from the Jackson Laboratory and analyzed with the above probes. Both the appearance of an additional fragment for probes located close to the junction (Fig. 3a) and the altered dosage for probes located centrally within the repeat unit (Fig. 3b) were unique to C57BL/Wlds mice, even among closely related C57 strains. This finding, together with the location of the rearrangement within the Wld candidate genetic interval, is strong evidence that this genomic rearrangement is the Wlds mutation.
Figure 3.

The genomic rearrangement is unique to C57BL/Wlds. (a) A BamHI genomic Southern blot was hybridized with a probe located close to the proximal boundary, giving a C57BL/Wlds-specific shifted band. (b) A BlpI Southern blot was hybridized with 269/L, located centrally within the repeat unit, and with 5P1 as a single-copy control. Closely related C57 strains are shown in a and a representative group of other strains in b. Additional strains tested and not shown were 129/J, AKR/J, BALB/cJ, BDP/J, BUB/BnJ, CAST/Ei, C3H, DBA, HRS/J, I/LnJ, LT/Sv-A<y>/A, MA/MyJ, and NZW/LacJ. In both assays, the Wlds allele was unique.
An 85-kb Triplication in Wlds Mice.
Pulsed-field gel electrophoresis was used to determine the total length of the extra repeat(s) in C57BL/Wlds (Fig. 4). All probes lying either within the repeat unit or a short distance to either side detect a single NotI fragment of approximately 220 kb in C57BL/6J and a single SmaI fragment of 180 kb, indicating that neither enzyme cuts within the repeat unit. In five of seven C57BL/Wlds mice analyzed (see below), these single fragments increased in size to approximately 390 and 350 kb, respectively. Results from a representative animal, WldA, are shown in Fig. 4. These C57BL/Wlds mice therefore have an insertion of approximately 170 kb. However, a phage and plasmid contig generated from BAC 269-F8 and P1 320-I15 established the length of the repeat unit as 85 kb (data not shown). This finding suggests that there must be two extra copies of the repeat unit in these C57BL/Wlds mice to account for the 170-kb insertion.
Figure 4.
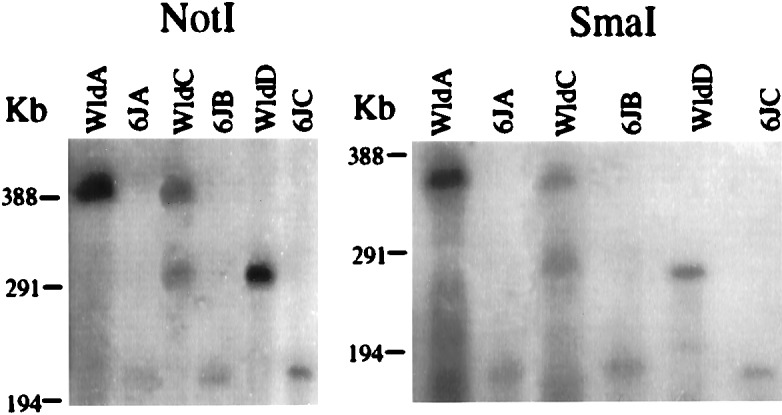
Identification of 170- and 85-kb insertions in C57BL/Wlds. Pulsed-field gel analysis of C57BL/Wlds and C57BL/6J genomic DNA digested with NotI or SmaI. After electrophoresis the gel was Southern-blotted and the DNA was hybridized with the probe 271/L, located centrally within the repeat unit (Fig. 1). WldA is representative of five of seven DNAs analyzed, whereas Wlds C and D were unique.
To confirm the presence of two extra repeats, the number of extra copies was estimated by PhosphorImager quantification of signal from genomic Southern blots. According to the model in Fig. 2d the haploid copy number of the unaltered fragment in Fig. 2c should be one, and the relative intensity of the C57BL/Wlds-specific fragment should indicate the number of extra copies. Visual inspection of this autoradiograph indicates that the shifted band is indeed more intense than the constant band, suggesting that there is more than one extra copy. PhosphorImager analysis of two representative animals, WldA (the same as in Fig. 4) and WldB, confirmed this (Table 1). Because the efficiency of Southern transfer is lower for larger DNA fragments, upward-shifted bands give an underestimate of the number of extra copies (at 1.6–1.7) and downward-shifted bands an overestimate (at 2.6–2.7). Hence, the likely number of extra copies in these animals is two.
