

Summary
The ability to repair membrane damage is conserved across eukaryotic cells, and is necessary for the cells to survive a variety of physiological and pathological membrane disruptions. Membrane repair is mediated by rapid Ca2+-triggered exocytosis of various intracellular vesicles, such as lysosomes and enlargeosomes, which lead to the formation of a membrane patch that reseals the membrane lesion. Recent findings suggest a crucial role for dysferlin in this repair process in muscle, possibly as a Ca2+ sensor that triggers vesicle fusion. The importance of membrane repair is highlighted by the genetic disease, dysferlinopathy, in which the primary defect is the loss of Ca2+-regulated membrane repair due to dysferlin deficiency. Future research on dysferlin and its interacting partners will enhance the understanding of this important process, and provide novel avenues to potential therapies.
Keywords: C2 domains, cardiomyopathy, dysferlin, membrane fusion, membrane repair, muscular dystrophy
Introduction
The plasma membrane is a biological barrier between the extracellular and intracellular environments and is essential to the maintenance of cell integrity. Damage to this barrier leads to cell death; however, plasma membrane disruptions occur physiologically, and even frequently, in a variety of cells such as the milk-secreting epithelial cells of the mammary gland [1] and the myocytes of both skeletal [2] and cardiac muscle [3]. For more than half a century, it has been known that animal cells can survive after experimental perforation of their cell membranes (holes of >1000 μm2). It is now widely accepted that an active membrane repair mechanism is conserved in many types of mammalian cells, and that it serves to reseal membrane lesions. Rapid Ca2+-triggered endomembrane exocytosis is a crucial step in the membrane resealing process [4,5] that enables cells to survive routine membrane disruptions.
Muscular dystrophies are a heterogeneous group of progressive muscle wasting disorders of genetic origin. A large number of muscular dystrophy genes encode components of the dystrophin-glycoprotein complex (DGC) that are directly or indirectly involved in linking the cytoskeleton to the surrounding basement membrane. Disruption of this link renders the muscle membrane abnormally susceptible to contraction-induced injury, and the accumulation of muscle membrane damage ultimately leads to muscle necrosis and weakness. This highlights the importance of maintaining plasma membrane integrity for normal physiological function and long-term survival of muscle cells. Several additional forms of muscular dystrophy arise from defects unrelated to the DGC. For example, although dysferlin is not a DGC component [6], mutations in dysferlin gene are responsible for three clinically distinct muscular dystrophies: limb-girdle muscular dystrophy type 2B (LGMD2B), which is first characterized by proximal muscle weakness at onset [7]; Miyoshi myopathy (MM), a distal muscle disorder that preferentially affects the gastrocnemius muscle [7,8]; and a distal anterior compartment myopathy that is distinct from MM, and progresses rapidly through the anterior tibial muscles [9].
Recent work has shown that loss of dysferlin compromises Ca2+-dependent membrane repair in skeletal muscle [6,10]. Wild-type muscle fibers, in the presence of Ca2+, can efficiently exclude the membrane impermeable dye FM 1–43 after laser wounding, but the dye continuously fills the muscle fiber through the wounding site when Ca2+ is omitted in the bath solution (Figure 1), suggesting the wild-type muscle fiber possesses an efficient Ca2+-dependent membrane resealing mechanism [6]. Dysferlin-null muscle fibers fail to exclude the dye entry even in the presence of Ca2+ (Figure 1), strongly suggesting the Ca2+-dependent membrane repair requires dysferlin [6]. Loss of dysferlin-mediated membrane repair results in progressive myonecrosis [6,10]. In this review, we discuss recent advances in our understanding of dysferlin-mediated membrane repair in disease processes.
Figure 1. Defective Ca2+-dependent membrane repair in dysferlin-deficient skeletal muscle cells.
