

Abstract
Glial growth factor 2 (GGF2) is a neuronal signal that promotes the proliferation and survival of the oligodendrocyte, the myelinating cell of the central nervous system (CNS). The present study examined whether recombinant human GGF2 (rhGGF2) could effect clinical recovery and repair to damaged myelin in chronic relapsing experimental autoimmune encephalomyelitis (EAE) in the mouse, a major animal model for the human demyelinating disease, multiple sclerosis. Mice with EAE were treated with rhGGF2 during both the acute and relapsing phases. Clinically, GGF2 treatment delayed signs, decreased severity, and resulted in statistically significant reductions in relapse rate. rhGGF2-treated groups displayed CNS lesions with more remyelination than in controls. This correlated with increased mRNA expression of myelin basic protein exon 2, a marker for remyelination, and with an increase in the CNS of the regulatory cytokine, interleukin 10, at both the RNA and protein levels. Thus, a beneficial effect of a neurotrophic growth factor has been demonstrated on the clinical, pathologic, and molecular manifestations of autoimmune demyelination, an effect that was associated with increased expression of a T helper 2 cytokine. rhGGF2 treatment may represent a novel approach to the treatment of multiple sclerosis.
Most therapeutic strategies in the human demyelinating disease, multiple sclerosis (MS), have been based on modulating the immune response, and the animal model of MS, experimental autoimmune encephalomyelitis (EAE), has been particularly useful for testing potential therapeutic agents (1, 2). EAE, a CD4+ T helper 1 (Th1) T cell-mediated disease of the central nervous system (CNS), involves autosensitization to myelin antigens and in the SJL mouse is a chronic relapsing neurologic disease with inflammatory demyelinated CNS lesions highly reminiscent of MS (3–5). Relatively unexplored in demyelinating conditions has been the therapeutic potential of neurotrophic factors with known regulatory effects on the myelinating cell. Prominent among these factors are members of the neuregulin family of soluble and transmembrane proteins belonging to the epidermal growth factor superfamily (6). Recombinant human glial growth factor 2 (rhGGF2) is a secreted isoform of neuregulin with documented stimulatory effects on oligodendrocytes and Schwann cells (7–10). Because oligodendrocytes are a major target in the MS lesion and because remyelination occurs in MS and EAE (3, 4), it was hypothesized that administration of rhGGF2 to animals with EAE might ameliorate the disease. This report presents the findings from a large series of experiments in which rhGGF2 was given to mice at different stages of adoptively transferred chronic relapsing EAE. The results have shown that not only does rhGGF2 have marked beneficial effects at the acute and relapsing phases of the disease but it is also associated with structural and molecular evidence for enhanced remyelination of CNS lesions in long-term rhGGF2-treated animals.
MATERIALS AND METHODS
Induction of EAE.
Female SJL/J mice (The Jackson Laboratory), 5–12 weeks of age, were maintained in an National Institutes of Health/American Association for the Accreditation of Laboratory Animal Care-approved facility. Donor mice were immunized with 0.4 mg of bovine myelin basic protein (MBP) (Sigma) in incomplete Freund’s adjuvant containing 60 μg of Mycobacterium tuberculosis H37Ra (Difco). Lymph nodes were removed 10 days later, and cells were cultured in the presence of 50 μg of MBP for 3 days, after which 3 × 107 lymph node cells were injected into the tail vein of naive syngeneic recipients. Onset of signs occurred 6–10 days posttransfer (dpt). Animals were graded as follows: 1, floppy tail; 2, hindlimb weakness; 3, one limb plegic; 4, plegia of 2 limbs; and 5, moribund or death.
Treatment with rhGGF2.
Groups of mice were treated with rhGGF2 in a blinded fashion under code at doses ranging from 0.02 to 2 mg/kg (see Baseline Studies), at various timepoints postsensitization in 11 experiments, both before acute onset and during the chronic phase. Controls received injections of formulation buffer (vehicle) containing 20 mM NaOAc, 100 mM arginine, 1% mannitol, and 100 mM Na2SO4, pH 6.5. In experiment 1, the subcutaneous (s.c.) route of administration was found to be more effective than the i.v. route (see Table 1). Subsequent experiments used s.c. injections. For experiments that dealt with acute phase treatment only, injections were given daily. In all others, animals were treated on alternate days. To examine for systemic effects, 7 normal mice were given 10 s.c. injections of rhGGF2 (2.0 mg/kg) and 7 were given vehicle.
Table 1.
CNS pathology of mice treated with GGF2 via the s.c. or i.v. route
| Treatment | Pathology | Clinical score*
|
||
|---|---|---|---|---|
| 8 dpt | 19 dpt | 42 dpt | ||
| Vehicle | Inflamm. | 3.4* | 2.5 | 2.3 |
| Demyel. | 2.6 | 2.6 | 1.9 | |
| 2.0 mg GGF2 s.c. | Inflamm. | 1.1 | 2.2 | 1.8 |
| Demyel. | 0.4 | 1.6 | 0.7 | |
| 2 mg GGF2 i.v. | Inflamm. | 2.2 | 2.4 | † |
| Demyel. | 1.0 | 1.7 | † | |
| 1 mg GGF2 i.v. | Inflamm. | 3.0 | 2.3 | 2.4 |
| Demyel. | 2.2 | 1.9 | 2.2 | |
Animals scored on scale of 0–5. Each score represents the mean of 18 slides per animal from 9 levels of the neuraxis.
