

Abstract
The distributions of scores for autistic behaviours obtained from the Autism Diagnostic Observation Scale-Generic (ADOS-G) were investigated in 147 males and females affected with the full mutation in the Fragile X mental retardation 1 (FMR1) gene, in 59 individuals with the premutation, and in 42 non-fragile X relatives, aged 4–70 years. The scores representing Communication and Social Interaction were continuously distributed across the two fragile X groups, and they were significantly elevated compared with the non- fragile X controls. Strong relationships were found between both these scores and FMRP deficits, but they became insignificant for Social Interaction, and the sum of Social Interaction and Communication scores, when FSIQ was included as another predictor of autism scores. Other significant predictors of these scores in both sexes were those executive skills which related to verbal fluency, and to the regulation and control of motor behaviour. Overall, our data have shown that cognitive impairment, especially of verbal skills, best explains the comorbidity of autism and fragile X. This implies some more fundamental perturbations of specific neural connections which are essential for both specific behaviours and cognition. We emphasize that FXS offers a unique molecular model for autism since FMRP regulates the translation of many other genes involved in synaptic formation and plasticity which should be natural targets for further exploration.
Keywords: fragile X, autism spectrum, ADOS-G, cognitive impairments, FMRP, predictors of autism behaviours
INTRODUCTION
Fragile X Syndrome (FXS) is the most common inherited form of intellectual disability, with cognitive and behavioural impairments of varying degree associated with distinct physical features (Hagerman, 2002). FXS is caused by an unstable mutation in the Fragile X mental retardation 1 (FMR1) X-linked gene (Verkerk et al., 1991), and involves the expansion of trinucleotide (CGG) repeats in the promoter region of this gene if transmitted from mothers to their offspring (Fu et al., 1991). Small expansions ranging from 55 to 200 CGG repeats (premutation) do not typically cause obvious developmental delay, but they tend to further expand into a ‘full mutation’ (>200 CGG repeats) if transmitted through a female (Fu et al., 1991). This usually leads to an inactivation of the FMR1 gene and gross deficit of its specific protein product, FMR1 protein (FMRP) (Pieretti et al., 1991), which is important for normal brain development (Irwin et al., 2000; Weiler & Greenough, 1999). FMRP is involved in synaptogenesis, especially in the cerebral cortex, cerebellum, and hippocampus, and more specifically, in synaptic pruning (Comery et al., 1997; Weiler et al., 1997; Weiler & Greenough, 1999). Hence developmental delay in FXS is primarily caused by the deficit of this protein.
FXS occurs in both sexes, but females are usually less affected because of the presence of a normal FMR1 gene on the second X chromosome which, if active, generates normal levels of FMRP. This is why only 50–71% of females with the full mutation demonstrate a significant cognitive deficit (de Vries et al., 1996; Loesch & Hay, 1988).
Behavioural features displayed by both male and female individuals affected with FXS, such as impairments in social interaction and communication, social anxiety, gaze avoidance, hand and finger mannerisms, repetitive and tangential speech, and other stereotypic behaviours (Hagerman, 2002; Lachiewicz et al., 1994; Miller et al., 1999; Sudhalter et al., 1990), resemble those seen in autism. These features may occur in a number of other conditions of known genetic background, such as tuberous sclerosis, phenylketonuria, and a wide range of chromosomal anomalies (Dykens & Volkmar, 1997; Fombonne et al., 1997; Gillberg, 1998), but they may also develop in the absence of any detectable cause (idiopathic autism). Therefore the psychiatric diagnosis of Autistic Disorder (AD) is heterogeneous in genetic etiology and is usually associated with general cognitive impairment. However, approximately 20% of individuals with idiopathic autism have an IQ in the normal range as measured by standard cognitive tests, and they are classified as ‘high functioning autism’, HFA (Fombonne, 2003).
Behavioural features in three domains, comprising impairments in social interaction, verbal and non-verbal communication, and restricted repetitive and stereotyped patterns of behaviour, interests, and activities, characterize autism (APA manual, 2000). A spectrum of autistic manifestations tends to occur in relatives of the probands affected with AD. The increased recurrence rate in siblings and MZ and DZ twins, as well as evidence from chromosomal, genome screen and linkage studies, have given evidence for significant multiple genetic factors predisposing to idiopathic autism (Muhle et al., 2004).
The frequency of autism amongst males with fragile X varies widely, from 18.5% in the first estimate by Brown et al. (1982), and ranging from 5% to 60% in subsequent studies depending on the diagnostic criteria used (Hagerman, 2002). The occurrence of autism diagnosed using stringent DSM criteria was between 15–28% across a wide range of ages (Baumgardner et al., 1995; Cohen, 1995; Hagerman et al., 1986; Reiss & Freund, 1990; Turk & Graham, 1997). The percentage of autism is lower (averaging 4%) in females with FXS (Bailey et al., 1993; Hagerman, 2002), although the rate of these behaviours in females is significantly higher than in controls (Mazzocco et al., 1997). However, all these estimates were based on older standardized autism measures and DSM criteria, and on samples where a diagnosis of fragile X was not always accurate.
An important development in autism research has been the introduction of new diagnostic tools: the Autism Diagnostic Interview-Revised (ADI-R) (Lord et al., 1994) and the Autism Diagnostic Observation Scale- Generic (ADOS- G) (Lord et al., 1999). Both instruments have been validated across ages and severity of the disorder, and jointly provide the gold standard assessment of autism. The ADI-R, a semi-structured parent interview, assesses the presence and severity of early childhood symptoms of autism across the three domains, which essentially correspond to the diagnostic criteria in DSM. The ADOS-G is a semi-structured standardized assessment administered directly to an individual, and it is complimentary to the ADI-R in providing diagnostic classification (Lord et al., 2000). Using both the ADI-R and ADOS-G assessment tools, the frequency of autism amongst a sample of (primarily) male American children affected with FXS, aged 21 to 48 months, was 33% (Rogers et al., 2001). Similar rates were determined in older boys with FXS using the ADI-R (Kaufman et al., 2004). Our own study, in a large Australian sample of male and female children and adults with FXS, showed that 18% of males and 10% of females with the full mutation met the AD criteria on both the ADI-R and ADOS-G, and that 67 % of males and 23 % of females in this category met either the AD criteria on at least one of these two tests, or autism spectrum disorder (ASD) criteria on the ADOS (Clifford et al., 2006). Our results also showed that 14 % of males and 5 % of females carrying the FMR1 premutation met the criteria for ASD on the ADOS.
In the current study we explore the distributions of the major ADOS domain scores across the whole range of CGG repeat expansions in the FMR1 gene (full mutation and premutation included) in an unselected sample of fragile X males and females. This quantitative approach allows us not only to consider a wide range of autism-related behaviours, but also to test hypotheses concerning possible mechanisms underlying these behaviours in individuals affected with FXS. Thus rather than being concerned with diagnostic issues, the aim in this study was, firstly, to assess the relationships between the major ADOS-G domain scores and the levels of FMRP depletion, which is a direct result of mutation in the FMR1 gene, and highly specific to FXS. Secondly, we examine the relationship between the major ADOS-G domain scores and standard and higher cognitive function deficits, which are common amongst different neurodevelopmental disorders and not unique to FXS.