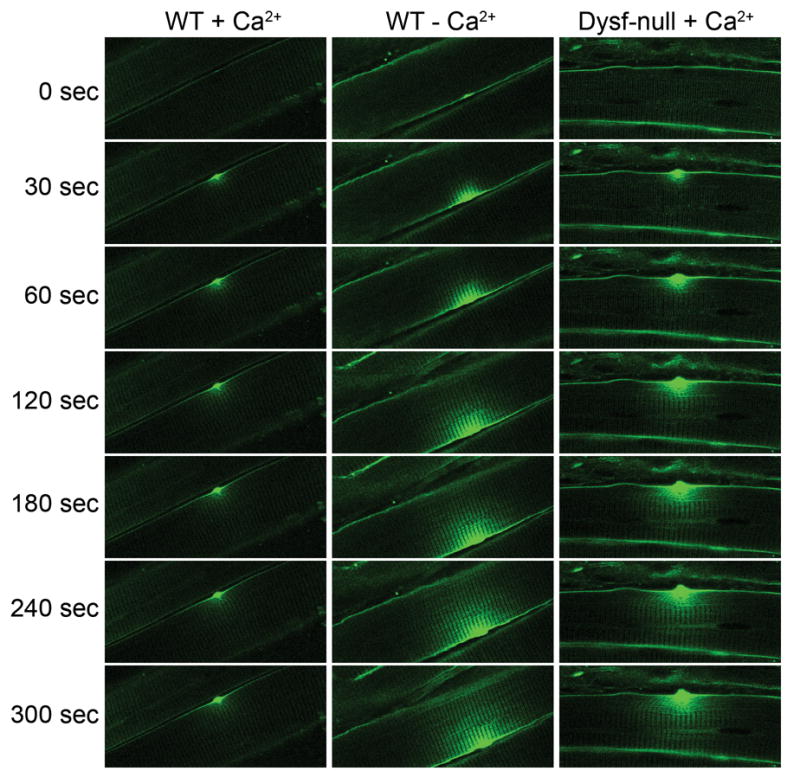
The sarcolemma of skeletal muscle fibers isolated from wild-type (WT) and dysferlin-deficient (Dysf-null) mice was wounded by full-power irradiation with a mode-locked infrared laser, in the presence of the membrane-impermeable dye FM 1–43. Fluorescence images were then taken every 10 seconds. In wild-type muscle fiber in the presence of Ca2+, dye influx ceased by 30–60 seconds after laser-induced disruption of the sarcolemma (left panel), suggesting that the site of damage had been resealed. In wild-type muscle in the absence of Ca2+, dye influx through the site of disruption continued over the entire time course of the experiment (middle panel), indicating a failure in the resealing mechanism. Likewise, in the dysferlin-null muscle fiber, dye filling occurred over the entire course of experiment (right panel), indicating that resealing failed, even in the presence of Ca2+.
Ferlin-1-like proteins
Soon after the initial identification of dysferlin as the product of a gene mutated in MM, a new family of mammalian proteins, named “ferlin-1 like proteins”, was predicted based on structural similarity and sequence homology (Figure 2). These include dysferlin or Fer1L1 [7,8]; otoferlin or Fer1L2 [11,12]; myoferlin or Fer1L3 [13,14]; Fer1L4; Fer1L5 and Fer1L6. Each protein contains multiple C2 domains, anchors to the membrane via a single carboxy-terminal transmembrane domain (Figure 2A), and shows sequence homology to the Caenorhabditis elegans ferlin-1 (FER-1) gene. FER-1 expression is restricted to primary spermatocytes, where it is essential for maturation of motile spermatozoa. In homozygous FER-1 mutants, spermatid membranous organelles (MO) fail to fuse with the plasma membrane during spermatogenesis, resulting in non-motile spermatozoa and infertility [15]. A single amino acid substitution in any of three FER-1 C2 domains is sufficient to disrupt MO fusion by altering the Ca2+ sensitivity of this protein [16]. A phylogenetic tree constructed from the alignment of individual mammalian ferlin C2 domain sequences shows that, in all ferlins, a given C2 domain is more similar to other C2 domains at similar positions in the other proteins than to C2 domains in different positions of the same protein [16] (Figure 2B). These data suggest that multiple C2 domains may be required for protein activity.
Figure 2. Structural characteristics of the ferlin-1-like proteins.

FER-1 and mammalian ferlin-1-like proteins are aligned, with their domain composition indicated (A). Most ferlins have five to seven C2 domains, and a single carboxyl transmembrane domain. FER-1, dysferlin (Dysf), myoferlin (Myof) and Fer1L5 have FerA and FerB domains, and nested DysfN and DysfC domains, which are not present in otoferlin (Otof) and other ferlins. The black dots on dysferlin domains indicate the missense mutations that lead to muscular dystrophy. A phylogenetic tree (B) was constructed based on the alignment of individual ferlin C2 domain sequences. The tree shows that the C2 domains at any similar positions of distinct proteins are more similar to each other than to those within the same protein.
Dysferlin is a 230 kDa protein that contains seven C2 domains. It is found in a variety of tissues including skeletal and cardiac muscle, kidney, placenta, lung, and brain, and it is most highly expressed in skeletal and cardiac muscle [7]. Interestingly, a single missense mutation in any of five dysferlin C2 domains (C2A, B, D, E and G) has been reported to cause muscular dystrophy [17] (Figure 2A), again suggesting non-redundancy among individual C2 domains. But mutations in these C2 domains may also lead to dysferlin misfolding and thus degradation [17,18]. The dysferlin C2A domain binds phospholipids in a Ca2+-dependent manner [19], consistent with its role in skeletal muscle membrane repair [6,10]. This novel function is also supported by ultrastructural observations of dysferlin-deficient skeletal muscle: subsarcolemmal regions are characterized by prominent aggregations of small vesicles of unknown origin [6,20,21]; the sarcolemma itself shows many gaps and microvilli-like projections rather than being continuous and smooth [6,21]; and the basal lamina is multilayered in some regions [21]. In addition, dysferlin deficiency delays myoblast fusion/maturation in vitro [22], suggesting that dysferlin may also participate in muscle differentiation/regeneration.