Table 1.
Estimates of the number of extra copies of the repeat unit in three Wlds mice
| Mouse | Lower estimate based on upward-shifted BlpI band | Upper estimate on downward-shifted EcoRV band |
|---|---|---|
| Wld A | 1.58 | 2.56 |
| Wld B | 1.67 | 2.69 |
| Wld C | 1.16 | 1.62 |
Identification of a Duplication Allele.
Although most C57BL/Wlds mice analyzed were homozygous for the triplication, two were found to have a different genotype (Fig. 4 and Table 1). WldD had a single shifted NotI or SmaI fragment approximately 85 kb larger than the corresponding C57BL/6J fragment, and WldC had a mixture of the 170-kb shifted fragment and the 85-kb shifted fragment. These genotypes are unlikely to be completely different rearrangements from those in WldA and B, because frequent-cutter restriction fragments were always identical in size (data not shown). Therefore, the most likely interpretation is that the 85-kb shifted band represents a duplication allele and that WldC is either a heterozygote for the 85-kb and 170-kb genotypes or a somatic mosaic. Quantification of the signal from WldC confirmed that the number of extra copies is between one and two (Table 1). Unfortunately there was insufficient DNA available from WldD to analyze the copy number in this mouse.
Sequences at the Boundaries of the Repeat Unit.
Clones spanning the proximal boundary of the repeat unit were isolated by genomic walking and the approximate location of the boundary deduced from hybridization patterns. Probe 59 (Fig. 5a) was found to lie within 1 kb of the proximal boundary because it detected a Wlds-specific PstI fragment of approximately 1 kb in addition to a constant fragment of 3 kb (data not shown). Primers 54 and 59 were used in inverse PCR (20) to amplify across the junction formed between adjacent repeat units (Fig. 2d), and the inverse PCR sequence was used to isolate clones spanning the distal boundary (Fig. 5b). The sequence of the 312-bp inverse PCR product (Fig. 5 a and b, lower line) indicates the point at which the junction diverges from the normal sequences. There are, of course, two junctions in C57BL/Wlds but it is unlikely that there are major differences between them because there is only one shifted hybridization band and only one distinguishable inverse PCR product.
Figure 5.

Sequencing of the boundaries of the repeat unit. (a) Sequence of the proximal boundary of the repeat unit showing the normal sequence (upper line) and the inverse PCR product (lower line), representing the inner junctions formed in Wlds. The two sequences can be aligned exactly, except for 19 bp (angled) that are unique to the inverse PCR product. These 19 bp are derived from the distal end of the repeat unit, having been juxtaposed in C57BL/Wlds by the formation of the junction (Fig. 2d). PstI sites shown left and right are the sites of circularization during inverse PCR. The 8-bp sequence shared between the proximal and distal boundaries is boxed. Sequences of PCR primers, represented by horizontal arrows, are 54 = 5′-CGTTGGCTCTAAGGACAGCAC-3′ and 59 = 5′-CGCCTCTTTGATCCCTACAGA-3′. (b) Sequence of the distal boundary of the repeat unit showing the approximate site of the boundary and the 8-bp motif common to the distal and proximal boundaries (boxed). The inverse PCR product diverges at a slightly different point from the normal distal and proximal sequences. This, and the possibility that one or two bases may be coincidentally shared between sequences, makes it difficult to define the exact site of each boundary.
A minisatellite array was identified immediately within the distal boundary of the repeat unit (Fig. 5b). Sequence compressions have prevented complete sequencing of the array, but five invariant 22-bp repeats (CCCTGCAGCCCCCACCCCTCTC) were sequenced at the distal end and five more variant repeats were identified. The full length of the array is approximately 700 bp in C57BL/10. A probe containing this minisatellite detected 13 genomic BlpI fragments at low stringency but only this single locus at high stringency (data not shown), indicating the presence of similar, but not identical, sequences elsewhere in the mouse genome. A moderate degree of polymorphism also was found but there were no further differences between Wlds and C57BL/6J. Interestingly, an 8-bp GC-rich sequence (GCCCCCAC) occurs immediately adjacent to the proximal boundary and forms part of the minisatellite repeat at the distal boundary. Recombination between these short sequences could have played an important part in the mutation mechanism.
