

Abstract
Stargardt disease-3 (STGD3) is a juvenile dominant macular degeneration caused by mutations in elongase of very long chain fatty acid-4. All identified mutations produce a truncated protein which lacks a motif for protein retention in endoplasmic reticulum, the site of fatty acid synthesis. In these studies of Stgd3-knockin mice carrying a human pathogenic mutation, we examined two potential pathogenic mechanisms: truncated protein-induced cellular stress and lipid product deficiency. Analysis of mutant retinas detected no cellular stress but demonstrated selective deficiency of C32-C36 acyl phosphatidylcholines. We conclude that this deficit leads to the human STGD3 pathology.
Keywords: Stargardt disease-3, Retina, Very long chain fatty acid, Phosphatidylcholine, Unfolded-protein response, Mass spectrometry
1. Introduction
Macular degenerations are a heterogenous group of retinal disorders that are a major cause of blindness in developed countries. The most common pathology is age-related macular degeneration (AMD) which affects approximately 30% of people over the age of 75. Besides this late form, there are early-onset hereditary macular pathologies. It is assumed that understanding their pathogenesis will help design therapies not only for the juvenile pathologies but also for AMD.
Stargardt disease-3 (STGD3) is a juvenile dominant macular dystrophy caused by three independent mutations in the gene which codes for elongase of very long chain fatty acids-4 (ELOVL4) [1-4]. All mutated alleles produce a truncated ELOVL4 protein that lacks a C-terminal di-lysine motif for endoplasmic reticulum (ER) retention. Such protein truncation favors two potential pathogenic mechanisms: (a) deficiency of ELOVL4 lipid products subsequent to reduced ELOVL4 protein levels in the ER, the site of long chain fatty acid synthesis, and (b) cellular stress induced by the misplaced truncated ELOVL4 protein. Experimental data supporting both mechanisms has been obtained from studies of cultured cells which express an ELOVL4 transgene containing a human STGD3-pathogenic 5-bp deletion. Contrary to wt ELOVL4, the mutated protein was shown to leave the ER [5,6] and to upregulate the cellular levels of BiP (also known as GRP78) and CHOP (also known as GADD153) [7], two ER chaperon proteins known to be increased during an unfolded-protein response.
Recently, we generated an animal model of STGD3, the Stgd3-knockin mice that carry the human pathogenic 5-bp deletion in the mouse Elovl4 gene [8]. We have shown that Stgd3-heterozygous mice reproduce early retinal features of the human STGD3 pathology: lipofuscin accumulation and reduced visual functions [9]. Additionally, Stgd3-homozygous mice show complete absence of epidermal acylceramides but normal levels of non-acylceramides [8], a finding replicated in several other homozygous Elovl4-mutant mice [10-12]. Unique to acylceramides are extremely long chain C28-C36 fatty acids. This suggests that (a) Elovl4 synthesizes C28-C36 fatty acids and (b) the STGD3 mutations abolish their synthesis and synthesis of lipids which contain C28-C36 acyl residues.
Retinas have no acylceramides but they do have a unique group of phosphatidylcholines (PCs) which contain residues of polyunsaturated C28-C36 fatty acids [13,14]. Therefore, we hypothesize that the STGD3 mutations reduce the levels of these PCs in the mouse retina. Such a deficit may play an important role in the STGD3 pathogenesis since C28-C36 acyl PCs have been shown to (a) be the only lipid group of photoreceptor outer segments which contains C28-C36 acyl residues [13,14], (b) bind strongly to rhodopsin [13], and (c) regulate activities of photo-transduction proteins [15]. In this study of mouse retina, we demonstrate not only the presence of C32-C36 acyl PC deficiency but also the absence of an unfolded-protein response in the retinas of Stgd3-heterozygous mice.
2. Materials and methods
2.1. Mice
Stgd3 mice were generated, housed and cared for in accordance with institutional guidelines as described previously [8].
2.2. Analysis of retinal lipids by mass spectrometry
A pair of mouse retinas was extracted at room temperature for 1 hr using 0.3 ml of a 1:1 (v:v) mixture of chloroform:methanol. Particulates were removed by centrifugation, and the extracted lipids were analyzed by electrospray mass spectrometry (MS) as described in the Supplementary Data. For each analyzed lipid sample, a total of five consecutive MS sample runs were recorded, and their mean value was used for quantitative analysis.