Myoferlin shares the highest sequence homology to dysferlin. It is present at the sarcolemma of skeletal muscle but, unlike dysferlin, it is also enriched in the nucleus [14]. Although both myoferlin and dysferlin are expressed in skeletal muscle, they seem to participate in distinct cellular events. In contrast to dysferlin, myoferlin functions in myoblast fusion during muscle differentiation/maturation and myoferlin-null mice show muscle atrophy [23]. Also, a lack of compensatory overexpression of myoferlin in muscles with dysferlinopathy [24], supports the view that myoferlin and dysferlin have few overlapping functions. So far, myoferlin has not been linked to any human disease.
Otoferlin is predominately expressed in the cochlea, vestibule and brain, although a low level of expression is seen in other tissues including lung, kidney, skeletal muscle and heart [12]. Mutations in otoferlin are responsible for non-syndromic deafness known as DFNB9 in humans [11,12]. Otoferlin was recently shown to be essential for a late step of synaptic vesicle exocytosis and may act as the major Ca2+ sensor triggering synaptic vesicle-plasma membrane fusion at the inner hair cell ribbon synapse [25].
The Fer1L4, Fer1L5 and Fer1L6 proteins are predicted from the human and mouse genomic sequences but have not yet been characterized. In light of the association of dysferlin and otoferlin with human diseases, it is reasonable to presume that these additional ferlin-1-like proteins may also be involved in human pathologies.
Mechanism of dysferlin-mediated membrane repair
Recent progress toward elucidating the mechanisms involved in Ca2+-dependent membrane repair has led to the proposal of two mechanisms: the lipid flow promotion hypothesis and the patch hypothesis.
According to the lipid flow promotion hypothesis, hydrophobic lipids at the free edge of the membrane disruption site are energetically unfavorable in the aqueous environment, creating “line tension”. The line tension may promote automatic lipid flow over this site to fuse to the complementary free membrane end, closing the disruption. The fact that the plasma membrane adheres to an underlying cortical cytoskeleton, which generates ‘membrane tension’ [26] opposing the ‘line tension’ (reviewed in [27]) argues against this. However, if the membrane disruption were capable of somehow reducing the ‘membrane tension’, membrane resealing through ‘line tension’-driven lipid flow would be a viable mechanism. Notably membrane disruption has been observed to cause a rapid Ca2+-dependent reduction in ‘membrane tension’ [25]. Consistent with the lipid flow hypothesis, treatment of damaged cells with surface active agents thought to reduce the ‘membrane tension’, also enhances resealing and cell survival [28,29]. With regard to the observed reduction in membrane tension in response to membrane disruption, this may result from depolymerization of the cortical cytoskeleton by Ca2+-activated calpain proteolytic activity (Figure 3), as supported by the finding that the typical μ- and m-calpains (also called calpain-1 and -2, respectively) are required for efficient membrane repair [30–32]. However, this membrane repair mechanism might only be applicable to relatively small disruptions (<1 μm diameter) [33].
Figure 3. Schematic model of dysferlin-mediated membrane repair.

In normal muscle fiber, dysferlin is localized to the sarcolemma and cytoplasmic vesicles, and interacts with annexins A1 and A2 (A). Membrane disruption causes calcium flooding into the muscle fiber and creates a zone of high calcium around the disruption site (B). The high calcium activates proteases such as calpain, which leads to the cleavage of cortical cytoskeleton proteins and thereby reduces membrane tension (C). The high calcium also triggers the aggregation of dysferlin-carrying repair vesicles likely involving annexins and the migration of these vesicles towards the disruption site, where they fuse with one another and with the plasma membrane in the presence of localized high levels of calcium (B, C, D). Dysferlin then triggers vesicle fusion with the plasma membrane, probably through SNAREs. Fusion of the repair vesicles with the plasma membrane results in a membrane patch being inserted across the site of membrane disruption, and thereby reseals the disrupted plasma membrane (E).
In the patch hypothesis for membrane repair, Ca2+ flooding through a membrane disruption is thought to evoke local vesicle-vesicle and vesicle-plasma membrane fusion events. As a result, a population of large vesicles accumulates underneath the disruption site, eventually creating a ‘patch’ of new membrane across the membrane gap via vesicle-vesicle and vesicle-membrane fusion (Figure 3). In support of this possibility, abnormally large vesicles rapidly accumulate, in a Ca2+-dependent manner, at sites of membrane damage in several systems: endothelial cells [5], sea urchin eggs [34] and nerve axons [35]. These findings indicate that Ca2+-triggered vesicle migration and vesicle-vesicle fusion events take place immediately following disruption of the plasma membrane.