SAMPLE AND METHODS
The participants
All aspects of this study were approved by the ethics committees of La Trobe University and the Royal Children’s Hospital in Melbourne, and the Institutional Review Board of the University of California at Davis. Participants and/or their parents signed written informed consent. The Australian subjects were recruited from the register of 59 extended families, who participated in a major Australia-USA fragile X genotype-phenotype relationship study supported by an NIH grant over a long time period. These families were originally ascertained through clinical admissions of probands diagnosed with FXS caused by the full mutation, or, on rare occasions, with the premutation carrier status, at the Genetic Health Services at the Royal Children’s Hospital in Melbourne. The families originally ascertained through clinical admissions underwent cascade genetic testing for fragile X involving other family branches, at La Trobe University, in collaboration with Kimball Genetics, Denver, USA. The American families were recruited, in a similar manner, through the Fragile X Research and Treatment Center at the M.I.N.D. Institute at the University of California at Davis in Sacramento. All willing members of each family were assessed at each investigative site (country).
A total of 152 Australian participants aged 5 to 70 years, all white and of European descent, were included in this study. Sixty-four individuals (33 males, 31 females) were carriers of the full mutation FMR1 allele, 50 were premutation carriers (7 males, 43 females), and 38 were control relatives with non-expanded alleles (29 males, 9 females). The American sample included 110 individuals between 4 and 50 years of age, consisting of 97 full mutation individuals (83 males, 14 females), 9 premutation carriers (6 male, 3 female), and 4 male controls. Of the American population, 10.4% were Hispanic, 5.6% were either mixed ethnicity, African American or American Indian, and 84% were white and non-Hispanic. Rare individuals with serious associated medical conditions (such as high fever, serious infections, post-surgical, cancer) were not tested, but those with corrected vision and hearing impairments or minor medical problems were included. The FMR1 allele status was established using a specific DNA test.
FMRP and DNA assays
The DNA and FMRP assays were conducted at Kimball Genetics, Inc (Denver, CO) for the Australian sample and at the University of California at Davis for the American sample.
FMRP assays were performed on blood smears, which were made within 24 hours of the blood draw using 20 μl of peripheral blood on each microscopic slide. FMRP immunostaining was performed using an indirect alkaline phosphatase technique (Tassone et al., 1999; Willemsen et al., 1995). For each slide, 200 lymphocytes were scored, and the percent of lymphocytes expressing FMRP was determined. Scoring was performed in a blinded fashion with respect to DNA results and clinical/intellectual status. FMRP expression and distributions in fragile X Australian and American samples combined are detailed in Loesch et al. (2004).
Genomic DNA was isolated from 10 ml of peripheral blood samples using standard methodology (Purgene kit, Gentra Systems, Inc., Minneapolis, MN). Both Southern blots and polymerase chain reaction (PCR) analyses were performed either as detailed in Taylor et al. (1994), and Loesch et al. (2002), or as described in Tassone et al. (2004). PCR analysis using primer c and f was as reported in Saluto et al. (2005).
The DNA results were used as a diagnostic tool in order to classify individuals into premutation, full mutation, and non-expanded (control) groups, for analysis of the distributions of the major ADOS domain scores, and between-group comparisons. The analysis of relationship of ADOS scores with the cognitive scores and FMRP levels was conducted only in the full mutation group, which included fully methylated low functioning individuals, as well as full mutation/premutation mosaic, and partially unmethylated/methylation mosaic males. The inclusion of the higher functioning full mutation males helped in obtaining relatively smooth distributions of FMRP levels in this category.
Autism Measures
Autism Diagnostic Observation Schedule-Generic (ADOS-G)
Apart from a short interview with the caregiver/participant, this instrument uses developmentally appropriate social and play-based interactions designed to elicit spontaneous behaviours across several autism domains, which are scored across different developmental levels and chronological ages. Two major domains: (1) Language and Communication (COM), and (2) Reciprocal Social Interaction (RSI), which are most characteristic of autism behaviours (Lord et al., 1999), were included in this study, together with their sum (CSIT). The remaining three: Stereotypic Behaviour and Restricted Interests (SBRI), Anxiety (ANX), and Imagination and Creativity (IC), which do not contribute to the overall diagnostic score (Lord et al., 1999) were omitted from this presentation. Each item in the respective domains is given a score from 0 to 3, but for the diagnostic algorithm a score of 3 is normally converted to score 2. However, in this study, which was not concerned with diagnostic thresholds, the full range of scores from 0 to 3 was considered (with 0 indicating no evidence of abnormality and 3 indicating markedly abnormal behaviour). The scores for individual items were summed for each domain to provide a measure of severity of autism-related behaviours.
The raters administering the ADOS-G at each investigative site were trained to a reliability of 85% or higher item agreement on the full range of scores with the training tapes originating from Cathy Lord’s group (who were involved in designing this instrument; Lord, Rutter, DiLavore et al., 1999), and the training was conducted in both centres by the members of this group. Because of obvious cognitive impairments associated with FXS, especially in males, the examiners were blinded to the FMR1 allele status only in those individuals (approximately one-third of the total sample) who were not cognitively impaired.
All study participants were administered the ADOS-G, regardless of their genetic or psychological status, and the sample sizes are shown in Table 1. In some proportion of these individuals however, there are missing data on one or more predictors of the ADOS, and the information of sample sizes for individual predictors in the full mutation group are shown in Table 2.
Table 1.
Differences between the fragile X and non-fragile categories and between males and females, in ADOS major domain scores. Means and standard deviations (SD) for two major ADOS-G domain scores, and a summary score, in males and females separately and combined, in the premutation and full mutation groups, and non-fragile X normal controls.
| Controls | Premutation | Full mutation | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Variable | N | Mean | SD | N | Mean | SD | N | Mean | SD |
| Language & Communication (COM) | |||||||||
| Total | 42 | 0.712 | 1.17 | 59 | 0.973 | 1.73 | 161 | 2.73 | 2.26 |
| F | 9 | 0.332 | 0.71 | 46 | 0.853 | 1.79 | 45 | 1.69* | 2.45 |
| M | 33 | 0.822 | 1.26 | 13 | 1.383 | 1.50 | 116 | 3.13 | 2.05 |
| Reciprocal Social Interaction (RSI) | |||||||||
| Total | 42 | 1.692 | 2.31 | 59 | 2.343 | 2.91 | 161 | 6.13 | 4.00 |
| Females | 9 | 1.112 | 1.36 | 46 | 1.703* | 2.48 | 45 | 4.40* | 4.40 |
| Males | 33 | 1.85 1,2 | 2.50 | 13 | 4.623 | 3.28 | 116 | 6.80 | 3.81 |
| Communication & Social Interaction Total (CSIT) | |||||||||
| Total | 42 | 2.402 | 3.27 | 59 | 3.323 | 4.49 | 161 | 8.86 | 5.78 |
| Females | 9 | 1.442 | 1.94 | 46 | 2.573* | 4.20 | 45 | 6.11* | 6.04 |
| Males | 33 | 2.671,2 | 3.52 | 13 | 6.003 | 4.58 | 116 | 9.93 | 5.33 |
Significant differences (p-values < 0.05) between: normal vs premutation
normal vs full mutation
premutation vs full mutation
and females vs males within each group (*).
Table 2.
Comparison between males and females with full mutation in means of all the predictors of ADOS-G measures included in the regression models: deficit of FMR1 specific protein (FMRP), cognitive Wechsler scores (FSIQ, PIQ, VIQ), and higher executive function test scores (BDS, STROOP (Interference), and COWATT and COWATTA of COWAT).
| Females | Males | |||||
|---|---|---|---|---|---|---|
| N | Mean | SD | N | Mean | SD | |
| FMRP | 33 | 59.9* | 21.0 | 82 | 12.6 | 11.6 |
| FSIQ | 42 | 72.0* | 17.5 | 90 | 54.8 | 12.1 |
| PIQ | 39 | 74.0* | 18.0 | 73 | 57.2 | 11.4 |
| VIQ | 39 | 75.5* | 16.2 | 74 | 56.1 | 11.1 |
| BDS | 27 | 14.1* | 4.22 | 18 | 3.55 | 4.02 |
| STROOP | 31 | 48.7 | 7.15 | 10 | 51.5 | 6.24 |
| COWATT | 34 | 21.8* | 9.23 | 28 | 7.32 | 5.05 |
| COWATA | 35 | 14.7* | 6.07 | 40 | 7.83 | 4.16 |
Significant male-female differences (p-values < 0.05), using either two-sample t-test or Mann-Whitney statistics.