2.3. Western blot analysis of BiP protein
Dissected mouse retinas were homogenized and extracted proteins resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis. Proteins were transferred to Immobilon-P membranes (Millipore, Bedford, MA, USA). The membranes were first incubated overnight with phosphate buffered saline containing 5% non-fat milk powder and 0.05% Tween 20, and then for 3 hrs with rabbit anti-Grp78 (Bip) antibody (Stressgen, MI, USA), washed and incubated for 1 hr with peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Jackson Immunoresearch, West Grove, PA, USA). Washed blots were developed with the ECL plus Western Blotting Detection System (Amersham Biosciences, Piscataway, NJ, USA) and exposed to Hyperfilm ECL (Amersham Biosciences).
2.4. RT-PCR analysis of XBP1 mRNA
Total RNA was isolated from mouse tissue samples using RNA Stat-60 (Tel-Test, Friendswood, TX, USA). RT-PCR analysis of XBP1 mRNAs was performed using the Superscript III Synthesis System (Invitrogen, Carlsbad, CA, USA). The RT reaction was primed with the XBP1 antisense primer 5’-AGTTCCTCCAGACTAGCAGACT-3’ which was subsequently also used to amplify XBP1 sequences. Sense PCR primers were as follows: 5’-CTGGAGCAGCAAGTGGTGGA-3’ (primer set #1) and 5’-GAGAACCACAAACTCCAGCTAGA-3’ (set #2). Both primer sets were designed to amplify sequence encompassing a stress-induced alternatively spliced 29-N sequence of mouse XBP1 mRNA.
2.5. Nuclease protection assay for Elovl4 mRNAs
Total RNA was extracted and mRNAs were analyzed as described previously [8].
2.6. Statistical analysis
Data were analyzed for statistical significance by a two-tailed, paired Student’s t test.
3. Results and discussion
3.1. Identification of C32-C36 acyl phosphatidylcholines in mouse retinal extracts
To test the hypothesis that the STGD3 mutation reduces retinal levels of C28-C36 acyl PCs, we performed analysis of mouse retinal lipids using positive ion mode MS. In this mode, PC species are easily ionized and analyzed due to the presence of a positively charged quaternary amine [16]. Since Stgd3-homozygous mice die within hours of birth, at a time when their retinas are not developed, the current studies were limited to analysis of Stgd3-heterozygous mice which display retinal morphology and a lifespan comparable to that of wt littermates [8].
MS analysis of retinal lipids generated more than twenty mass peaks of mass-to-charge ratio (m/z) between 700 and 1100 (Fig. 1). The same major mass peaks are observed in the retinal samples from wt and Stgd3-heterozygous mice. Based on m/z value and monoisotopic mass, fourteen peaks can be assigned to known PC species shown either in Fig. 1 or as follows: 732.6 Da, [PC32:1 + H]+; 758.6 Da, [PC34:2 + H]+; 782.6 Da, [PC34:0 + H]+; 786.6 Da, [PC36:2 + H]+; 788.6 Da, [PC36:1 + H]+; and 810.6 Da, [PC38:4 + H]+.
Fig. 1.

MS analysis of retinal lipids in 5 mM ammonium acetate from (A) a 1-month-old wt and (B) Stgd3-heterozygous mouse.
In order to confirm the assignments of PC 34:1, PC 44:12, PC 54:12, PC 56:12 and PC 58:12, their mass peaks were further analyzed by tandem MS. When subjected to MS fragmentation, all five PC species produced only one major fragment peak (m/z 184.0) (Supplementary Data) which corresponds to positively charged phosphocholine (monoisotopic mass 184.07 Da).