With regard to the kinds of vesicles that might be involved in membrane repair by the patch process, accumulated evidence suggests that yolk granules [34], lysosomes [36], enlargeosome [37] or vesicles generated by endocytosis of axolemma [35] are utilized dependent on their abundance in different cells. Lysosomes are widely present and competent for Ca2+-dependent regulated exocytosis, and thus considered an ideal candidate for membrane repair. Indeed, inhibition of lysosome exocytosis has been reported to inhibit membrane resealing in NRK, CHO, L6E9, 3T3, and primary skin fibroblast cells [36], suggesting that Ca2+-regulated exocytosis of lysosomes is likely involved in membrane repair. However, whether lysosomes are dispensable for membrane repair is a matter of debate. One group reported that a small molecule vacuolin-1 blocked the Ca2+-dependent exocytosis of lysosomes induced by ionomycin or plasma membrane wounding, without affecting the process of resealing [38], while the inhibitory effect of vacoulin-1 on Ca2+-regulated exocytosis of lysosomes was not confirmed by the other group [39]. The reason for this discrepancy is not yet clear. The specific organelles providing membrane for resealing in skeletal muscle remain to be determined. Skeletal muscle undergoes frequent damage, thus requiring frequent membrane resealing. If lysosomes were the predominant donor membranes for resealing, lysosomal enzymes would be expected to accumulate in the extracellular space, and this would likely have deleterious effects on the muscle. It is conceivable that skeletal muscle may use whatever Ca2+-regulated exocytic vesicles available for resealing.
Membrane fusion requires several membrane proteins that could potentially be required for membrane resealing. These include SNARE proteins, a family of transmembrane proteins essential for most intracellular membrane fusion processes, and synaptotagmins (Syt), transmembrane proteins with two highly conserved C2 domains that serve as calcium sensors in the regulation of vesicle exocytosis in neurons and other cell types [39]. Several lines of evidence support that SNAREs are involved in membrane repair. Membrane resealing in both sea urchin eggs and fibroblasts is inhibited by injections of botulinum and tetanus toxins [40], which cleave various SNARE proteins and are known to inhibit Ca2+-mediated synaptic vesicle exocytosis. Also, antibodies to syntaxin-1 and Syt-1 inhibit resealing in crayfish giant axons [41], and antibody- or peptide-based inhibition of Syt-VII blocked Ca2+-dependent exocytosis of lysosomes in permeablized fibroblasts [42]. Moreover, Syt-VII has been implicated in the plasma membrane repair of skeletal muscle, since Syt-VII-deficient mice developed inflammatory myopathy with extensive fibrosis, high serum creatine kinase levels and progressive muscle weakness [43]. Furthermore, otoferlin has recently been shown to bind SNAP25, and syntaxin-1 to trigger synaptic vesicle exocytosis in inner hair cells, which do not express Syt-I [25]. Since dysferlin shares high homology to otoferlin, it is reasonable to hypothesize that dysferlin might also work through SNAREs to regulate vesicle fusion in membrane repair.
Recent work provides insights into the mechanism of synaptotagmin-potentiated membrane fusion. In response to Ca2+, the C2A and C2B domains of synaptotagmin-1 insert into the target membranes [44,45], induce high positive curvature of the membranes and thus lower the activation energy barrier for membrane fusion [46]. As mentioned earlier, dysferlin and other ferlin proteins also contain C2 domains and at least some of these C2 domains bind phospholipids in response to Ca2+. It is very intriguing to examine whether dysferlin triggers membrane fusion by the same mechanism.
Dysferlin interactors that may be involved in membrane repair
Recent studies of dysferlin interacting proteins have provided new mechanistic insight into dysferlin function. Dysferlin normally associates with annexins A1 and A2 in a Ca2+ and membrane injury-dependent manner [10]. In dysferlinopathy patients, expression of annexins A1 and A2 is elevated compared to controls, and annexin expression levels are significantly correlated with clinical severity scores [47]. These data suggest that annexins A1 and A2 play a role in dysferlin-mediated membrane resealing in skeletal muscle. Indeed, a requirement for annexin A1 in membrane repair was recently confirmed in HeLa cells [48]. Although the mechanism of annexin action in membrane repair is currently unclear, the findings that annexins (a) bind phospholipids in a Ca2+-dependent manner, (b) initiate vesicle aggregation in vitro, and (c) interact with the actin cytoskeleton [49] suggest an involvement in vesicle-vesicle fusion for ‘patch’ formation and vesicle movement (Figure 3). It would be interesting to examine model systems in which endogenous annexins A1 and A2 are deleted. One might expect that the loss of these proteins would lead to defective membrane repair in skeletal muscle and to muscular dystrophy, or that it would exacerbate the phenotype of dysferlin deficiency.
Caveolin-3 and calpain-3 are both muscle-specific proteins that are responsible for distinct forms of muscular dystrophy (LGMD1C and LGMD2A, respectively), and both have also been found to interact with dysferlin [50,51]. Whether these interactions are important in the membrane repair pathway remains unclear. However, patients deficient for caveolin-3 have been reported to exhibit a reduction or mis-localization of dysferlin in caveolinopathy patients [50,52], and patients deficient for dysferlin have reduced levels of calpain-3 [51]. Thus, these diseases may well involve overlapping or associated pathogenic mechanisms.