No survivors in this i.v. group. Lower dosing via the i.v. route and s.c. administration at all dose levels tested did not result in any deaths.
Neuropathology.
At selected timepoints during and after treatment, representative mice from each group were taken. A total of 75 mice (6 acute, 69 chronic) from 11 experiments were sampled for analysis. From two to five timepoints were examined from each experiment. In addition, 8 of the 14 normal mice given 10 injections of rhGGF2 (2.0 mg/kg) or vehicle, were sampled to assess for possible effects of rhGGF2 on the normal CNS. All animals sampled for neuropathology were perfused with 2.5% glutaraldehyde in PO4 buffer. CNS tissue was removed and thin slices from 10 levels of the neuraxis were postfixed in cold 1% OsO4 for 1 h, dehydrated, and embedded in epoxy resin. One micron epoxy sections stained with toluidine blue were examined in a blinded fashion by light microscopy by two investigators. Based on the degree of CNS involvement, a score of 0–5 was determined for inflammation, demyelination, remyelination, and Wallerian degeneration, according to established criteria (3). For electron microscopy, thin sections were placed on copper grids, stained, and examined in a Siemens 101.
Immunocytochemistry.
Representative mice (n = 43) from 7 of the 11 experiments were perfused with PBS, the CNS was removed, and slices from cerebral hemispheres, cerebellum, and cervical, thoracic, and lumbar spinal cord were embedded in OCT. Frozen sections were fixed in acetone for 10 min and stained using the avidin-biotin-peroxidase complex technique (Vector Laboratories). Overnight incubations at 4°C were performed with the following antibodies: goat anti-mouse tumor necrosis factor α (TNFα) (Santa Cruz Biotechnology), at 1:100 dilution; rat anti-mouse interferon γ (PharMingen), at 1:100; rat anti-mouse IL-4 (PharMingen), at 1:200; rat anti-mouse IL-10 (PharMingen), at 1:100; rat anti-mouse F4/80 (Serotec) at 1:10; and monoclonal anti-cyclic nucleotide phospho-hydrolase (Sigma), at 1:20. Slides were scored in a blinded fashion by two observers on a scale from 0 to 4 based on the density of labeled cells.
In Situ Hybridization.
Synthetic deoxyribonucleotide probes (48-mers from Oligo Therapeutics, Wilsonville, OR), were designed as described (11) using the murine antisense sequence (5′-CTTGTACATGTGGCACAGCCCAGGACGGCTGCGGGCATGAGAGGGCAG) to detect MBP exon 2 mRNA expression. The murine exon 2 sense sequence (5′-CTGCCCTCTCATGCCCGCAGCCGTCCTGGGCTGTGCCACATGTACAAG) was used as control. Frozen sections of CNS were taken from a total of 11 EAE-sensitized animals (7 treated; 4 vehicle), sampled up to 117 dpt. For control purposes, the CNS of 4 normal adult mice (2 vehicle- and 2 rhGGF2-treated) and four 15-day-old pups were studied. Sections were fixed in 4% paraformaldehyde for 20 min, washed in PBS, diethyl pyrocarbonate (DEPC)-treated double-distilled water, and triethanolamine. After dehydration and air-drying, they were prehybridized and hybridized according to Jordan et al. (12). Oligonucleotide probes were labeled at their 3′ ends with [α-[35S]thio]-dATP by terminal deoxynucleotidyltransferase (Boehringer Mannheim), to a specific activity of 1 × 109 cpm/μg and used at a concentration of 3 × 107 cpm/ml. Slides were dipped in 1:1 Kodak NTB2 emulsion/water at 45°C and exposed at 4°C for 14 days. After processing, slides were counterstained with cresyl violet.
PCR Analysis of Cytokine RNA.
Representative mice (n = 6) from treated and control groups were sampled by a blinded investigator at 11, 20, and 63 dpt, which represented the acute, remission, and chronic phases of the disease, respectively. To maintain the integrity of the study, only one animal from each group was sacrificed at each timepoint for these analyses. Lumbar spinal cord tissue from each animal was removed after perfusion with PBS and was frozen and embedded in OCT. Thirty adjacent 20-μm sections were pooled from each block (representing 0.6 mm of spinal cord) and total RNA was prepared by using a Purescript kit (Gentra Systems). Total RNA (1 μg) was reverse-transcribed and cDNA amplified [reverse transcription (RT)–PCR kit; Perkin–Elmer] by using primer pairs specific for IL-10 (CLONTECH no. 5494–1), TNFα (CLONTECH no. 5468–1), or a housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (5′-GTGAAGGTCGGAGTCAACG, upstream; 5′-GAGATGATGACCCTTTTGGC, downstream). DNA amplification products were run on 8% polyacrylamide gels (NOVEX, San Diego) along with DNA markers. Gels were then stained with ethidium bromide and the products were visualized by transillumination.
Relapse Rate Tabulation and Statistical Analysis.
The cumulative relapse index was tabulated as the total number of relapses in each group divided by the number of relapsing and remitting animals in that group. An event was scored as a relapse when it showed an increase of at least one full point of the clinical score that was maintained for at least two observations occurring after 21 dpt until the end of the study. The percent reduction of relapse index was calculated by [100% − (cumulative relapse index of rhGGF2-treated animals/control animals) × 100]. Statistical significance of reduction in relapse rate compared with vehicle-treated animals was done using the Mann–Whitney U test (P < 0.05).