To gain more detailed information on the PC structures, we further analyzed retinal lipids in the presence of LiCl salt. This analysis detected the presence of Li-adducts for all PC species (Supplementary Data) seen previously as the protonated PCs (Fig.1). Five individual [PC + Li]+ peaks from the MS spectra were subsequently subjected to tandem MS analysis. Li-adducts of PCs are more extensively fragmented than are the protonated PCs, thus yielding more pertinent structural information [16,17]. Comparison of the acquired m/z values with the calculated monoisotopic mass of the expected fragmentation products (Supplementary Data) allowed for a final identification of the analyzed lipids as PC species that contain residues of either: (a) relatively short chain fatty acids (PC 16:0/18:1), or (b) two molecules of docosahexaenoic acid (DHA) (PC 22:6/22:6), or (c) one C32-C36 fatty acid and one DHA molecule (PC 32:6/22:6, PC 34:6/22:6 and PC 36:6/22:6).
In bovine photoreceptor outer segments, the unique C28-C36 acyl PC species is the only lipid group that contains residues of such extremely long chain fatty acids [13,14]. Majority of these fatty acids belong to n-3 series with the dominant acyl chain length of C30-C34 [13]. All identified retinal C28-C36 fatty acids are polyunsaturated, mostly with six double bonds, less often with five or four. On the contrary, C28-C36 acyl chains of epidermal acylceramides are either saturated or monounsaturated [8,10-12]. These findings show that (a) Elovl4 elongates fatty acids of all levels of saturation and (b) a tissue type and its lipid metabolizing proteins determine which fatty acids are available as substrates for Elovl4-mediated elongation.
3.2. C32-C36 acyl phosphatidylcholines are deficient in the retinas of Stgd3-heterozygous mice
When the MS spectra of retinal lipids from Stgd3-heterozygous mice are compared with the spectra of their wt littermates, a majority of the corresponding peaks are similar (Fig. 1). The only peaks that have their heights reduced in the mutant mouse samples belong to C32-C36 acyl PCs. In order to quantify the decrease, the peak heights of C32-C36 acyl PCs were normalized to the height of the PC 22:6/22:6 peak from the same spectrum. Therefore, PC 22:6/22:6 served as an internal standard with its peak height assigned an arbitrary unit of 1. Using this approach, we found that the retinal level of PC 32:6/22:6 was 0.305 ± 0.009 (mean ± S.D., n = 5 animals) in wt and 0.212 ± 0.007 (n = 5) in Stgd3-heterozygous mice; the PC 34:6/22:6 level was 0.385 ± 0.039 in wt and 0.158 ± 0.012 in mutant mice; and the PC 36:6/22:6 level was 0.101 ± 0.029 in wt and 0.034 ± 0.004 in mutant mice.
In the mutant retinas the levels of PC 32:6/22:6, PC 34:6/22:6 and PC 36:6/22:6 were, therefore, significantly reduced (P < 0.001) down to 69%, 41% and 34% of the wt levels, respectively. The loss of PC with the longer acyl chain length (C34:6 or C36:6) was significantly greater (P < 0.001) than the loss of PC containing the shorter C32:6 acyl chain, suggesting that the length of a fatty acid product is determined by the Elovl4 activity. A similar quantitative outcome was achieved when comparative analysis of the C32-C36 acyl PC peaks was performed using mass peaks for lipids other than PC 22:6/22:6 as internal standards.
Retinal PCs were analyzed using positive ion mode which is less suitable for analysis of other phospholipid species since they, when ionized, preferentially form negatively charged ions. Therefore, analysis of mouse retinal lipids was also performed using negative mode MS. As previously reported by others [13,14], residues of C28-C36 fatty acids were not detected in phosphatidylethanolamines, phosphatidylserines or phosphatidylinositols. Additionally, our preliminary quantitative analysis of these phospholipids found no significant differences between the wt and mutant retinal lipid samples. These findings collaborate with the results of previous epidermal lipid studies [8,10-12]. They showed that the Elovl4 mutations reduce the levels of lipids that contain C28-C36 acyl residues (acylceramides), but not the levels of lipids that lack these acyl groups (non-acylceramides).
In summary, the MS analysis of lipids demonstrates that the STGD3 mutation causes selective deficiency of C32-C36 acyl PCs in mouse retinas. Two previous studies, which analyzed DHA in different STGD3-knockin mouse models, reported no changes in retinal DHA levels [18,19]. This shows that the role of exteremely long chain C28-C36 fatty acids in retinal physiology is independent of that of DHA.