DISCUSSION
A tandem triplication of an 85-kb repeating unit has been identified within the Wlds candidate genetic interval on distal mouse chromosome 4, and it is unique to C57BL/Wlds. Therefore, this is a strong candidate for the Wlds mutation. Three mechanisms are proposed through which this mutation might affect the Wld gene, although others can be envisaged. First, the gene and all its regulatory regions may be located entirely within the triplicated region. In this case the increased dosage of the gene may cause an increase in its rate of transcription, and consequently an increase in the steady-state level of the protein. Second, if the gene crosses one or both boundaries of the triplicated region its coding sequence is likely to be disrupted. This mutation might truncate the protein product by introducing a premature stop codon, or it could generate a hybrid protein with a novel function. Third, the coding region of the gene may be brought under the action of a new regulatory element by the DNA junctions formed in this rearrangement (Fig. 2d). Such a position effect might increase the rate of transcription even more than 3-fold and conceivably could affect genes lying just outside the triplicated region. Given an average spacing of one gene per 30–60 kb in the mouse (based on 50,000 to 100,000 genes in 3 × 109 bp), this 85-kb region of genomic DNA is likely to code for more than one gene. Therefore, to identify the Wld gene it will be necessary to construct a detailed transcript map of the triplicated region and to use a functional model to test whether candidate gene(s) can confer the dominant Wlds phenotype on wild-type neurons. These studies also will be necessary to provide formal proof that the triplication is the Wlds mutation.
There are very few examples of germ-line triplication, and little is known about the mechanism by which they might arise. There are some rare triplications within the α-globin gene cluster (21) and a tandem triplication has been observed in the Drosophila double Bar mutant (22–24). In the mouse it has been predicted that a triplication should be formed as a reciprocal genotype when the 70-kb pink-eyed unstable (pun) duplication reverts to wild type (25). However, double Bar occurs at a lower than expected frequency, and no pun triplication has ever been observed. In these two cases it has been suggested that a triple dosage of a gene(s) within the repeated region confers a growth disadvantage. In contrast, any gene lying within the Wlds triplication does not harm the mouse in any discernible way, even in homozygotes. Indeed, although there may be some instability (see below) the triplication genotype appears to have been predominant in the Wlds population for many generations.
As with the formation of double Bar, it is likely that the Wlds triplication arose in a two-step process, because an initial duplication would predispose to homologous (but unequal) recombination within the 85-kb repeat unit. This model raises the question of further repeat instability including partial or complete reversion. Interestingly, a duplication genotype was observed and there are two plausible explanations for this. Either there are a few surviving copies of the proposed original mutation, or partial reversion has occurred (triplication back to duplication). The pink-eyed unstable (pun) duplication (25) provides a precedent for tandem repeat reversion in the mouse but to confirm that reversion occurs in C57BLWlds it would be necessary to demonstrate the de novo appearance of a duplication from parents (or a breeding population) that carry only a triplication. The triplication is, however, sufficiently stable to have been the predominant genotype in the Wlds population for several years, because the mice carrying a triplication date from early 1994, 1996, and 1997, whereas the duplication dates from 1995. It will be interesting to determine whether there is any phenotypic difference between mice carrying the duplication and the triplication, although so far the duplication has been observed only in postmortem tissue.
The sequences surrounding the boundaries of the repeat unit suggest a mutation mechanism involving recombination between short regions of homology (sometimes called “illegitimate” or “nonhomologous” recombination). The sequence 5′-GCCCCCAC-3′ lies at the proximal boundary and is also part of the 22-bp minisatellite repeat at the distal end. An 8-bp motif should occur approximately once every 64 kb in a random sequence so it seems unlikely to be a coincidence that it should be placed so prominently next to each boundary of the triplicated region. Minisatellites have been shown to be highly recombinogenic (26) and involved in genomic rearrangements leading to cancer (27). Short stretches of sequence homology are sufficient for recombination and have been observed in several other chromosomal rearrangements: the sequences at each end of the mouse pink-eyed unstable (pun) duplication match eight of nine bp from the consensus sequence of DNA gyrase sites (25); and the sequence TACTCTA occurs at both deletion junctions in Hunter syndrome (28). In contrast, there is no extended region of homology between the two ends of the Wlds repeat unit such as that which occurs in the CMT1A duplication (19, 29). The evidence is that probes lying just inside the Wlds repeat detect single-copy and different DNA fragments (except, of course, for the Wlds-specific fragment that is formed by the junction between repeat units) (Fig. 2c).