Dysferlin also binds to AHNAK [53], a previously reported marker of enlargeosomes [37]. Again, it is not known whether the association of AHNAK and dysferlin is relevant for membrane repair. In fact, both proteins redistribute to the cytosol during skeletal muscle regeneration [53], suggesting that they may act together in membrane fusion events that are necessary during regeneration rather than in events crucial to membrane repair.
Affixin, an integrin-linked kinase focal adhesion protein, has also been reported to localize to the sarcolemma and to co-immunoprecipitate with dysferlin [54]. Affixin immunoreactivity is reduced at the sarcolemma of MM and LGMD2B muscles, and also in other muscle diseases including LGMD1C. In all these cases, affixin and dysferlin show quite similar changes in their expression patterns, including a reduction in sarcolemmal staining (with or without cytoplasmic accumulations, depending on the specific disease forms) [54]. Given what is known about affixin, such an interaction could potentially coordinate cytoskeletal reorganization required for efficient vesicle trafficking during the resealing process. However, such a role has not been established, nor has the functional significance of this interaction been addressed.
Defective membrane repair and disease
As described above, defective dysferlin-mediated membrane repair is responsible for three clinically distinct forms of muscular dystrophy in humans. The cause of the observed disease heterogeneity remains unclear. Interestingly, the same mutations in dysferlin gene have been found to cause different disease manifestation even within the same family [55,56], suggesting that other genetic factors may affect the clinical symptoms in the patients. But these distinct symptoms share overlapping features: age of onset, slow disease progression and early, marked elevation of serum CK.
Recent work has shown that dysferlin deficiency greatly reduces the membrane repair efficiency of cardiac muscle, with the aged dysferlin-null mice manifesting hallmarks of cardiomyopathy, e.g. elevated serum cardiac troponin T levels, cardiac necrosis and cardiac fibrosis [57]. Although echocardiography recordings from ~1-year-old dysferlin-null mice failed to detect abnormalities in cardiac physiology, mechanical stress disturbed ventricular function sufficiently to unmask the cardiac phenotype of these mice [57]. The importance of maintaining an efficient dysferlin-mediated membrane repair system for cardiac muscle is further supported by the fact that dysferlin deficiency greatly accelerates the progression of cardiomyopathy in mdx mice (which carry a point mutation in the dystrophin gene), where skeletal and cardiac muscle are highly susceptible to membrane damage [57]. Although cardiomyopathy is not commonly reported in human dysferlinopathy patients, a 57-year-old woman with dysferlin deficiency presented with cardiomyopathy after more than 20 years of progressive muscle wasting [58], suggesting that dysferlinopathy patients are indeed prone to develop cardiomyopathy, but with a late onset. A systematic examination of cardiac involvement in dysferlinopathy patients has been initiated, and an early report has shown that a significant proportion (9 out of 11) of these patients (ages 19 to 48) have elevated serum cardiac troponin T levels (S Yilmazer et al., abstract in Neuromuscul Disord 2006, 16:S110). Similarly, dysferlin-null mice show an increase in serum cardiac troponin T levels at early ages (eg. 30 weeks old) although they do not present clear cardiac muscle abnormality, by either histological or echocardiography analyses. Further examination of cardiomyopathy in older dysferlin-deficient patients is therefore necessary.
Conclusions
Membrane resealing is an emergency response that is highly conserved among different species and cell types. It is mediated by rapid Ca2+-triggered exocytosis of intracellular vesicles in animal cells, which form a patch at the disruption site (Figure 3). Skeletal and cardiac muscles are mechanically active tissues that are often subjected to injury; thus, they require a robust membrane resealing mechanism, and may therefore have developed a specialized mechanism for this purpose. Dysferlin plays a critical role in the membrane repair of both skeletal and cardiac myocytes. The dysferlin-mediated resealing response in muscle likely involves the SNARE family of proteins and may also require the participation of other Ca2+-activated proteins such as calpains and annexins (Figure 3). At this time, it is not clear specifically which SNARE proteins might be involved. Although both Syt-VII-regulated membrane repair and dysferlin-mediated membrane repair seem to be active in skeletal muscle, it remains unclear whether they regulate fusion of the same vesicle pools in the same step or different steps or different vesicle pools. Future experiments addressing these questions will significantly advance our understanding of the detailed mechanism of muscle membrane repair.