RESULTS
Baseline Studies.
For treatment with rhGGF2, an initial starting dose of 2 mg/kg was tested, a dose based on data obtained from a rat sciatic nerve crush model in which regenerative efficacy was seen in a bioassay (11). Low doses used were arrived at from data generated from in vitro experiments that showed low doses of GGF2 to be mitogenic for oligodendrocytes (8). Initial studies on mice sensitized for EAE showed that s.c. injection of rhGGF2 was more effective than i.v. (Table 1). The s.c. route also gave a longer half-life of rhGGF2 in plasma (15 h by s.c.; 1.1 h by i.v., as tested in the rat), which worked effectively when repeated dosing was required. Also, for control purposes, groups of normal mice were given injections of rhGGF2 (2.0 mg/kg) or vehicle over a 20-day period to examine for systemic effects and changes in expression of exon 2 MBP, a marker of early myelination and remyelination (see below). No clinical effects were observed and the CNS revealed no changes. As a positive control for exon 2 MBP mRNA expression, CNS tissue from 4 normal mice sampled at 15 days of age (i.e., during myelination), were examined by in situ hybridization.
rhGGF2 Delays Acute EAE in a Dose-Dependent Manner and Reduces Disease Severity.
rhGGF2 was examined for its effect on the induction of EAE when administered at three doses (0.2, 0.6, and 2.0 mg/kg) vs. vehicle, by daily (days 1–10) s.c. administration (Fig. 1a). The results showed a dose-dependent decrease in severity of clinical signs that peaked between 17 and 20 dpt in the rhGGF2 group, vs. 10–11 dpt in controls. In addition, delay in clinical onset was dose-dependent, with the highest dose (2.0 mg/kg) effecting the greatest delay. The mean peak clinical score for all rhGGF2 groups was ≈50% lower than that of the vehicle-treated group. Analysis of the CNS of controls sampled at 11 dpt revealed lesions typical of acute EAE that comprised widespread inflammation and demyelination and in some animals, Wallerian degeneration (Fig. 2a). In contrast, the CNS of rhGGF2-treated animals sampled at corresponding timepoints displayed markedly reduced lesion activity (Fig. 2b; Table 1).
Figure 1.

Effect of treatment on clinical course of EAE in SJL/J mice. (a) Mice treated daily by s.c. injections days 1–10 posttransfer (dpt) with vehicle and 0.2, 0.6, or 2.0 mg/kg rhGGF2. Mean clinical score plotted for each group (n = 6). Differences between 2.0 mg/kg rhGGF2 and vehicle groups significant (P < 0.001). (b) Same animals as in a retreated on alternate days in the chronic phase of EAE from 33–53 dpt as follows: vehicle group retreated with vehicle; 2.0 mg/kg rhGGF2 group, with 0.02 mg/kg rhGGF2. Mean clinical score observed for each group (n = 3) until 67 dpt. Differences significant (P < 0.001). (c) Animals (n = 6) treated on alternate days from 21 to 79 dpt, using vehicle, 0.02 or 0.2 mg/kg. Observations conducted until 116 dpt. Mean clinical score plotted for each group (n = 6). (d) The cumulative relapse index of animals shown (c) that displayed a relapsing and remitting pattern of disease (n = 5, vehicle; n = 6, 0.02 mg/kg; and n = 6, 0.2 mg/kg). The cumulative relapse index for each group after 116 dpt was 1.40 (vehicle), 0.33 (0.02 mg/kg) and 0.66 (0.2 mg/kg). The reduction in relapse rate for each rhGGF2-treated compared with vehicle-treated group (100%) was 76% (0.02 mg/kg) and 53% (0.2 mg/kg). (P < 0.05, Mann–Whitney U test). (e) Treatment from 9 dpt, the peak of acute clinical signs, until 40 dpt on alternate days using vehicle, 0.02 or 1.0 mg/kg rhGGF2. Mean clinical score plotted for each group (n = 13 at 0 dpt) until end of observation period (n = 6, at day 66, and n = 4, at day 81). (f) Relapse index tabulated for animals in (e) that displayed a relapsing and remitting pattern (n = 10) observed from 21- 81 dpt. The cumulative relapse index for each group of relapsing-remitting animals at 81 dpt was 1.1 (vehicle), 0.50 (0.02 mg/kg) and 0.55 (1.0 mg/kg). The reduction in relapse rate for each rhGGF2 compared with vehicle-treated group (100%) was 58% (0.02 mg/kg) and 54% (1.0 mg/kg). Difference in cumulative relapse index between vehicle and each rhGGF2 treatment group significant (P < 0.05, Mann–Whitney U test).
Figure 2.