Finally, the physical mapping resources reported here and the genomic rearrangement itself will facilitate the mapping and identification of a number of disease genes that map either in distal mouse chromosome 4 or in the region of conserved synteny in humans, 1p36. These include Charcot-Marie-Tooth type 2A (16), neuroblastoma (30), diabetes susceptibility (Idd9) (31), and resistance to plasmacytoma (Pctr2) (32).
In summary, an 85-kb tandem triplication found within the candidate genetic interval of distal mouse chromosome 4 is likely to be the Wlds mutation, and this mutation could affect gene function by a mechanism involving gene dosage, gene disruption, or a position effect. This unusual mutation appears to have been generated by a mechanism involving nonhomologous recombination at a minisatellite and instability in the copy number of the repeat unit.
Acknowledgments
We are grateful to the Resource Centre Primary Database (RZPD, Berlin) and to Dr. Roger Cox for the provision of P1 clones. This work was supported by Bristol-Myers Squibb and by the award of a Blaschko Visiting Research Scholarship to L.C.
ABBREVIATIONS
- BAC
bacterial artificial chromosome
- P1
P1 artificial chromosome
- Wlds
slow Wallerian degeneration mutant
Footnotes
References
- 1.Waller A. Philos Trans R Soc London. 1850;140:423–429. [Google Scholar]
- 2.Beuche W, Friede R L. J Neurocytol. 1984;13:767–796. doi: 10.1007/BF01148493. [DOI] [PubMed] [Google Scholar]
- 3.Schlaepfer W W, Hasler M B. Brain Res. 1979;168:299–309. doi: 10.1016/0006-8993(79)90171-9. [DOI] [PubMed] [Google Scholar]
- 4.Glass J D, Brushart T M, Griffin J W. J Neurochem. 1994;62:2472–2475. doi: 10.1046/j.1471-4159.1994.62062472.x. [DOI] [PubMed] [Google Scholar]
- 5.Buckmaster E A, Perry V H, Brown M C. Eur J Neurosci. 1995;7:1596–1602. doi: 10.1111/j.1460-9568.1995.tb01155.x. [DOI] [PubMed] [Google Scholar]
- 6.Lunn E R, Perry V H, Brown M C, Rosen H, Gordon S. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 7.Perry V H, Lunn E R, Brown M C, Cahusac S, Gordon S. Eur J Neurosci. 1990;2:408–413. doi: 10.1111/j.1460-9568.1990.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 8.Perry V H, Brown M C, Lunn E R, Tree P, Gordon S. Eur J Neurosci. 1990;2:802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x. [DOI] [PubMed] [Google Scholar]
- 9.Glass J D, Brushart T M, George E B, Griffin J W. J Neurocytol. 1993;22:311–321. doi: 10.1007/BF01195555. [DOI] [PubMed] [Google Scholar]
- 10.Sagot Y, Dubois-Dauphin M, Tan S A, De Bilbao F, Aebischer P, Martinou J C, Kato A C. J Neurosci. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sagot Y, Tan S A, Hammang J P, Aebischer P, Kato A C. J Neurosci. 1996;16:2335–2341. doi: 10.1523/JNEUROSCI.16-07-02335.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown M C, Lunn E R, Perry V H. J Neurobiol. 1992;23:521–536. doi: 10.1002/neu.480230507. [DOI] [PubMed] [Google Scholar]
- 13.Chen S, Bisby M A. J Comp Neurol. 1993;333:449–454. doi: 10.1002/cne.903330310. [DOI] [PubMed] [Google Scholar]