Acknowledgments
We thank Drs. Steven A. Moore, Aaron Beedle, Daniel Beltran, Yvonne Kobayashi, Rolf Turk and other Campbell lab members for critical reading and comments. Research in the authors’ laboratory was supported in part by a Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant 1 U54 NS053672, the Muscular Dystrophy Association, and U.S. Department of Defense Grants W81XWH-05-1-0079. K.P.C. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aumuller G, Wilhelm B, Seitz J. Apocrine secretion--fact or artifact? Ann Anat. 1999;181:437–446. doi: 10.1016/S0940-9602(99)80020-X. [DOI] [PubMed] [Google Scholar]
- 2.McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am J Pathol. 1992;140:1097–1109. [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke MS, Caldwell RW, Chiao H, Miyake K, McNeil PL. Contraction-induced cell wounding and release of fibroblast growth factor in heart. Circ Res. 1995;76:927–934. doi: 10.1161/01.res.76.6.927. [DOI] [PubMed] [Google Scholar]
- 4.Bi GQ, Alderton JM, Steinhardt RA. Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol. 1995;131:1747–1758. doi: 10.1083/jcb.131.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyake K, McNeil PL. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol. 1995;131:1737–1745. doi: 10.1083/jcb.131.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **6.Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. Using an elegant membrane repair assay, the authors demonstrate that skeletal muscle possesses Ca2+-regulated membrane repair and establish dysferlin as the first component of membrane repair machinery in skeletal muscle.
- 7.Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20:37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- 8.Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 9.Illa I, Serrano-Munuera C, Gallardo E, Lasa A, Rojas-Garcia R, Palmer J, Gallano P, Baiget M, Matsuda C, Brown RH. Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol. 2001;49:130–134. [PubMed] [Google Scholar]
- *10.Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH., Jr Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003;278:50466–50473. doi: 10.1074/jbc.M307247200. The authors show that dysferlin interacts with annexins A1 and A2, and is required for muscle membrane repair following membrane injury by Ca2+ imaging.
- 11.Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21:363–369. doi: 10.1038/7693. [DOI] [PubMed] [Google Scholar]
- 12.Yasunaga S, Grati M, Chardenoux S, Smith TN, Friedman TB, Lalwani AK, Wilcox ER, Petit C. OTOF encodes multiple long and short isoforms: genetic evidence that the long ones underlie recessive deafness DFNB9. Am J Hum Genet. 2000;67:591–600. doi: 10.1086/303049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Britton S, Freeman T, Vafiadaki E, Keers S, Harrison R, Bushby K, Bashir R. The third human FER-1-like protein is highly similar to dysferlin. Genomics. 2000;68:313–321. doi: 10.1006/geno.2000.6290. [DOI] [PubMed] [Google Scholar]
- 14.Davis DB, Delmonte AJ, Ly CT, McNally EM. Myoferlin, a candidate gene and potential modifier of muscular dystrophy. Hum Mol Genet. 2000;9:217–226. doi: 10.1093/hmg/9.2.217. [DOI] [PubMed] [Google Scholar]
- 15.Achanzar WE, Ward S. A nematode gene required for sperm vesicle fusion. J Cell Sci. 1997;110(Pt 9):1073–1081. doi: 10.1242/jcs.110.9.1073. [DOI] [PubMed] [Google Scholar]
- 16.Washington NL, Ward S. FER-1 regulates Ca2+-mediated membrane fusion during C. elegans spermatogenesis. J Cell Sci. 2006;119:2552–2562. doi: 10.1242/jcs.02980. [DOI] [PubMed] [Google Scholar]
- 17.Therrien C, Dodig D, Karpati G, Sinnreich M. Mutation impact on dysferlin inferred from database analysis and computer-based structural predictions. J Neurol Sci. 2006;250:71–78. doi: 10.1016/j.jns.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16:618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 19.Davis DB, Doherty KR, Delmonte AJ, McNally EM. Calcium-sensitive phospholipid binding properties of normal and mutant ferlin C2 domains. J Biol Chem. 2002;277:22883–22888. doi: 10.1074/jbc.M201858200. [DOI] [PubMed] [Google Scholar]
- 20.Selcen D, Stilling G, Engel AG. The earliest pathologic alterations in dysferlinopathy. Neurology. 2001;56:1472–1481. doi: 10.1212/wnl.56.11.1472. [DOI] [PubMed] [Google Scholar]
- 21.Cenacchi G, Fanin M, De Giorgi LB, Angelini C. Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J Clin Pathol. 2005;58:190–195. doi: 10.1136/jcp.2004.018978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Luna N, Gallardo E, Soriano M, Dominguez-Perles R, de la Torre C, Rojas-Garcia R, Garcia-Verdugo JM, Illa I. Absence of dysferlin alters myogenin expression and delays human muscle differentiation “in vitro”. J Biol Chem. 2006;281:17092–17098. doi: 10.1074/jbc.M601885200. [DOI] [PubMed] [Google Scholar]
- 23.Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, Hadhazy M, McNally EM. Normal myoblast fusion requires myoferlin. Development. 2005;132:5565–5575. doi: 10.1242/dev.02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inoue M, Wakayama Y, Kojima H, Shibuya S, Jimi T, Oniki H, Nishino I, Nonaka I. Expression of myoferlin in skeletal muscles of patients with dysferlinopathy. Tohoku J Exp Med. 2006;209:109–116. doi: 10.1620/tjem.209.109. [DOI] [PubMed] [Google Scholar]
- **25.Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040.This work demonstrates that otoferlin interacts with SNARE proteins in a Ca2+-dependent manner at the afferent synapses of cochlear inner hair cells to trigger exocytosis of neurotransmitter. The authors also show that the deafness in otoferlin-deficient mice is caused by an almost complete loss of calcium-mediated exocytosis in the inner hair cells but without disruption to the synaptic structure or calcium ion influx.