Standard cognitive and executive function measures
Wechsler Intelligence Scales
Intellectual functioning was assessed by the Wechsler intelligence test appropriate for chronological age. Individuals under the age of 6 years were tested using the WPPSI-R or the WPPSI-III (Wechsler, 2002), those aged between 6 and 16 years, using the WISC-III, and those over the age of 16, using the WAIS-III (Wechsler, 1997).
Controlled Oral Word Association Test (COWAT)
The COWAT assesses verbal fluency (Spreen & Benton, 1977), and is considered a useful measure of abstract thinking; it requires the ability not only to generate verbal lists but to monitor responses and avoid repetitions and perseveration (Lezak, 1995). Three phonemic (letter) fluency trials (F, A, and S) were administered, and the scores were collapsed into a total score (COWATT). Category (animals) fluency (COWATA) was also assessed. Analyses were based on raw scores adjusted for age.
Stroop Colour and Word Test (STROOP)
The STROOP measures selective attention and cognitive flexibility (Golden & Freshwater, 2002). The interference measure of the STROOP was specifically used here to examine divided attention and the ability to inhibit automatic responding. Age adjusted T-scores, with a mean of 50 and a standard deviation of 10, were used in calculating the interference score on this test. Age corrections were applied to the scores for participants younger than 17 years, according to the test manual specifications.
Behavioural Dyscontrol Scale (BDS)
The BDS, developed and validated by Grigsby & Kaye (1996), is particularly useful in a population with intellectual disability, because it is brief and the instructions are clear and simple. It consists of nine items that are designed to assess an individual’s ability to initiate deliberate motor behaviour, and examines executive regulation and the control of such behaviour. The tenth item is a measure of the participant’s insight into their performance.
Statistical analysis
In order to capture the full range of the ADOS domain scores in the two categories of FMR1 allele expansion and in non-expanded (control) individuals, the data were collected from all available members of families with fragile X. The three ADOS scores (COM, RSI, CSIT) met the criteria for a continuous distribution, but because of a large number of zero scores (from 16% for CSIT to 33% for COM) these distributions were strongly negatively skewed in both male and female samples (Fig 1) and thus not amenable for transformation to normality. We therefore used the Poisson regression model, which gave a good fit to the data. For traits where the mean was not equal to the variance, a negative binomial regression model was used as in Cameron & Trivedi (1988).
Figure 1.
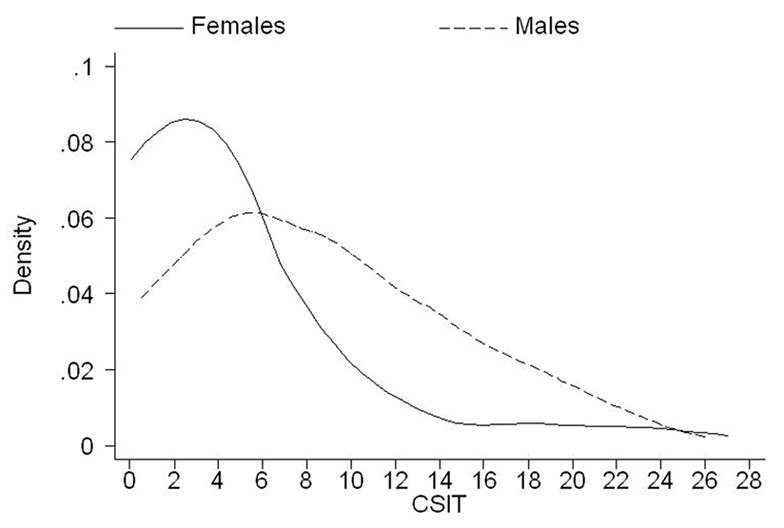
Distribution of CSIT (ADOS-G) score in premutation and full mutation individuals, separately for males and females (Australian and American data combined). Kernel density estimates were used to smooth the distributions.
The regression analysis, following Hosmer & Lemeshow (1989), was carried out using STATA (version 7), first considering each of the three individual ADOS scores as the response variable, and a single predictor. We then included other predictors in the regression models, such as FSIQ, age, and the investigative site, whenever these predictors were found to have a significant effect on the response variable. Because the choice of ADOS modules are primarily based on expressive speech level, age was included as one of the predictors as it showed a significant effect on some ADOS domain scores. Considering the unique genetics of fragile X, this effect is likely to result largely from generational differences in severity of cognitive involvement (anticipation), so that more seriously cognitively affected individuals come from the younger generations. Indeed, this effect was no longer significant for a majority of ADOS scores once FSIQ was included as another predictor in the regression model. If this was not the case and the age effect was directly relevant to autistic behaviour, then both FSIQ and age, or age alone, were included in the final model for the respective ADOS domains. Another predictor, which was found to be significant for most of the domain scores, was the investigative site where the data for this study had been collected, with a small shift towards higher scores in the American sample. Considering the fact that all the raters were trained by the same group and using the same training tapes, it is unlikely that this shift could be attributed to the centres’ ADOS-G ratings. The more likely source of this effect is a difference in recruitment between the investigative sites already noted in our earlier collaborative works (e.g. Loesch al., 2003a & b), with the larger proportion of subjects recruited through clinical admissions, relative to those recruited through cascade testing of families, in the American sample. Indeed, a shift toward more severe involvement in the American sample was also noted across the cognitive measures. The results of analysis in both samples combined (with the investigative site predictor included) were highly consistent with similar results obtained from the Australian sample of males and females only.
Simple statistical comparisons of the differences in the ADOS scores (COM, RSI, and CSIT) between the three fragile X status categories were conducted using negative binomial regression. The two-sample t-test, or the Mann-Whitney test, was used (where appropriate) for comparisons of the predictors of the ADOS domains between males and females in the individual fragile X categories.
RESULTS
ADOS-G standard statistics
Means for the two major ADOS domains (COM, RSI), and their sum (CSIT) are listed in Table 1, and the distributions of the CSIT scores in males and females in the combined premutation and full mutation sample are illustrated in Figure 1. It is evident from Table 1 that in males, the means for each of the domain scores are generally higher in the full mutation group compared to either the premutation or normal groups. It is important to note that the RSI and CSIT scores are also significantly higher in premutation males compared to male controls with the normal allele. Females are, predictably, less affected in both the full mutation and premutation categories (Table 1 and Figure 1), and both males and females with the premutation are less affected than their full mutation counterparts (Table 1). There are also significant differences between sexes in all the mean scores in the full mutation group.
Relationships of ADOS-G with other phenotypic measures
Means for all the predictors of ADOS scores considered in this study are given in Table 2, for the full mutation males and females separately, and differences in the means between males and females are also indicated. The data in Table 2, as well as the results of the regression analyses in Tables 3, 4 and 5, are presented only for the individuals with the full mutation, because our preliminary analysis in the small samples of individuals with the premutation and normal allele showed no significant or consistent relationships between the ADOS-G measures and these predictors. Moreover, the data on these relationships in the affected individuals with FXS are of primary interest in consideration of the possible origins of autistic behaviours in this condition.