Histopathology of spinal cord vehicle- and rhGGF2-treated mice; 1 μm epoxy sections; toluidine blue stain. (Bar = 10 μm.) (a) Acute EAE. Experiment 3: Vehicle treatment 1–10 dpt; sampled 11 dpt; clinical grade 4. An area of subpial spinal cord displays extensive inflammation. Note the many myelin-laden macrophages and scattered demyelinated axons (arrows). (×875.) (b) Acute EAE. Matching level of spinal cord to a, treated with rhGGF2 (2 mg/kg) 1–10 dpt; sampled 11 dpt; clinical grade 0.5. A matching area of spinal cord displays no lesion activity. Oligodendrocytes difficult to discern. (×875.) (c) Chronic relapsing EAE. Experiment 4: Vehicle 31–55 dpt; sampled 60 dpt; clinical grade 3. A chronic gliotic lesion contains macrophages and displays many demyelinated axons (arrows). Oligodendrocytes difficult to discern. (×875.) (d) Chronic EAE. rhGGF2 (2.0 mg/kg) 31–55 dpt; sampled 60 dpt; clinical grade 3.5; spinal cord. Despite the comparable clinical grade of this matching animal to that shown in (c), note the large amount of CNS remyelination (large arrows). Some demyelinated axons (small arrows) lie toward the subpial surface. Numerous oligodendrocytes (∗) identified by their rounded nuclei and clumped heterochromatin, can be seen. (×875.) (e) Chronic relapsing EAE. Experiment 7: Vehicle 21–79 dpt; sampled day 81; clinical grade 3. A subpial lesion from the L7 spinal cord level displays intense fibrous astrogliosis, fibrotic blood vessels, and numerous demyelinated axons (arrows), but no obvious oligodendrocytes. (×875.) (f) Chronic relapsing EAE. rhGGF2 (0.2 mg/kg and 0.02 mg/kg) days 21–79; sampled day 81; clinical grade 2. Matching animal to e, same level of spinal cord. Note the more compact, less gliotic parenchyma and presence of remyelinated CNS fibers (arrows). A few oligodendrocytes (∗) are present. (×875.)
rhGGF2 Reduces Clinical Severity in Mice Retreated During the Chronic Phase of EAE.
Animals that received rhGGF2 during the acute phase (1–10 dpt) Fig. 1a, were retreated at a lower concentration from 33–53 dpt, during the chronic phase. The 2.0 mg/kg group was retreated with a hundredfold lower dose (0.02 mg/kg) and these animals maintained a lower clinical score than controls (P < 0.001) (Fig. 1b). In a separate experiment, animals with severe EAE (grade 3.5) were treated with rhGGF2 (or vehicle) from 31–55 dpt and were retreated from 75–103 dpt (data not shown). These animals displayed no differences in clinical course. This suggested that treatment had commenced at a timepoint too late to have any effect on the clinical score or that the acute phase was so severe that clinical signs were irreversible. Nevertheless, analysis of tissue samples from representative animals from each group of rhGGF2-treated mice showed evidence of enhanced remyelination (see Fig. 2 c–f).
rhGGF2 Treatment During the Chronic Phase of EAE Reduces Relapse Rate.
rhGGF2 treatment by s.c. administration (three times per week) was initiated in the chronic phase of disease at 21 dpt and continued through 79 dpt. Doses included 0.02 and 0.2 mg/kg. Controls received vehicle only. The experiment was terminated after 116 dpt. Under these conditions, animals receiving either 0.02 or 0.2 mg/kg showed reduction in mean clinical score throughout and following the period of treatment (Fig. 1c). Both rhGGF2-treated groups had a similar significant decrease in mean clinical score from 25–75 dpt (2.17 ± 0.14; 2.11 ± 0.19) when compared with control groups (2.51 ± 0.27) - P < 0.01. rhGGF2 treatment at both doses elicited a decrease in the number of relapses when compared with the vehicle-treated group (P < 0.05; Fig. 1d). The effect on relapse number was similar with both doses of rhGGF2. Reductions in number of relapses were maintained for up to 36 days after cessation of rhGGF2 treatment, until termination of the experiment.
rhGGF2 Treatment at Peak of EAE Reduces Relapses but Has Little Effect on Mean Clinical Score.
In one experiment, rhGGF2 treatment was commenced at the peak of acute clinical signs (9 dpt) and was continued until 40 dpt. In this instance, although there were no significant differences in mean clinical score over 81 days of observation (Fig. 1e), there was a significant decrease in the relapse rate in rhGGF2-treated animals (Fig. 1f). The cumulative relapse index of the vehicle-treated group was 1.1 compared with 0.5 and 0.55 for the 0.02 mg/kg and 1.0 mg/kg rhGGF2-treated groups, respectively. This represented a 58% and 54% reduction in relapse rate relative to the control group (P < 0.05, Mann–Whitney U test).
rhGGF2 Treatment Enhances CNS Remyelination.
Histopathologic analysis was made of CNS tissue prepared from a total of 69 EAE mice with clinical scores representative of each treatment group. Samples were taken at several timepoints from each treatment regimen, e.g., at day 60 after treatment from 31–55 dpt; day 53 after treatment from 1–10 dpt and from 33–53 dpt; and on day 64 after treatment from 1–40 dpt. Histopathology was read in a blinded manner and revealed in six separate experiments, a clear-cut enhancement of CNS remyelination in the rhGGF2 treatment groups. Evidence of remyelination was detectable at a very low level in vehicle-treated animals but was most apparent in optic nerve and lower spinal cord of rhGGF2-treated mice (Figs. 2 c–-f; 3 a and b). Remyelination consisted of large collections of disproportionately thinly myelinated axons in affected areas (Fig. 2 d and f). This appearance differed from that seen in controls that showed widespread demyelination, macrophage activity, Wallerian degeneration and fibrous astrogliosis (Fig. 2 c and e). CNS remyelination was most prominent in animals treated during the chronic phase of the disease, i.e., between 31–55 dpt, 21–79 dpt (Figs. 2 d and f), and 47–56 dpt and 1–40 dpt (data not shown). CNS remyelination was present in tissue from animals treated with all doses of rhGGF2 tested (0.02, 0.2, 0.8, and 2.0 mg/kg), and when compared with controls (n = 5 and n = 4, respectively), the enhanced remyelination was statistically significant (1.05 ± 0.26 vs. 0.5 ± 0.14; P < 0.007, Student’s t test). Ultrastructurally, increased remyelination was most apparent in optic nerve and spinal cord (Figs. 3 a and b).