- 26.Raucher D, Sheetz MP. Characteristics of a membrane reservoir buffering membrane tension. Biophys J. 1999;77:1992–2002. doi: 10.1016/S0006-3495(99)77040-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chernomordik LV, Melikyan GB, Chizmadzhev YA. Biomembrane fusion: a new concept derived from model studies using two interacting planar lipid bilayers. Biochim Biophys Acta. 1987;906:309–352. doi: 10.1016/0304-4157(87)90016-5. [DOI] [PubMed] [Google Scholar]
- 28.Togo T, Alderton JM, Bi GQ, Steinhardt RA. The mechanism of facilitated cell membrane resealing. J Cell Sci. 1999;112(Pt 5):719–731. doi: 10.1242/jcs.112.5.719. [DOI] [PubMed] [Google Scholar]
- 29.Togo T, Krasieva TB, Steinhardt RA. A decrease in membrane tension precedes successful cell-membrane repair. Mol Biol Cell. 2000;11:4339–4346. doi: 10.1091/mbc.11.12.4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie XY, Barrett JN. Membrane resealing in cultured rat septal neurons after neurite transection: evidence for enhancement by Ca(2+)-triggered protease activity and cytoskeletal disassembly. J Neurosci. 1991;11:3257–3267. doi: 10.1523/JNEUROSCI.11-10-03257.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *31.Mellgren RL, Zhang W, Miyake K, McNeil PL. Calpain is required for the rapid, calcium-dependent repair of wounded plasma membrane. J Biol Chem. 2007;282:2567–2575. doi: 10.1074/jbc.M604560200. Using calpain-deficient fibroblasts, the authors demonstrate that calpain is required for Ca2+-regulated membrane repair, possibly through disruption of cortical cytoskeleton
- 32.Howard MJ, David G, Barrett JN. Resealing of transected myelinated mammalian axons in vivo: evidence for involvement of calpain. Neuroscience. 1999;93:807–815. doi: 10.1016/s0306-4522(99)00195-5. [DOI] [PubMed] [Google Scholar]
- 33.McNeil PL, Steinhardt RA. Plasma membrane disruption: repair, prevention, adaptation. Annu Rev Cell Dev Biol. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. [DOI] [PubMed] [Google Scholar]
- 34.McNeil PL, Vogel SS, Miyake K, Terasaki M. Patching plasma membrane disruptions with cytoplasmic membrane. J Cell Sci. 2000;113(Pt 11):1891–1902. doi: 10.1242/jcs.113.11.1891. [DOI] [PubMed] [Google Scholar]
- 35.Eddleman CS, Ballinger ML, Smyers ME, Fishman HM, Bittner GD. Endocytotic formation of vesicles and other membranous structures induced by Ca2+ and axolemmal injury. J Neurosci. 1998;18:4029–4041. doi: 10.1523/JNEUROSCI.18-11-04029.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106:157–169. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 37.Cocucci E, Racchetti G, Podini P, Rupnik M, Meldolesi J. Enlargeosome, an exocytic vesicle resistant to nonionic detergents, undergoes endocytosis via a nonacidic route. Mol Biol Cell. 2004;15:5356–5368. doi: 10.1091/mbc.E04-07-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerny J, Feng Y, Yu A, Miyake K, Borgonovo B, Klumperman J, Meldolesi J, McNeil PL, Kirchhausen T. The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep. 2004;5:883–888. doi: 10.1038/sj.embor.7400243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sudhof TC, Rizo J. Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron. 1996;17:379–388. doi: 10.1016/s0896-6273(00)80171-3. [DOI] [PubMed] [Google Scholar]
- 40.Steinhardt RA, Bi G, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- 41.Detrait E, Eddleman CS, Yoo S, Fukuda M, Nguyen MP, Bittner GD, Fishman HM. Axolemmal repair requires proteins that mediate synaptic vesicle fusion. J Neurobiol. 2000;44:382–391. doi: 10.1002/1097-4695(20000915)44:4<382::aid-neu2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 42.Martinez I, Chakrabarti S, Hellevik T, Morehead J, Fowler K, Andrews NW. Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol. 2000;148:1141–1149. doi: 10.1083/jcb.148.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS, Andrews NW. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol. 2003;162:543–549. doi: 10.1083/jcb.200305131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hui E, Bai J, Chapman ER. Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys J. 2006;91:1767–1777. doi: 10.1529/biophysj.105.080325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrick DZ, Sterbling S, Rasch KA, Hinderliter A, Cafiso DS. Position of synaptotagmin I at the membrane interface: cooperative interactions of tandem C2 domains. Biochemistry. 2006;45:9668–9674. doi: 10.1021/bi060874j. [DOI] [PubMed] [Google Scholar]