Table 3.
Relationships between ADOS test scores and three Wechsler scores in the full mutation males and females separately and combined. Regression coefficients (β) and P-values (p) were estimated using simple (unadjusted) models, but changes in p-values estimated from more complex models are also indicated (as specified below).
| FSIQ | VIQ | PIQ | ||||
|---|---|---|---|---|---|---|
| ADOS Domain scores | Regression coefficient (β) | p | Regression coefficient (β) | p | Regression coefficient (β) | p |
| COM | ||||||
| Total | −0.035** | < 0.001 | −0.039** | < 0.001 | −0.033** | < 0.001 |
| Females | −0.063** | < 0.001 | −0.072** | < 0.001 | −0.046** | < 0.001 |
| Males | −0.017 | 0.007 | −0.016** | 0.015 | −0.013** | 0.088 |
| RSI | ||||||
| Total | −0.031 | < 0.001 | −0.034 | < 0.001 | −0.031 | < 0.001 |
| Females | −0.040 | < 0.001 | −0.046 | < 0.001 | −0.035 | < 0.001 |
| Males | −0.023** | < 0.001 | −0.022 | < 0.001 | −0.022 | < 0.001 |
| CSIT | ||||||
| Total | −0.032 | < 0.001 | −0.036 | < 0.001 | −0.032 | < 0.001 |
| Females | −0.046* | < 0.001 | −0.052* | < 0.001 | −0.038* | < 0.001 |
| Males | −0.022 | 0.001 | −0.020 | < 0.001 | −0.019 | < 0.001 |
Adjusted for * age or ** country, in complex models, with changes in p values after adjustment indicated as follows:
Underlined are p values remaining or becoming significant (< 0.05); bold are p values remaining or becoming insignificant (> 0.05).
Table 4.
Relationships between ADOS major domain scores and FMRP levels (in %) in the full mutation males and females separately and combined. Regression coefficients (β) and P-values (p) were estimated from simple (unadjusted) models (FMRP ONLY), and from complex models (FMRP+FSIQ), where FSIQ score was included as an additional predictor.
| FMRP+ FSIQ | ||||||
|---|---|---|---|---|---|---|
| FMRP only FMRP | FMRP | FSIQ | ||||
| ADOS | Regression coefficient (β) | p | Regression coefficient (β) | p | Regression coefficient (β) | p |
| COM | ||||||
| Total | −0.022 | < 0.001 | −0.011 | 0.009 | −0.026 | 0.001 |
| Females | −0.034 | < 0.001 | −0.019 | 0.006 | −0.055 | < 0.001 |
| Males | −0.025 | 0.001 | −0.019 | 0.028 | −0.012 | 0.134 |
| RSI | ||||||
| Total | −0.011 | < 0.001 | −0.001 | 0.785 | −0.028 | < 0.001 |
| Females | −0.013 | 0.053 | −0.003 | 0.618 | −0.041 | < 0.001 |
| Males | −0.011 | 0.049 | 0.001 | 0.866 | −0.021 | 0.001 |
| CSIT | ||||||
| Total | −0.013 | < 0.001 | −0.004 | 0.176 | −0.028 | < 0.001 |
| Females | −0.018 | 0.009 | −0.006 | 0.265 | −0.045 | < 0.001 |
| Males | −0.015 | 0.005 | −0.004 | 0.477 | −0.018 | 0.003 |
Table 5.
Relationships between major ADOS domain scores and BDS, STROOP, and COWAT test scores in the full mutation males and females separately and combined. Regression coefficients (β) and P-values (p) were estimated using simple models, but changes in p-values from complex models are also indicated (as specified below).
| BDS | STROOP | COWATT | COWATA | |||||
|---|---|---|---|---|---|---|---|---|
| ADOS | Regression coefficient (Β) | p | Regression coefficient (β) | p | Regression coefficient (β) | p | Regression coefficient (β) | p |
| COM | ||||||||
| All | −0.147 | < 0.001 | −0.016* | 0.556 | −0.079* | < 0.001 | −0.080* | < 0.001 |
| Female | −0.129* | 0.002 | −0.022* | 0.338 | −0.076* | 0.001 | −0.133 | < 0.001 |
| Male | −0.294 | 0.012 | −0.017 | 0.726 | −0.078 | 0.007 | 0.004 | 0.887 |
| RSI | ||||||||
| All | −0.089* | < 0.001 | −0.014* | 0.507 | −0.038* | < 0.001 | −0.051* | 0.001 |
| Female | −0.088* | 0.050 | −0.023* | 0.378 | −0.035* | 0.056 | −0.058* | 0.018 |
| Male | −0.105 | 0.001 | 0.002** | 0.954 | −0.041* | 0.077 | 0.019* | 0.317 |
| CSIT | ||||||||
| All | −0.099* | < 0.001 | −0.013* | 0.493 | −0.047* | < 0.001 | −0.059* | < 0.001 |
| Female | −0.098* | 0.018 | −0.022* | 0.383 | −0.043* | 0.011 | −0.065* | 0.003 |
| Male | −0.130 | < 0.001 | −0.002** | 0.955 | −0.052* | 0.010 | 0.015* | 0.440 |
Adjusted for * FSIQ or ** age, in complex models, with changes in p values after adjustment indicated as follows:
Underlined are p values remaining or becoming significant (< 0.05); bold are p values remaining or becoming insignificant (> 0.05).
The estimates from simple (unadjusted) models, including the regression coefficient (β) and P-values (P) for 2-sided tests are listed in the Tables 3, 4 and 5, and any change in the significance of the coefficients after including additional predictors, wherever appropriate, is also indicated for individual measures.
Standard cognitive scores and FMRP levels
The standard cognitive (Wechsler) measures used as predictors of ADOS test items were FSIQ, PIQ, and VIQ scores, and the results of the regression analyses are presented in Table 3. All the relationships between FSIQ, PIQ, and VIQ, and the ADOS scores, are significant for females and males (with the exception of PIQ in males), and for the sexes combined, both before and after adjustment for either age or investigative site.
FMRP deficit represents the severity of FMR1-specific genetic change and, unlike the size of CGG repeat expansion, it can conveniently be interpreted in a uniform fashion in both sexes. In the simple model (that is, without FSIQ adjustment; see Table 4 ‘FMRP only’), the level of autistic manifestations as represented by the major ADOS domain scores, COM, RSI, and the total CSIT, is significantly related to the FMRP deficit in full mutation males and females (with the exception of RSI in females where the relationship is marginal). This relationship for the total (CSIT) score is also illustrated in Figure 2 for the full mutation male and female samples separately and combined. There are obvious differences in the strength of the relationship between males and females which, as for the FSIQ scores, may be attributed to the narrow range of FMRP levels in the males with the full mutation compared with females, where the normal allele on the second X has an effect in normalizing the distribution of these levels.
Figure 2.