3.3. An unfolded-protein response is not detectable in Stgd3 mice
Pathogenesis of many genetic diseases is associated with defective folding of a mutant protein which induces an unfolded-protein response [20]. This protective compensatory mechanism is characterized by (a) a selective increase in the expression of a unique group of stress-response proteins, (b) an overall decrease in the expression of other genes, (c) an enhanced degradation of cellular proteins, and (d) an induction of apoptosis if the overload of unfolded proteins is not resolved.
Here, we present data that are consistent with the conclusion that an unfolded-protein response is not induced in Stgd3 mice. First of all, the retinal levels of BiP protein, a master regulator of the cellular stress response [20,21], are not increased in Stgd3-heterozygous mice relative to their wt littermates (Fig. 2A). Secondly, neither spliced nor unspliced form of XBP1 mRNA was detected by RT-PCR analysis in the retinas of wt and Stgd3-heterozygous mice (Fig. 2B). In the presence of ER stress, XBP1 is upregulated and its mRNA is alternatively spliced, generating a short form of XBP1 mRNA which codes for a potent transcriptional activator for many genes of the unfolded-protein response [20]. XBP1 mRNA was detected in the epidermis of both neonatal Stgd3-homozygous and wt mice but only as the unspliced XBP1 mRNA (Fig. 2B). The latter findings argue against induction of an unfolded-protein response in tissues of Stgd3 mice, especially given that the Stgd3-homozygous mice carry the mutation in both Elovl4 alleles and express high levels of Stgd3 mRNA in the epidermis [8].
Fig. 2.

Absence of an unfolded-protein response in Stgd3 mice. (A) Western blot analysis of BiP protein in mouse retinas of wt and Stgd3-heterozygous mice. (B) RT-PCR analysis shows absence of PCR products derived from the stress-induced, alternatively spliced form of XBP1 mRNA (379 bp and 349 bp for the primer sets #1 and 2, respectively) in the retinas of 3-month-old Stgd3-heterozygous mice and in the skin of neonatal Stgd3-homozygous mice. A PCR product derived from the unspliced form of XBP1 mRNA (405 bp and 375 bp for the primer sets #1 and 2, respectively) was detected in the skin but not in the retina.
No decrease in the levels of Elovl4 mRNA expression in Stgd3-heterozygous retinas (Fig. 3) further supports lack of an unfolded protein response. The absence of photoreceptor death in Stgd3-heterozygous retinas [8] is also consistent with the absence of cellular stress.
Fig. 3.
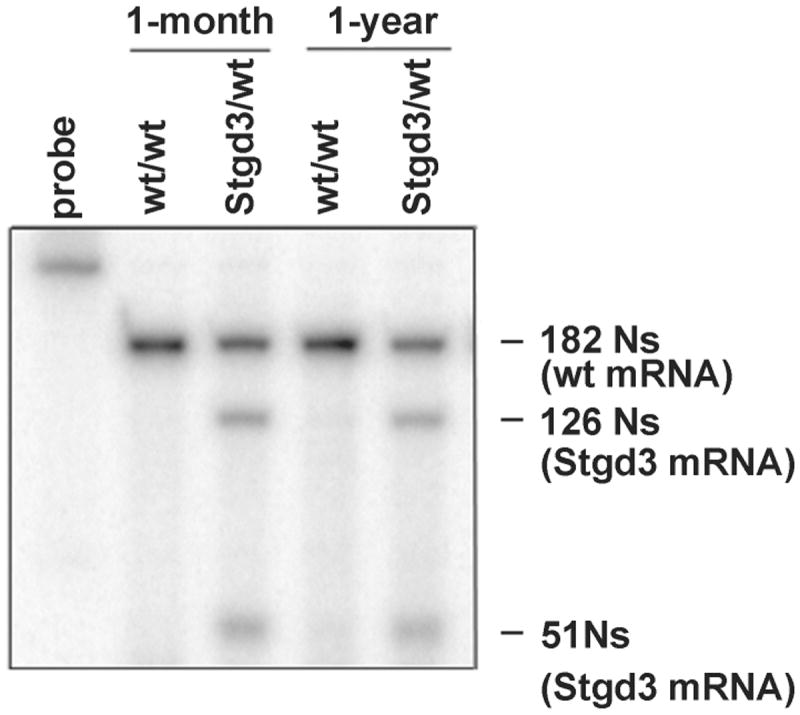
Elovl4 mRNA expression in eyecups from 1-month and 1-year-old wt and Stgd3-heterozygous mice. S1-nuclease protection assay detects a 182-N fragment of a 322-N probe protected by wt Elovl4 mRNA, and two fragments (126 Ns and 51 Ns) protected by mutated Elovl4 (Stgd3) mRNA. The molar amount of Elovl4 mRNA in wt mouse eyecups is equal to the combined molar amounts of wt and Stgd3 mRNAs in Stgd3-heterozygous eyecups.