- **46.Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. The authors found that, in response to Ca2+ binding, synaptotagmin-1 induces high positive curvature in target membranes upon C2-domain membrane insertion and thus lowers the activation energy barrier of SNARE-mediated membrane fusion
- 47.Cagliani R, Magri F, Toscano A, Merlini L, Fortunato F, Lamperti C, Rodolico C, Prelle A, Sironi M, Aguennouz M, et al. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum Mutat. 2005;26:283. doi: 10.1002/humu.9364. [DOI] [PubMed] [Google Scholar]
- *48.McNeil AK, Rescher U, Gerke V, McNeil PL. Requirement for annexin A1 in plasma membrane repair. J Biol Chem. 2006;281:35202–35207. doi: 10.1074/jbc.M606406200. This study for the first time shows directly that annexin A1 is required for efficient membrane repair
- 49.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 50.Matsuda C, Hayashi YK, Ogawa M, Aoki M, Murayama K, Nishino I, Nonaka I, Arahata K, Brown RH., Jr The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum Mol Genet. 2001;10:1761–1766. doi: 10.1093/hmg/10.17.1761. [DOI] [PubMed] [Google Scholar]
- 51.Anderson LV, Harrison RM, Pogue R, Vafiadaki E, Pollitt C, Davison K, Moss JA, Keers S, Pyle A, Shaw PJ, et al. Secondary reduction in calpain 3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy (primary dysferlinopathies) Neuromuscul Disord. 2000;10:553–559. doi: 10.1016/s0960-8966(00)00143-7. [DOI] [PubMed] [Google Scholar]
- 52.Capanni C, Sabatelli P, Mattioli E, Ognibene A, Columbaro M, Lattanzi G, Merlini L, Minetti C, Maraldi NM, Squarzoni S. Dysferlin in a hyperCKaemic patient with caveolin 3 mutation and in C2C12 cells after p38 MAP kinase inhibition. Exp Mol Med. 2003;35:538–544. doi: 10.1038/emm.2003.70. [DOI] [PubMed] [Google Scholar]
- 53.Huang Y, Laval SH, van Remoortere A, Baudier J, Benaud C, Anderson LV, Straub V, Deelder A, Frants RR, den Dunnen JT, et al. AHNAK, a novel component of the dysferlin protein complex, redistributes to the cytoplasm with dysferlin during skeletal muscle regeneration. Faseb J. 2007;21:732–742. doi: 10.1096/fj.06-6628com. [DOI] [PubMed] [Google Scholar]
- 54.Matsuda C, Kameyama K, Tagawa K, Ogawa M, Suzuki A, Yamaji S, Okamoto H, Nishino I, Hayashi YK. Dysferlin interacts with affixin (beta-parvin) at the sarcolemma. J Neuropathol Exp Neurol. 2005;64:334–340. doi: 10.1093/jnen/64.4.334. [DOI] [PubMed] [Google Scholar]
- 55.Weiler T, Bashir R, Anderson LV, Davison K, Moss JA, Britton S, Nylen E, Keers S, Vafiadaki E, Greenberg CR, et al. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s) Hum Mol Genet. 1999;8:871–877. doi: 10.1093/hmg/8.5.871. [DOI] [PubMed] [Google Scholar]
- 56.Illarioshkin SN, Ivanova-Smolenskaya IA, Greenberg CR, Nylen E, Sukhorukov VS, Poleshchuk VV, Markova ED, Wrogemann K. Identical dysferlin mutation in limb-girdle muscular dystrophy type 2B and distal myopathy. Neurology. Dec 26;55(12):1931–3. doi: 10.1212/wnl.55.12.1931. [DOI] [PubMed] [Google Scholar]
- **57.Han R, Bansal D, Miyake K, Muniz VP, Weiss RM, McNeil PL, Campbell KP. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J Clin Invest. 2007;117(7):1805–1813. doi: 10.1172/JCI30848. Using a modified membrane repair assay, the authors demonstrate that cardiac muscle also possesses Ca2+-regulated membrane repair, and dysferlin is important for this process. Defective dysferlin-mediated membrane repair renders the heart prone to stress-induced ventricular injury and accelerates the progression of cardiomyopathy in mdx mice
- 58.Kuru S, Yasuma F, Wakayama T, Kimura S, Konagaya M, Aoki M, Tanabe M, Takahashi T. [A patient with limb girdle muscular dystrophy type 2B (LGMD2B) manifesting cardiomyopathy] Rinsho Shinkeigaku. 2004;44:375–378. [PubMed] [Google Scholar]