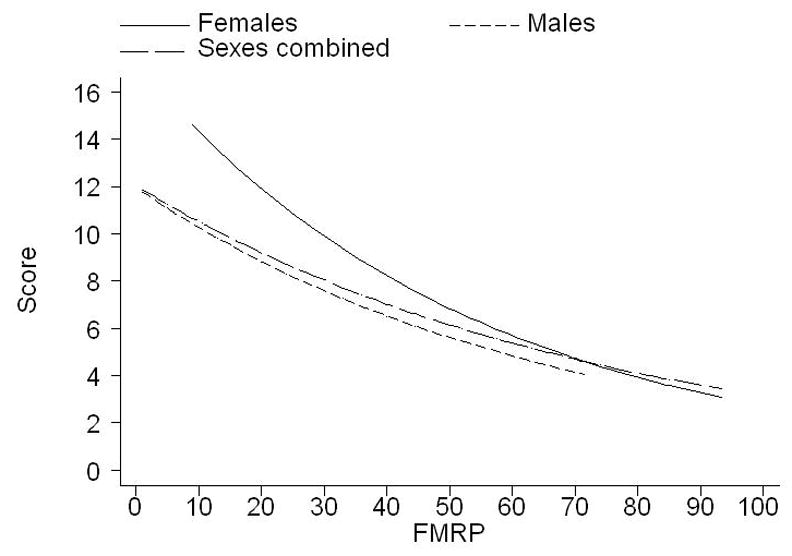
Illustration of the relationship between CSIT score of ADOS (SCORE) and FMRP levels in the full mutation category for females and males separately and combined (Australian and American data combined). The relationship is represented by plots of fitted values based on negative binomial regression of CSIT score on FMRP levels.
It was of special interest to establish if FSIQ, representing the level of intellectual impairment, or FMRP, representing the level of FMR1 dysfunction, is more strongly related to the ADOS domain scores in the individuals with full mutation. The results (in Table 4) show that, if both FMRP and FSIQ predictors are included (‘FMRP+FSIQ’), the effect of FMRP remains significant only for COM in both males and females. In contrast, the effect of FSIQ in this complex model is significant for all scores in all the samples (with the exception of COM in males). In conclusion, the major autism characteristics, although they show strong relationships with deficits of FMRP, are predominantly associated, in both sexes, with the level of intellectual impairment, when both FMRP and FSIQ are included in the regression model.
Executive function measures
The results of regression of the ADOS domain scores on the three executive function test scores (BDS, STROOP, and COWAT) are shown in Table 5. In the simple model (i.e., without FSIQ adjustment), the BDS total score is significantly related to all the ADOS domains (except for RSI in females where the relationship is marginal); COWATA is related to all domains in the female, and sexes combined, samples; COWATT is related to COM and CSIT in the male and female samples, separately and combined, and to RSI only in the sexes combined. In contrast, STROOP is not correlated with any ADOS domains. However, as indicated in Table 5 (bold figures), some of the relationships between ADOS and BDS, COWATT and COWATA become insignificant when FSIQ is included as another predictor in the complex model. The exceptions are: CSIT, which remains significantly related to BDS and COWATT in the male and combined samples, RSI, which remains significantly related to BDS in the male sample; and COM, which remains significantly related to both BDS and COWATT, in the male and combined samples, and to COWATA, in the female and combined samples.
DISCUSSION
Although there is strong evidence for an association between the autism spectrum and fragile X, the underlying cause of this co-morbidity is not known. In order to shed some light on this issue, we explored the distributions of two major aspects of autistic behaviour, as assessed by the ADOS-G, in two categories of fragile X allele status, full mutation and premutation, compared with the non-fragile X controls. We also explored the relationship of these distributions with standard and higher cognitive abilities and genetic changes in the full mutation (FXS) males and females. This is the first study where the relationship between the scores representing major autistic behaviours and a deficit of a specific product of the FMR1 gene causing fragile X has been determined in males and females with FXS. Moreover, consistent with some earlier findings that fragile X individuals manifesting autistic behaviours were more cognitively affected (Bailey et al., 1998, 2000; Denmark et al., 2003; Rogers et al., 2001), we provide evidence for this relationship using a quantitative approach with a gold standard instrument, the ADOS-G.
By using the distribution of test scores, instead of diagnostic thresholds to separate AD and non-AD categories, we have assumed that the problem of co-morbidity extends far beyond the limits imposed by strict diagnostic criteria for AD. This assumption is justified considering the results from earlier studies, which have shown that elements of autistic behaviours, such as perseveration of speech, hand stereotypes, tactile defensiveness, and poor eye contact, occur in the majority of individuals with FXS (Bailey et al., 1998; Denmark et al., 2003; Hagerman, 2002). The continuum of autistic manifestations has been further supported by the results of earlier studies reporting the occurrence of ASD in some proportion of premutation carriers (Aziz et al., 2003; Clifford et al., 2006; Farzin et al., 2006; Goodlin-Jones et al., 2004).
In this study we illustrate a broad spectrum of autistic behaviours, by presenting a continuity of distributions of the major ADOS-G domain scores within the full mutation category and overlapping with those in the premutation category. We therefore suggest that studies of co-morbidity of autism with other defined developmental abnormalities, including fragile X, would be more effective if they extended beyond a clear-cut distinction between ‘autistic’ and ‘non-autistic’ categories, and considered the distributions of full range autism behaviour scores, as well as diagnostic thresholds based on algorithm-processed scores. Such an approach, which allows for more analytical options, has recently been adopted by Kaufman et al. (2004), who have demonstrated strong relationships between the severity of autism in FXS assessed by the ADI-R, and adaptive behaviour as assessed by the Vineland Adaptive Behaviour Scales (Sparrow et al., 1984).
The most important outcome of our analysis based on regression is the unprecedented finding that the two major domain scores of the ADOS-G and their sum are strongly related, in the FXS sample, to the level of FMRP deficit, which is a unique feature of this syndrome, and reflects the level of dysfunction of the FMR1 gene. Cognitive deficits are a characteristic, although unlike FMRP, not unique, feature of the fragile X phenotype, and our results also showed highly significant correlations between the IQ scores and the major ADOS-G domain scores, in both males and females with full mutation. Considering the close relationships between IQ scores and FMRP deficits in FXS (Loesch et al., 2004), it was of interest to ascertain if cognitive status or FMRP levels were stronger predictors of the level of autistic behaviours considered in this study. Our results have clearly shown that, overall, the level of cognitive impairment is a major correlate of autistic behaviours in fragile X syndrome.
It was of particular interest to identify those specific cognitive deficits which were the strongest predictors of autistic behaviours in FXS. Our results from regression of the ADOS scores on the executive function test scores showed that verbal fluency (COWATT) had, unlike the STROOP measure, a significant effect on the COM domain, and the sum of COM and RSI, in males and sexes combined, independent of an overall cognitive deficit represented by FSIQ scores. A relationship of these scores with BDS, which examines motor control, may be explained by the effect of motor dysfunction, which is a common feature of autism (Ghaziuddin & Butler, 1998; Manjiviona & Prior, 1995).
In addition, some evidence for the proposal that specific cognitive deficits are closely associated with autistic behaviour in FXS is available from our (unpublished) results of regression of Wechsler subtest scores (adjusted for FSIQ) on the ADOS-G major domain scores, in a smaller (Australian only) sub sample of males and females combined. This analysis demonstrated a significant effect of verbal comprehension, vocabulary and matrix reasoning on the COM domain of the ADOS.
Relationships between autism diagnosis and the level of intellectual impairment in FXS have been reported in earlier studies. Bailey et al. (1998) and Demark et al. (2003) found that individuals with FXS with a lower IQ were more likely to be diagnosed with autism, and that all of the non-verbal children with a diagnosis of FXS fell in the autistic range on the CARS (Bailey et al., 1998). Moreover, Philofsky et al. (2004) found that significant comprehension deficits were related to autism in boys with FXS. Using a quantitative approach, Kaufman et al. (2004) also found a significant relationship between the degree of autistic manifestations, assessed by ADI-R major domain scores, and language comprehension in a pre-selected sample of boys with FXS and autism.