Figure 3.

Ultrastructure of CNS of vehicle- and rhGGF2- treated mice. (Bar = 1 μm.) (a) L7 spinal cord, vehicle, 31–55 dpt; sampled 60 dpt; clinical grade 3, same animal as Fig. 2c. Note extensive fibrous astrogliosis, Wallerian degeneration, myelin debris, and nerve fiber loss. Meningeal surface (m) above. (×6,650.) (b) Matching level of spinal cord to a, rhGGF2 (2.0 mg/kg) 31–55 dpt; sampled 60 dpt; clinical grade 3.5, same animal as Fig. 2d. Thinly remyelinated CNS fibers surround an oligodendrocyte. (×6,650.)
rhGGF2-Treatment Affects Th2-Type Cytokine Immunoreactivity in CNS Tissue.
A panel of Th1- and Th2-type cytokines (TNFα and interferon γ; IL-4 and IL-10, respectively), known from previous studies to be involved in EAE (2), was examined by immunocytochemistry for expression within the CNS from treated and control groups at various stages of chronic relapsing EAE. Of these, only IL-4 and IL-10 levels changed with rhGGF2 treatment. In animals treated from 1–10 dpt, increased immunoreactivity for IL-4 was seen whereas IL-10 levels were higher in the vehicle-treated group. However, at more chronic timepoints, IL-4 levels in both groups were comparable whereas IL-10 was consistently higher in GGF2-treated animals (Figs. 4 a and b). These findings correlated with PCR data for cytokine RNA expression (see below). Microglial cell reactivity was greatly increased in controls, whereas rhGGF2-treated mice showed only background levels of F4/80 staining (data not shown).
Figure 4.
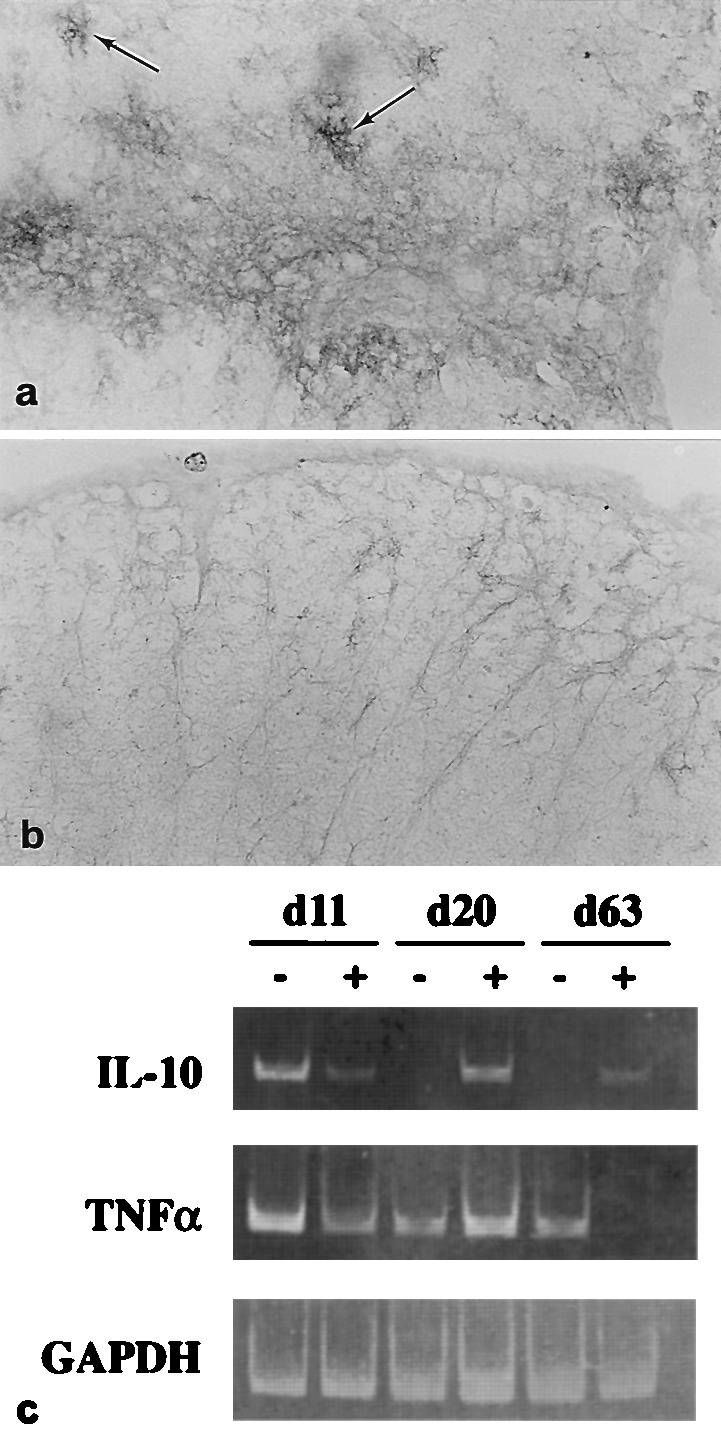
Th2-type cytokine expression and GGF2-treatment. (a) Lumbar spinal cord; rhGGF2-mouse (1.0 mg/kg; 9–40 dpt), sampled 42 dpt shows high level immunoreactivity for IL-10 on perivascular and parenchymal astrocytes (arrows) bordering a penetrating blood vessel. (×250.) (b) A comparable area of lumbar spinal cord from a control mouse at 42 dpt displays low level reactivity for IL-10. (×250.) (c) reverse transcription–PCR analysis of CNS tissue. Representative mice (n = 6) from acute (11 dpt), remission (20 dpt) and chronic (63 dpt) phases of EAE, from vehicle (−) and rhGGF2-treated (+) groups for analysis of cytokine gene expression. At 11 and 20 dpt, the rhGGF2-treated animals were from the 2.0 mg/kg group, whereas at 63 dpt, animals from the 0.8 mg/kg group were selected. RNA from spinal cord was analyzed by reverse transcription–PCR using specific primers for IL-10, TNFα, or glyceraldehyde-3-phosphate dehydrogenase. DNA amplification products analyzed by gel electrophoresis were 455 bp, 354 bp, and 356 bp, respectively. Note increased levels of IL-10 RNA in rhGGF2-treated animals at days 20 and 63 dpt, and the marked decrease of TNFα at 63 dpt in the same group.
rhGGF2-Treatment Is Associated with RNA Up-Regulation of a Th2 Cytokine.
By reverse transcription–PCR, IL-10 was detected in the spinal cord of both vehicle- and rhGGF2-treated animals at day 11, at the peak of acute EAE (Fig. 4c). However, only rhGGF2-treated animals retained detectable levels of IL-10 sequences during remission from the acute phase and during the chronic phase (at 20 and 63 dpt). A noticeable effect on TNFα was apparent only at 63 dpt, where levels seemed to decline dramatically in the rhGGF2-treated animal as compared with the vehicle-treated animal.
rhGGF2 Treatment Correlates with Enhanced Expression of Exon 2 MBP.