3.4. Pathogenic events of, and potential treatments for STGD3
Based on the current and previously published results, we propose the following pathway of STGD3-pathogenic events:
STGD3 mutations result in deletion of the ELOVL4 protein motif for ER retention [1-4].
Truncated ELOVL4 protein leaves the ER [5,6] and carries with it associated wt ELOVL4 protein [7,22].
Reduced ELOVL4 protein levels in the ER cause decreased synthesis of ELOVL4 products: C24-C36 fatty acids and lipids containing C28-C36 acyl residues [8,10-12]. In the retina, this results in C32-C36 acyl PC deficiency (this study).
Retinal deficit of C32-C36 acyl PCs changes properties of phototransduction proteins [13,15], reducing visual functions and increasing lipofuscin accumulation as seen in both STGD3 patients [9] and Stgd3 mice [8].
In animals, a diet rich in n-3 fatty acids has been shown to increase the amounts of retinal polyunsaturated C28-C36 fatty acids [23] which are synthesized from eicosapentaenoic acid, but not from DHA [14]. These findings suggest that the retinal C28-C36 acyl PC deficiency may be corrected by supplementation with polyunsaturated C28-C36 fatty acids or their precursors. Such treatments have the potential to benefit STGD3 patients (a) by reducing effects of the decreased ELOVL4 synthetic capacity and (b) by compensating for potentially inadequate dietary intake of n-3 fatty acids.
Supplementary Material
Supplementary Data associated with this article can be found, in the online version, at
Acknowledgments
This work was supported by an NIH EY 15409 grant and a grant from the David M. Crowley Foundation (to WK), by a grant to the Department of Ophthalmology from Research to Prevent Blindness, and by the Intramural Research Program of NIDA, NIH. A.S. Woods thanks the Office of National Drug Control Policy for instrumentation funding.
Abbreviations
- AMD
age-related macular degeneration
- DHA
docosahexaenoic acid
- ELOVL4
elongase of very long chain fatty acid-4
- ER
endoplasmic reticulum
- MS
mass spectrometry
- m/z
mass-to-charge ratio
- N
nucleotide
- PC
phosphatidylcholine
- STGD3
Stargardt disease-3
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang K, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- 2.Edwards AO, Donoso LA, Ritter R., 3rd A novel gene for autosomal dominant Stargardt-like macular dystrophy with homology to the SUR4 protein family. Invest Ophthalmol Vis Sci. 2001;42:2652–2663. [PubMed] [Google Scholar]
- 3.Bernstein PS, et al. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest Ophthalmol Vis Sci. 2001;42:3331–3336. [PubMed] [Google Scholar]
- 4.Maugeri A, et al. A novel mutation in the ELOVL4 gene causes autosomal dominant Stargardt-like macular dystrophy. Invest Ophthalmol Vis Sci. 2004;45:4263–4267. doi: 10.1167/iovs.04-0078. [DOI] [PubMed] [Google Scholar]
- 5.Ambasudhan R, et al. Atrophic macular degeneration mutations in ELOVL4 result in the intracellular misrouting of the protein. Genomics. 2004;83:615–625. doi: 10.1016/j.ygeno.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Karan G, Yang Z, Zhang K. Expression of wild type and mutant ELOVL4 in cell culture: subcellular localization and cell viability. Mol Vis. 2004;10:248–253. [PubMed] [Google Scholar]
- 7.Karan G, et al. Loss of ER retention and sequestration of the wild-type ELOVL4 by Stargardt disease dominant negative mutants. Mol Vis. 2005;11:657–664. [PubMed] [Google Scholar]
- 8.McMahon A, et al. Retinal pathology and skin barrier defect in mice carrying a Stargardt disease-3 mutation in elongase of very long chain fatty acids-4. Mol Vis. 2007;13:258–272. [PMC free article] [PubMed] [Google Scholar]