The evidence provided above, combined with the fact of a widespread occurrence of autism in a number of chromosomal (Fombonne et al., 1997; Gillberg, 1998; Muhle et al., 2004) and genetic (Dykens & Volkmar, 1997; Fombonne et al., 1997; Muhle et al., 2004) mental retardation syndromes, favour the conclusion that there may be a neural mechanism common to all co-morbid forms of autism, leading to both specific intellectual impairments and autism behaviours. However, there have been no systematic studies, to our knowledge, concerned with the assessment of the relationship between the level of cognitive deficits in various forms of co-morbid autism to support or refute this claim. The concept of common mechanism may also apply to idiopathic, including high functioning, autism where, even in an absence of obvious intellectual impairment, there may exist some more subtle cognitive deficits, such as verbal perception, that closely correlate with autism manifestations.
In recent years, there has been an increasing interest in identification of more specific cognitive deficits in ‘high functioning’ idiopathic autism (HFA). Goldstein et al. (1994) demonstrated impairment in processing the meaning of complex sentences in HFA. Direct evidence for the presence of a deficit in processing and integration of information in HFA has recently been presented by Just et al. (2004), where these deficits were linked to impaired functional connectivity between the hippocampus and amygdala. In support of these findings, deficits in Theory of Mind (ToM), resulting from social perceptual disabilities, and thought to underpin abnormal social behaviour in autism (Shaw et al., 2004; Tager-Flushberg et al., 1998; Tager-Flushberg & Sullivan, 2000), have been linked to executive function impairments (Shaw et al., 2004).
The notion that some specific neurodevelopmental deficit/s that may correlate with autistic manifestation in both co-morbid and idiopathic forms of autism is also supported by numerous examples of an overlap between the different forms of autism in brain developmental anomalies. Hypoplasia of the cerebellar vermis occurs in a large subset of individuals with idiopathic autism (Courchesne et al., 1994), as well as in individuals affected with FXS (Mostofsky et al., 1998; Reiss et al., 1991a, 1991b), where a relationship between autism behaviours and decrease in cerebellar vermal lobules VI and VII region has been found in affected females (Mazzocco et al.,1997). Moreover, the relevance of a dysfunction of the amygdala, and of the associated frontal-subcortical circuits, to co-morbidity of autism with either FXS (Hessl et al., 2004) or tuberous sclerosis (Asano et al., 2001; Smalley, 1998), have been postulated on the basis of the occurrence of the same type of anomaly in idiopathic autism. On the other hand, developmental insults to the amygdala have been linked to deficits in ToM (Tager-Flushberg & Sullivan, 2000).
It is thus possible that the final pathway in autistic brain development may be universal irrespective of the etiology per se, and it may involve particular circuitry or circuitries which are related to sophisticated language and/or social skills and the integration of information (Happe, 2003; Just et al., 2004; Sabbagh, 2004). The data presently available provide strong support to the hypothesis that impairments in some cognitive skills and autistic behaviours are both a consequence of perturbations of the same neural connections. However, future multidisciplinary studies of the dynamics of autistic development may provide more specific information as to whether some specific cognitive deficits, especially in the area of verbal comprehension, may be more fundamental, and a significant contributor to autistic behaviours. Although the studies based on FXS, or other syndromes associated with a widespread insult to the brain, and thus global intellectual impairment, are of limited use in directly addressing this issue, we have succeeded in demonstrating that cognitive impairment, especially in verbal abilities, was the main predictor of communication and social interaction in FXS, followed by FMRP deficits. This defines an entry point into the complex network of interdependent processes through which autistic development arises and proceeds (Herbert, 2004). FXS offers a unique molecular model for autism since FMRP regulates the translation of many other genes that can influence both IQ and social impairments, perhaps through deficits in synaptic plasticity or other neurobiological mechanisms that may be common in other disorders that cause autism. Therefore those proteins whose mRNAs are bound by FMRP should be natural targets for further exploration.
Fragile X offers yet another opportunity for investigation of the relationships of the higher cognitive abilities that may be linked to a ‘core’ deficit in autism, such as more sophisticated language and social skills, with the level of autistic manifestations. This is because the carriers of the fragile X premutation, as well as at least half of the females with FXS, are not intellectually disabled, but they often manifest some higher order cognitive function deficits (Loesch et al., 2002; Loesch et al., 2003), as well as a range of autism behaviours. By considering a continuous distribution of autistic manifestations, subtle, more primary deficits may be detected in such cases since they are not masked by a widespread developmental disturbance seen in FXS full mutation males. Indeed, Mazzocco et al. (1993) found an association between impairment in executive functioning and autistic behaviours in females with FXS without obvious intellectual impairment, who are therefore testable for these skills. Unfortunately, we were not able to explore these associations in the present study because our sample of high functioning FXS females, and premutation carriers, was not sufficiently large.
Future studies based on a quantitative approach, and involving higher functioning individuals with FXS, as well as individuals with other developmental conditions co-morbid with autism, should also utilize recent developments in imaging techniques (Barnea-Goraly et al., 2003; Herbert et al., 2004), which provide the unique opportunity to correlate the particular circuitries defined at the anatomical or molecular level, with both cognitive and behavioural impairments in different forms of autism.
Acknowledgments
We thank the study participants and their family for their contribution.
Grant sponsor: This study was supported by the National Institute of Child Health and Human Development grant HD36071.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- APA. Diagnostic and Statistical Manual of Mental Disorders. 4. American Psychiatric Association; Washington DC: [Google Scholar]
- Asano E, Chugani DC, Muzik O, Behen M, Janisse J, Rothermel R, Mangner TJ, Chakraborty PK, Chugani HT, Bailey DB, Jr, et al. Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction. Neurology. 2001;57:1269–1277. doi: 10.1212/wnl.57.7.1269. [DOI] [PubMed] [Google Scholar]
- Aziz M, Stathopulu E, Callias M, Taylor C, Turl J, Oostra B, Willemsen R, Patton M. Clinical features of boys with fragile X premutations & intermediate alleles. Am J Med Gen. 2003;121B:119–127. doi: 10.1002/ajmg.b.20030. [DOI] [PubMed] [Google Scholar]