Forms of MBP gene containing exon 2 are selectively expressed during development (12, 13) and in baseline studies, CNS sections from normal 15-day-old mice (n = 4) showed high levels of transcript. More recently, expression of exon 2 MBP isoforms has been linked to CNS remyelination in EAE and MS (14, 15). We analyzed exon 2 MBP by in situ hybridization on frozen sections of spinal cord tissue from two normal adult mice treated with vehicle (10 injections over 20 days), two normals similarly treated with rhGGF2 (2.0 mg), 7 rhGGF2-treated and 4 vehicle-treated mice with EAE sampled at various timepoints after treatment up to 117 dpt. All 4 normal treated mice showed no exon 2 MBP mRNA. CNS tissue sections from vehicle-treated mice with EAE displayed low levels of exon 2 MBP transcript whereas sections from rhGGF2-treated EAE mice revealed higher levels over large areas of white matter corresponding to regions most prone to lesion development and remyelination (Figs. 5 a–d). The sense probe was used as the negative control and showed no reactivity.
Figure 5.

In situ hybridization for MBP exon 2 message.(a) Bright field microscopy shows punctate lesion activity in a vehicle-treated mouse sensitized for EAE. Vehicle; 1–40 dpt; sampled 64 dpt; clinical grade 2.5. No counterstain. (×20.) (b) Same section as in a, in situ hybridization, developed for anti-sense MBP exon 2 message. Background levels of MBP exon 2 mRNA seen throughout the spinal cord with some elevation over dorsal roots (above). (×20.) (c) Matching animal to a treated with rhGGF2 (0.8 mg/kg) 0–40 dpt; sampled 64 dpt; clinical grade 3.5. No obvious lesions apparent other than some inflammation over the spinal cord. No counterstain (×20.) (d) Same section as in c, in situ hybridization, developed for MBP exon 2 message. Note enhanced signal for MBP exon 2 over anterior and lateral columns, probably indicative of diffuse CNS remyelination. Some elevation of signal occurs over the dorsal roots and meninges. (×20.)
DISCUSSION
Current approaches to the management of MS involve attenuation of the immune response or treatment of symptoms. The administration of a growth factor that could potentially promote remyelination of axons and resolution of lesions is a promising alternate strategy for the treatment of MS. A similar approach was used previously with IGF-1 in an actively-induced, non-relapsing monophasic acute model of EAE in the rat, which did not demonstrate remyelination (16, 17). The present study tested rhGGF2 in a chronic relapsing model for MS, adoptively transferred EAE in the mouse, a model that displays an acute phase of clinical signs peaking at 7–11 dpt and subsequently, a pattern of relapsing-remitting signs in the chronic phase. Eleven separate experiments were conducted in which the time and dose parameters were varied to examine the effect of rhGGF2 on various phases of disease to simulate the clinical presentation of MS, a feat not possible in acute models. In comparison to control groups, rhGGF2 treatment (either as single or double regimens), consistently resulted in an improved clinical course, a reduction in relapse rate, lessened CNS pathology, and an increase in the amount of CNS remyelination. The degree of remyelination was statistically significant and correlated with an increase in exon 2 MBP transcript.