- 14.Lyon M F, Ogunkolade B W, Brown M C, Atherton D J, Perry V H. Proc Natl Acad Sci USA. 1993;90:9717–9720. doi: 10.1073/pnas.90.20.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coleman M P, Buckmaster E A, Ogunkolade B W, Tarlton A, Lyon M F, Brown M C, Perry V H. Mamm Genome. 1996;7:552–553. doi: 10.1007/s003359900287. [DOI] [PubMed] [Google Scholar]
- 16.Ben-Othmane K B, Middleton L T, Loprest L J, Wilkinson K M, Lennon F, Rozear M P, Stajich J M, Gaskell P C, Roses A D, Pericak-Vance M A, Vance J M. Genomics. 1993;17:370–375. doi: 10.1006/geno.1993.1334. [DOI] [PubMed] [Google Scholar]
- 17.Francis F, Zehetner G, Hoglund M, Lehrach H. Genet Anal Tech Appl. 1994;11:148–157. doi: 10.1016/1050-3862(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 18.Saiki R K, Scharf S, Faloona F, Mullis K B, Horn G T, Erlich H A, Arnheim N. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 19.Pentao L, Wise C A, Chinault A C, Patel P I, Lupski J R. Nat Genet. 1992;2:292–300. doi: 10.1038/ng1292-292. [DOI] [PubMed] [Google Scholar]
- 20.Silverman G A, Jockel J I, Domer P H, Mohr R M, Taillon-Miller P, Korsmeyer S J. Genomics. 1991;9:219–228. doi: 10.1016/0888-7543(91)90245-a. [DOI] [PubMed] [Google Scholar]
- 21.Vellegas A, Sanchez J, Carreno D L, Ropero P, Gonzalez F A, Espinos D, Penalver M A, Lozano M. Am J Hematol. 1995;49:294–298. doi: 10.1002/ajh.2830490407. [DOI] [PubMed] [Google Scholar]
- 22.Sturtevant A H. Genetics. 1925;10:117–147. doi: 10.1093/genetics/10.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller H J, Prokofieva-Belgovskaya A A, Kossikov K V. Acad Sci USSR. 1936;1:87–88. [Google Scholar]
- 24.Bridges C B. Science. 1936;83:210–211. doi: 10.1126/science.83.2148.210. [DOI] [PubMed] [Google Scholar]
- 25.Gondo Y, Gardner J M, Nakatsu Y, Durham-Pierre D, Deveau S A, Kuper C, Brilliant M H. Proc Natl Acad Sci USA. 1993;90:297–301. doi: 10.1073/pnas.90.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wahls W P, Wallace L J, Moore P D. Cell. 1990;60:95–103. doi: 10.1016/0092-8674(90)90719-u. [DOI] [PubMed] [Google Scholar]
- 27.Kaplanski C, Srivtanakul P, Wild C P. Int J Cancer. 1997;72:248–254. doi: 10.1002/(sici)1097-0215(19970717)72:2<248::aid-ijc8>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 28.Karsten S L, Lagerstedt K, Carlberg B M, Kleijer W J, Zaremba J, Van-Diggelen O P, Czartoryska B, Pettersson U, Bondeson M L. Genomics. 1997;43:123–129. doi: 10.1006/geno.1997.4811. [DOI] [PubMed] [Google Scholar]
- 29.Reiter L T, Murakami T, Koeuth T, Gibbs R A, Lupski J R. Hum Mol Genet. 1997;6:1595–1603. doi: 10.1093/hmg/6.9.1595. [DOI] [PubMed] [Google Scholar]
- 30.White P S, Maris J M, Beltinger C, Sulman-E, Marshall H N, Fujimori M, Kaufman B A, Biegel J A, Allen C, Hilliard C, et al. Proc Natl Acad Sci USA. 1995;92:5520–5524. doi: 10.1073/pnas.92.12.5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McAleer M A, Reifsnyder P, Palmer S M, Prochazka M, Love J M, Copeman J B, Powell E E, Rodrigues N R, Prins J B, Serreze D V, et al. Diabetes. 1995;44:1186–1195. doi: 10.2337/diab.44.10.1186. [DOI] [PubMed] [Google Scholar]
- 32.Potter M, Mushinski E B, Wax J S, Hartley J, Mock B A. Cancer Res. 1994;54:969–975. [PubMed] [Google Scholar]