- Bailey A, Bolton P, Butler L, le Couteur A, Murphy M, Scott S, Webb T, Rutter M. Prevalence of the fragile X anomaly amongst autistic twins and singletons. J Child Psychol Psychiatry. 1993;34:673–688. doi: 10.1111/j.1469-7610.1993.tb01064.x. [DOI] [PubMed] [Google Scholar]
- Bailey DB, Jr, Mesibov GB, Hatton DD, Clark RD, Roberts JE, Mayhew L. Autistic behavior in young boys with fragile X syndrome. J Autism Dev Disord. 1998;28:499–508. doi: 10.1023/a:1026048027397. [DOI] [PubMed] [Google Scholar]
- Bailey DB, Jr, Hatton DD, Mesibov GB, Ament N, Skinner M. Early development, temperament and functional impairment in autism and fragile X syndrome. J Autism Dev Disord. 2000;30:49–59. doi: 10.1023/a:1005412111706. [DOI] [PubMed] [Google Scholar]
- Barnea-Goraly N, Eliez S, Hedeus M, Menon V, White CD, Moseley M, Reiss AL. White matter tract alterations in fragile X syndrome: preliminary evidence from diffusion tensor imaging. Am J Med Genet. 2003;118B:81–88. doi: 10.1002/ajmg.b.10035. [DOI] [PubMed] [Google Scholar]
- Baumgardner TL, Reiss AL, Freund LS, Abrams MT. Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics. 1995;95:744–752. [PubMed] [Google Scholar]
- Brown WT, Jenkins EC, Friedman E, Brooks J, Wisniewski K, Raguthu S, French J. Autism is associated with the fragile-X syndrome. J Autism Dev Disord. 1982;12:303–308. doi: 10.1007/BF01531375. [DOI] [PubMed] [Google Scholar]
- Cameron AC, Trivedi PK. Regression Analysis of Count Data. Cambridge University Press; Cambridge: 1988. [Google Scholar]
- Channon S, Crawford S. The effects of anterior lesions on performance on astory comprehension test: left anterior impairment on a theory of mind-type task. Neuropsychologia. 2000;38(7):1006–17. doi: 10.1016/s0028-3932(99)00154-2. [DOI] [PubMed] [Google Scholar]
- Clifford S, Dissanayake C, Bui QM, Huggins RM, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2006 doi: 10.1007/s10803-006-0205-z. In Press. [DOI] [PubMed] [Google Scholar]
- Cohen IL. Behavioral profiles of autistic and non autistic fragile X males. Dev Brain Dysfunct. 1995;8:252–269. [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Townsend J, Saitoh O. The brain in infantile autism: Posterior fossa structures are abnormal. Neurology. 1994;44:214–223. doi: 10.1212/wnl.44.2.214. [DOI] [PubMed] [Google Scholar]
- Demark JL, Feldman MA, Holden JJA. Behavioral relationship between autism and fragile X syndrome. Am J Ment Ret. 2003;108:314–326. doi: 10.1352/0895-8017(2003)108<314:BRBAAF>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- de Vries BB, Wiegers AM, Smits AP, Mohkamsing S, Duivenvoorden HJ, Fryns JP, Curfs LM, Halley DJ, Oostra BA, van den Ouweland AL, et al. Mental status of females with an FMR1 gene full mutation. Am J Med Genet. 1996;58:1025–1032. [PMC free article] [PubMed] [Google Scholar]
- Dykens EM, Volkmar FR. Medical Conditions Associated with Autism. In: Cohen DJ, Volkmar FR, editors. Handbook of Autism and Pervasive Developmental Disorders. 2. John Wiley & Sons Inc; New York: 1997. pp. 388–410. [Google Scholar]
- Faraz F, Perry H, Hessl D, Loesch DZ, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman PJ, Hagerman R. Autism spectrum disorders and attention deficit hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006 doi: 10.1097/00004703-200604002-00012. In Press. [DOI] [PubMed] [Google Scholar]
- Fombonne E, Du Mazaubrun C, Cans C, Grandjean H. Autism and associated medical disorders in a French epidemiological survey. J Am Acad Child Adolesc Psychiatry. 1997;36:1561–1569. doi: 10.1016/S0890-8567(09)66566-7. [DOI] [PubMed] [Google Scholar]
- Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: An update. J Autism Dev Disord. 2003;33(4):365–382. doi: 10.1023/a:1025054610557. [DOI] [PubMed] [Google Scholar]
- Fu YH, Kuhl DP, Pizzuti A, Pieretti A, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RJ, Jr, Warren ST, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- Ghaziuddin M, Butler E. Clumsiness in autism and Asperger syndrome: A further report. J Int Disab Res. 1998;42:43–48. doi: 10.1046/j.1365-2788.1998.00065.x. [DOI] [PubMed] [Google Scholar]
- Gillberg C. Chromosomal disorders and autism. J Autism Dev Disord. 1998;28:415–425. doi: 10.1023/a:1026004505764. [DOI] [PubMed] [Google Scholar]
- Golden CJ, Freshwater SM. Stroop Color and Word Tests. Stoelting Co; Wood Dale: 2002. [Google Scholar]
- Goldstein G, Minshew NJ, Siegel DJ. Age differences in academic achievement in high-functioning autistic individuals. J Clin Exp Neuropsychol. 1994;16(5):671–80. doi: 10.1080/01688639408402680. [DOI] [PubMed] [Google Scholar]
- Goodlin-Jones B, Tassone F, Gane LW, Hagerman RJ. Autistic spectrum disorder and the fragile X premutation. J Dev Behav Pediatr. 2004;25:392–398. doi: 10.1097/00004703-200412000-00002. [DOI] [PubMed] [Google Scholar]
- Grigsby J, Kaye K. Behavioral Dyscontrol Scale Manual. 2. University of Colorado; Denver: 1996. [Google Scholar]
- Hagerman RJ, Jackson AW, Levitas A, Romland B, Braden M. An analysis of autism in fifty males with the fragile X syndrome. Am J Med Genet. 1986;23:359–374. doi: 10.1002/ajmg.1320230128. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ. Physical and Behavioral Phenotype. In: Hagerman RJ, Hagerman PJ, editors. Fragile X Syndrome: Diagnosis, Treatment and Research. 3. The Johns Hopkins University Press; Baltimore: 2002. pp. 3–109. [Google Scholar]
- Happe F. Cognition in autism: one deficit or many? Novartis Found Symp. 2003;251:198–207. [PubMed] [Google Scholar]
- Herbert MR. Neuroimaging in disorders of social and emotional functioning: what is the question? J Child Neurol. 2004;19(10):772–84. doi: 10.1177/08830738040190100701. [DOI] [PubMed] [Google Scholar]
- Herbert MR, Ziegler DA, Makris N, Makris N, Filipek PA, Kemper TL, Normandin JJ, Sanders HA, Kennedy DN, Caviness VS., Jr Localization of white matter volume increase in autism and developmental language disorder. Ann Neurol. 2004;55:530–540. doi: 10.1002/ana.20032. [DOI] [PubMed] [Google Scholar]
- Hessl D, Rivera SM, Reiss AL. The neuroanatomy and neuroendocrinology of fragile X syndrome. Ment Retard Dev Disabil Res Rev. 2004;10:17–24. doi: 10.1002/mrdd.20004. [DOI] [PubMed] [Google Scholar]
- Hosmer DW, Lemeshow S. Applied Logistic Regression. Wiley; New York: 1989. [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Just MA, Cherkassky VL, Keller TA, Minshew NJ. Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain. 2004;127:1811–1821. doi: 10.1093/brain/awh199. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, Cox C, Capone GT, Stanard P. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet. 2004;129A:225–234. doi: 10.1002/ajmg.a.30229. [DOI] [PubMed] [Google Scholar]
- Lachiewicz AM, Spiridigliozzi GA, Gullion CM, Ransford SN, Rao K. Aberrant behaviors of young boys with fragile X syndrome. Am J Ment Retard. 1994;98:567–579. [PubMed] [Google Scholar]
- Lezak MD. Neuropsychological assessment. 3. Oxford University Press; New York: 1995. [Google Scholar]
- Loesch DZ, Hay DA. Clinical features and reproductive patterns in fragile X female heterozygotes. J Med Genet. 1988;25:407–414. doi: 10.1136/jmg.25.6.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch DZ, Huggins RM, Bui QM, Epstein JL, Taylor AK, Hagerman RJ. Effect of the deficits of Fragile X mental retardation protein on cognitive status of fragile x males and females assessed by robust pedigree analysis. J Dev Behav Pediatr. 2002;23:416–423. doi: 10.1097/00004703-200212000-00004. [DOI] [PubMed] [Google Scholar]