The administration of rhGGF2 during the acute phase of disease from 1–10 dpt, before appearance of clinical signs, resulted in a dose-dependent decrease in both onset and severity of disease. Retreatment during the chronic phase with lower doses of rhGGF2 than those used in the acute phase, showed that a 100 fold lower dose maintained the suppression of the severity of clinical signs. When rhGGF2 treatment (0.02 or 0.2 mg/kg) was initiated from 21–79 dpt, during the chronic phase, there was a significant decrease in the average mean clinical score during that period, as well as a statistically significant reduction in relapse rate when compared with control animals.
The analysis of chronic relapsing EAE over such protracted periods (≈100 days) is made difficult by the asynchronous pattern and unpredictability of relapse events. This asynchrony can obscure the effect of treatment on mean clinical score. This is similar to the situation in MS and as a result, analysis of data from drug trials relies on relapse rate as the primary outcome measure. This parameter was particularly revealing in the present study where analysis of treatment groups clearly showed a dramatic reduction in relapse rate in the chronic phase of EAE after rhGGF2 treatment (Figs. 1 d and f), even when comparisons of mean clinical scores showed little difference between groups (Fig. 1e). Treatment initiated at the peak of clinical signs in the acute phase had little effect on mean clinical score, yet there was a significant reduction in relapse rate by using either 0.2 or 1.0 mg/kg. A dose-dependent effect on relapse rate was not detected. Therefore, rhGGF2 treatment of chronic EAE led to an improved clinical course, fewer relapses, and increased remyelination of demyelinated lesions.
Although both phases of disease were scored for clinical signs of paralysis, the neurologic impairment may have been caused by different mechanisms. In the acute phase, inflammation and edema were prominent whereas in the chronic phase, there was evidence of multiple demyelinated severely gliotic CNS lesions—see Fig. 2. rhGGF2 administration during the initial phase of disease attenuated and delayed the clinical severity of acute disease as well as inflammation, demyelination and eventually Wallerian degeneration, in comparison to controls. The down-regulatory effect of rhGGF2 on axonal degeneration was particularly striking during treatment of acute EAE and may be relevant to the treatment of active MS lesions in which axonal disease has recently been re-emphasized (18). Thus, the effect of rhGGF2 on the acute phase of disease was rapid and correlated with decreased microglial immunoreactivity. Although the effect of rhGGF2 on the immune system is unknown, the observation of a prolonged increase in expression of IL-10 seen by reverse transcription–PCR and an elevation of IL-10 and IL-4 immunoreactivity in treated mice, may invoke immunoregulatory mechanisms. There is currently little information on Th2-type cytokine expression in vivo in EAE although our findings are in accord with others (19) where an increase in IL-10 mRNA correlated with clinical remission. Whether the dramatic difference in TNFα RNA levels in long-term (63 dpt) animals and the elevation of IL-10 in rhGGF2-treated mice represented a Th1–Th2 shift, remains to be proven with greater numbers of animals. Nevertheless, the observed effect of rhGGF2 on the acute phase when administration was commenced on 1 dpt, was quite remarkable. In these animals, disease onset was clearly delayed (see Fig. 1a), and moreover, the resultant disease was less severe than control groups and did not progress. These findings suggest that rhGGF2 exerted a direct effect on either the induction or effector phase of the disease through a reduction in cell activation (lymphocytic or microglial) or modulation of cell traffic at the blood-brain barrier.
In the chronic phase of EAE, enhanced CNS remyelination in rhGGF2-treated animals was observed and may be the result of growth factor interactions. Neuregulin receptors are present on oligodendrocytes in vitro and treatment of precursor cells with rhGGF2 supports their survival in serum-free media, whereas treatment of more mature oligodendrocytes leads to a dedifferentiated state (8, 9). These activities may underlie the beneficial effect of rhGGF2 observed in the present studies on mice with EAE. For example, some oligodendrocytes probably survive the autoimmune attack and these mature survivors may have the potential to proliferate and remyelinate neighboring demyelinated axons, as is believed to be the case in MS (3, 4). Dedifferentiation and proliferation or maturation would be required to repopulate the lesioned area, followed by re-expression of myelin genes (15). These actions of rhGGF2 on oligodendrocytes would require access of the factor to the CNS, perhaps via a compromised blood-brain barrier, a known feature of EAE and MS (20, 21). Similar mechanisms have been proposed in other models showing enhanced remyelination after treatment e.g., guinea pig EAE treated with MBP/GalC mixtures (22), and after treatment with Igs in Theiler’s virus encephalomyelitis (23), and precursor oligodendrocytes have recently been detected in chronic MS lesions (24).
In terms of relevance to MS, the most significant finding of this study lies in our ability with rhGGF2 treatment to ameliorate both clinically and structurally animals with a chronic relapsing demyelinating condition. In this regard, rhGGF2 administered during the chronic phase of established disease resulted in a reduction in relapse rate that correlated with remyelination. Because relapse rate has traditionally been a primary endpoint in most clinical trials in MS, these data indicate that rhGGF2 might represent a useful addition to current available therapies, either alone or in combination with immunomodulatory agents. To what extent the above findings hold potential for the MS patient remains to be shown.