- 9.Donoso LA, et al. Autosomal dominant Stargardt-like macular dystrophy: founder effect and reassessment of genetic heterogeneity. Arch Ophthalmol. 2001;119:564–570. doi: 10.1001/archopht.119.4.564. [DOI] [PubMed] [Google Scholar]
- 10.Cameron DJ, et al. Essential role of Elovl4 in very long chain fatty acid synthesis, skin permeability barrier function, and neonatal survival. Int J Biol Sci. 2007;3:111–119. doi: 10.7150/ijbs.3.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W, et al. Depletion of ceramides with very long chain fatty acids causes defective skin permeability barrier function, and neonatal lethality in ELOVL4 deficient mice. Int J Biol Sci. 2007;3:120–128. doi: 10.7150/ijbs.3.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasireddy V, et al. Loss of functional ELOVL4 depletes very long-chain fatty acids (>=C28) and the unique {omega}-O-acylceramides in skin leading to neonatal death. Hum Mol Genet. 2007;16:471–482. doi: 10.1093/hmg/ddl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aveldano MI. Phospholipid species containing long and very long polyenoic fatty acids remain with rhodopsin after hexane extraction of photoreceptor membranes. Biochemistry. 1988;27:1229–1239. doi: 10.1021/bi00404a024. [DOI] [PubMed] [Google Scholar]
- 14.Suh M, Clandinin MT. 20:5n-3 but not 22:6n-3 is a preferred substrate for synthesis of n-3 very-long- chain fatty acids (C24-C36) in retina. Curr Eye Res. 2005;30:959–968. doi: 10.1080/02713680500246957. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell DC, et al. Effects of ROS disk membrane phospholipids with extremely long polyunsaturated acyl chains on visual signaling. ARVO Annual Meeting; Fort Lauderdale, Fl. 2007. p. 144. Abstract #2928. [Google Scholar]
- 16.Jackson SN, Wang HY, Woods AS. In situ structural characterization of phosphatidylcholines in brain tissue using MALDI-MS/MS. J Am Soc Mass Spectrom. 2005;16:2052–2056. doi: 10.1016/j.jasms.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 17.Hsu FF, Bohrer A, Turk J. Formation of lithiated adducts of glycerophosphocholine lipids facilitates their identification by electrospray ionization tandem mass spectrometry. J Am Soc Mass Spectrom. 1998;9:516–526. doi: 10.1016/S1044-0305(98)00012-9. [DOI] [PubMed] [Google Scholar]
- 18.Raz-Prag D, et al. Haploinsufficiency is not the key mechanism of pathogenesis in a heterozygous Elovl4 knockout mouse model of STGD3 disease. Invest Ophthalmol Vis Sci. 2006;47:3603–3611. doi: 10.1167/iovs.05-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vasireddy V, et al. Elovl4 5-bp-deletion knock-in mice develop progressive photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2006;47:4558–4568. doi: 10.1167/iovs.06-0353. [DOI] [PubMed] [Google Scholar]
- 20.Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- 21.Gulow K, Bienert D, Haas IG. BiP is feed-back regulated by control of protein translation efficiency. J Cell Sci. 2002;115:2443–2452. doi: 10.1242/jcs.115.11.2443. [DOI] [PubMed] [Google Scholar]
- 22.Grayson C, Molday RS. Dominant negative mechanism underlies autosomal dominant Stargardt-like macular dystrophy linked to mutations in ELOVL4. J Biol Chem. 2005;280:32521–32530. doi: 10.1074/jbc.M503411200. [DOI] [PubMed] [Google Scholar]
- 23.Xi ZP, Wang JY. Effect of dietary n-3 fatty acids on the composition of long-and very-long-chain polyenoic fatty acid in rat retina. J Nutr Sci Vitaminol (Tokyo) 2003;49:210–213. doi: 10.3177/jnsv.49.210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data associated with this article can be found, in the online version, at