- Loesch DZ, Huggins RM, Bui QM, Taylor AK, Pratt C, Epstein J, Hagerman RJ. Effect of the fragile X status categories and FMRP deficits on cognitive profiles estimated by robust pedigree analysis. Am J Med Genet. 2003;122A(1):13–23. doi: 10.1002/ajmg.a.20214. [DOI] [PubMed] [Google Scholar]
- Loesch DZ, Bui QM, Grigsby J, Butler E, Epstein J, Huggins RM, Taylor AK, Hagerman RJ. Effect of the fragile X status categories and the FMRP levels on executive functioning in fragile X males and females. Neuropsychology. 2003;17:546–657. doi: 10.1037/0894-4105.17.4.646. [DOI] [PubMed] [Google Scholar]
- Loesch DZ, Huggins RM, Hagerman RJ. Phenotypic variation and FMRP levels in fragile X. Ment Retard Dev Disabil Res Rev. 2004;10:31–41. doi: 10.1002/mrdd.20006. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: A Revised Version of a Diagnostic Interview for Caregivers of Individuals with Possible Pervasive Developmental Disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, DiLavore PC, Risi S. Autism Diagnostic Observation Schedule. Western Psychological Services; Los Angeles: 1999. [Google Scholar]
- Lord C, Risi S, Lambrecht L, Cook E, Leventhal B, DiLavore P, Pickles A, Rutter M. The Autism Diagnostic Observation Schedule-Generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;30:205–223. [PubMed] [Google Scholar]
- Manjiviona J, Prior M. Comparison of Asperger syndrome and high-functioning autistic children on a test of motor impairment. J Autism Dev Disord. 1995;25:23–39. doi: 10.1007/BF02178165. [DOI] [PubMed] [Google Scholar]
- Mazzocco MM, Pennington BF, Hagerman RJ. The neurocognitive phenotype of female carriers of fragile X: additional evidence for specificity. J Dev Behav Pediatr. 1993;14:328–335. [PubMed] [Google Scholar]
- Mazzocco MM, Kates WR, Baumgardner TL, Freund LS, Freund LS, Reiss AL. Autistic behaviors among girls with fragile X syndrome. J Autism Dev Disord. 1997;27:415–435. doi: 10.1023/a:1025857422026. [DOI] [PubMed] [Google Scholar]
- Miller LJ, McIntosh DN, McGrath J, Shyu V, Lampe M, Taylor AK, Tassone F, Neitzel K, Stackhouse T, Hagerman RJ. Electrodermal Responses to Sensory Stimuli in Individuals with Fragile X Syndrome: A Preliminary Report. Am J Med Genet. 1999;83:268–279. [PubMed] [Google Scholar]
- Mostofsky SH, Mazzocco MM, Aakalu G, Warsofsky IS, Denckla MB, Reiss AL. Decreased cerebellar posterior vermis size in fragile X syndrome: correlation with neurocognitive performance. Neurology. 1998;50:121–130. doi: 10.1212/wnl.50.1.121. [DOI] [PubMed] [Google Scholar]
- Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics. 2004;113:e472–486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- Philofsky A, Hepburn SL, Hayes A, Hagerman R, Rogers SJ. Linguistic and cognitive functioning and autism symptoms in young children with fragile X syndrome. Am J Ment Retard. 2004;109:208–218. doi: 10.1352/0895-8017(2004)109<208:LACFAA>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Freund L. Fragile X syndrome, DSM-III-R, and autism. J Am Acad Child Adolesc Psychiatry. 1990;29:885–891. doi: 10.1097/00004583-199011000-00007. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Aylward E, Freund LS, Joshi PK, Bryan RN. Neuroanatomy of fragile X syndrome: the posterior fossa. Ann Neurol. 1991a;29:26–32. doi: 10.1002/ana.410290107. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Freund L, Tseng JE, Joshi PK. Neuroanatomy in fragile X females: the posterior fossa. Am J Hum Genet. 1991b;49:279–288. [PMC free article] [PubMed] [Google Scholar]
- Rogers SJ, Wehner EA, Hagerman RJ. The behavioral phenotype in Fragile X: Symptoms of autism in very young children with Fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001;22:409–417. doi: 10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- Sabbagh MA. Understanding orbitofrontal contributions to theory-of-mind reasoning: implications for autism. Brain Cogn. 2004;55:209–219. doi: 10.1016/j.bandc.2003.04.002. [DOI] [PubMed] [Google Scholar]
- Saluto A, Brussino A, Tassone F, Pappi P, Arduino C, Hagerman PJ, Migone N, Brusco A. An enhanced PCR assay to detect pre- and full mutation alleles of the FMR1 gene. Journal of Molecular Diagnostics. 2005 doi: 10.1016/S1525-1578(10)60594-6. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Lawrence EJ, Radbourne C, Bramham J, Polkey CE, David AS. The impact of early and late damage to the human amygdala on ‘theory of mind’ reasoning. Brain. 2004;127(Pt 7):1535–48. doi: 10.1093/brain/awh168. [DOI] [PubMed] [Google Scholar]
- Smalley SL. Autism and tuberous sclerosis. J Autism Dev Disord. 1998;28:407–414. doi: 10.1023/a:1026052421693. [DOI] [PubMed] [Google Scholar]
- Sparrow SS, Balla DA, Cicchetti DV. Vineland Adaptive Behavior Scales Survey Form Manual. American Guidance Service; Circle Pines: 1984. [Google Scholar]
- Spreen O, Benton AL. Neurosensory Center Comprehensive Examination for Aphasia. University of Victoria Neuropsychology Laboratory; Victoria: 1977. [Google Scholar]
- Sudhalter V, Cohen IL, Silverman W, Wolf-Schein EG. Conversational analyses of males with fragile X, Down syndrome, and autism: comparison of the emergence of deviant language. Am J Ment Retard. 1990;94:431–441. [PubMed] [Google Scholar]
- Tager-Flusberg H, Boshart J, Baron-Cohen S. Reading the windows to the soul: evidence of domain-specific sparing in Williams syndrome. J Cogn Neurosci. 1998;10(5):631–9. doi: 10.1162/089892998563031. [DOI] [PubMed] [Google Scholar]
- Tager-Flusberg H, Sullivan K. A componential view of theory of mind: evidence from Williams syndrome. Cognition. 2000;76(1):59–90. doi: 10.1016/s0010-0277(00)00069-x. [DOI] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Garcia-Arocena D, Khandjian EW, Greco CM, Hagerman PJ. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet. 2004:41–43. doi: 10.1136/jmg.2003.012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Hagerman RJ, Iklé DN, Dyer PN, Lampe M, Willemsen R, Oostra BA, Taylor AK. FMRP expression as a potential prognostic indicator in fragile X syndrome. Am J Med Genet. 1999;84:250–261. [PubMed] [Google Scholar]
- Taylor AK, Safanda JF, Fall MZ, Quince C, Lang KA, Hull CE, Carpenter I, Staley LW, Hagerman RJ. Molecular predictors of cognitive involvement in female carriers of fragile X syndrome. JAMA. 1994;271:507–514. [PubMed] [Google Scholar]
- Turk J, Graham P. Fragile X syndrome, autism, and autistic features. Autism. 1997;1:175–197. [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu Yh, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Greenough WT. Synaptic synthesis of the Fragile X protein: possible involvement in synapse maturation and elimination. Am J Med Genet. 1999;83:248–252. doi: 10.1002/(sici)1096-8628(19990402)83:4<248::aid-ajmg3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Preschool and Primary Scale of Intelligence. The Psychological Corporation; San Antonio: 2002. [Google Scholar]
- Wechsler D. Administration and Scoring Manual. The Psychological Corporation; San Antonio: 1997. Wechsler Adult Intelligence Scale-Third Edition. [Google Scholar]
- Wechsler D. Wechsler Intelligence Scale for Children, Third Edition (WISC-III) The Psychological Corporation; San Antonio: 1991. [Google Scholar]
- Willemsen R, Mohkamsing S, de Vries B, van den Ouweland AM, Mandel JL, Galjaard H, Oostra BA. Rapid antibody test for fragile X syndrome. Lancet. 1995;345:1147–1148. doi: 10.1016/s0140-6736(95)90979-6. [DOI] [PubMed] [Google Scholar]