Acknowledgments
This work was supported in part by Health and Human Services Grants NS 08952, NS 11920, and NS 07098; the Sol Goldman Charitable Trust (NMSS RG 1001-I-9); a grant from Cambridge NeuroScience, Inc., and the Wollowick Family Foundation.
ABBREVIATIONS
- Th1 and Th2
T helper 1 and 2
- GGF2
glial growth factor
- rhGGF2
recombinant human GGF2, MBP, myelin basic protein
- MS
multiple sclerosis
- EAE
experimental autoimmune encephalomyelitis
- CNS
central nervous system
- dpt
days posttransfer
- IL
interleukin
- RT
reverse transcription
References
- 1.Goodkin D E. In: Multiple Sclerosis Clinical and Pathogenetic Basis. Raine C S, McFarland H F, Tourtellotte W W, editors. London: Chapman & Hall; 1997. pp. 307–324. [Google Scholar]
- 2.Brosnan C F, Racke M K, Selmaj K. In: Multiple Sclerosis Clinical and Pathogenetic Basis. Raine C S, McFarland H F, Tourtellotte W W, editors. London: Chapman & Hall; 1997. pp. 325–340. [Google Scholar]
- 3.Raine C S. In: Multiple Sclerosis Clinical and Pathogenetic Basis. Raine C S, McFarland H F, Tourtellotte W W, editors. London: Chapman & Hall; 1997. pp. 243–286. [Google Scholar]
- 4.Prineas J W, McDonald W I. In: Greenfield’s Neuropathology. Graham D I, Lantos P L, editors. Oxford: Oxford Univ. Press; 1997. pp. 813–896. [Google Scholar]
- 5.Mokhtarian F, McFarlin D E, Raine C S. Nature (London) 1984;309:356–358. doi: 10.1038/309356a0. [DOI] [PubMed] [Google Scholar]
- 6.Gassmann M, Lemke G. Curr Opin Neurobiol. 1997;7:87–92. doi: 10.1016/s0959-4388(97)80125-0. [DOI] [PubMed] [Google Scholar]
- 7.Marchionni M A, Goodearl A D, Chen M S, Bermingham-McDonogh O, Kirk C, Hendricks M, Danehy D, Misumi D, Sudhalfer J, Kobayashi D, et al. Nature (London) 1993;362:312–318. doi: 10.1038/362312a0. [DOI] [PubMed] [Google Scholar]
- 8.Canoll P D, Musacchio J M, Hardy R, Reynolds R, Marchionni M A, Salzer J L. Neuron. 1996;17:229–243. doi: 10.1016/s0896-6273(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 9.Milner R, Anderson H J, Rippon R F, McKay J S, Franklin R J, Marchionni M A, Reynolds R, ffrench-Constant C. Glia. 1997;19:85–90. doi: 10.1002/(sici)1098-1136(199701)19:1<85::aid-glia9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 10.Rutkowski J L, Kirk C J, Lerner M A, Tennekoon G I. Nat Med. 1995;1:24–25. doi: 10.1038/nm0195-80. [DOI] [PubMed] [Google Scholar]
- 11.Marchionni M A, Kirk C J, Isaacs I J, Hoban C J, Mahanthappa N K, Anton E S, Chen C, Wason F, Lawson D, Hamers F T P, et al. Cold Spring Harbor Symp Quant Biol. 1996;61:459–472. [PubMed] [Google Scholar]
- 12.Jordan C, Friedrich V, Dubois-Dalcq M. J Neurosci. 1989;9:248–257. doi: 10.1523/JNEUROSCI.09-01-00248.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamholz J, Toffeneti J, Lazzarini R A. J Neurosci Res. 1988;21:62–70. doi: 10.1002/jnr.490210110. [DOI] [PubMed] [Google Scholar]
- 14.Nagasato K, Farris R W, Dubois-Dalcq M, Voskuhl R R. J Neuroimmunol. 1997;72:21–25. doi: 10.1016/S0165-5728(96)00137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capello E, Voskuhl R R, McFarland H F, Raine C S. Ann Neurol. 1997;41:797–805. doi: 10.1002/ana.410410616. [DOI] [PubMed] [Google Scholar]
- 16.Yao D-L, Liu X, Hudson L D, Webster H d. Proc Natl Acad Sci USA. 1995;92:6190–6194. doi: 10.1073/pnas.92.13.6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao D-L, Liu X, Hudson L D, Webster H d. Life Sci. 1996;58:1301–1306. doi: 10.1016/0024-3205(96)00095-1. [DOI] [PubMed] [Google Scholar]
- 18.Trapp B D, Peterson J, Ransohoff R M, Rudick R, Mork S, Bo L. N Engl J Med. 1998;338:323–325. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy M K, Torrance D S, Picha K S, Mohler K M. J Immunol. 1992;149:2496–2505. [PubMed] [Google Scholar]
- 20.Kwon E E, Prineas J W. J Neuropathol Exp Neurol. 1994;53:617–624. doi: 10.1097/00005072-199411000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Claudio L, Raine C S, Brosnan C F. Acta Neuropathol. 1995;90:228–238. doi: 10.1007/BF00296505. [DOI] [PubMed] [Google Scholar]
- 22.Raine C S. Lab Invest. 1984;50:608–635. [PubMed] [Google Scholar]
- 23.Rodriguez M. Lab Invest. 1991;64:358–370. [PubMed] [Google Scholar]
- 24.Wolswijk G. J Neurosci. 1998;18(2):601–609. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]