

Abstract
Shotgun lipidomics is a rapidly developing technology, which identifies and quantifies individual lipid molecular species directly from lipid extracts of biological samples. Alterations in lipid molecular species in the brain induced by neurodegenerative diseases, such as Alzheimer’s disease (AD) could provide fundamental clues to disease pathogenesis. To date, the cause(s) leading to AD pathogenesis are still unknown and apolipoprotein E (apoE) allele 4 is the only known major risk factor for this devastating disease. By utilizing shotgun lipidomics, we have recently shown that a substantial and specific depletion of sulfatide (a class of specialized myelin sphingolipids) is present in postmortem brains from subjects at the earliest clinically recognizable stage of AD. In subsequent studies to identify the biochemical mechanisms underlying sulfatide depletion at this very mild stage of AD, we have found that apoE is associated with sulfatide transport and mediates sulfatide homeostasis in the nervous system through lipoprotein metabolism pathways and that alterations in apoE-mediated sulfatide trafficking can lead to sulfatide depletion in the brain. Thus, a working model related to the potential biochemical mechanisms underlying sulfatide depletion in AD can be derived based on these results. Collectively, the results obtained from lipidomic analyses of brain samples provide important insights into the biochemical mechanisms underlying AD pathogenesis.
Keywords: Alzheimer’s disease, apolipoprotein E, electrospray ionization, lipidomics, mass spectrometry, shotgun lipidomics, sulfatide metabolism
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and is the most common cause of dementia in the aging population. In the United States, its prevalence is ~10% of those over 65 and is ~50% in those over 85. Synapse loss, cholinergic system defects, and neuronal degeneration are some of the physiological sequelae of this devastating disease. The presence of neuritic plaques enriched with amyloid-β peptides and neurofibrillary tangles containing hyperphosphorylated tau protein represent well-established AD pathological hallmarks (Yankner 1996; Morishima-Kawashima and Ihara 2002). Many hypotheses regarding the fundamental causes of Alzheimer’s disease have been investigated. These include theories related to the importance of the amyloid-cascade, tau deposition, oxidative stress, inflammation (i.e., arthritis-of-the brain hypothesis), energy metabolism, and acetylcholine signaling defects, among others (Armstrong et al. 1996; Albers and Beal 2000; Cummings and Cole 2002; Mattson 2003; McGeer and McGeer 2003; Selkoe 2004; Hauptmann et al. 2006; Onyango and Khan 2006; Sivaprakasam 2006; Tabet 2006; Wyss-Coray 2006). Although tremendous progress has been made toward understanding Alzheimer’s disease, the true biochemical mechanism(s) underlying the pathogenesis of the disease still remain unknown.
To date, the only known major genetic risk factor for late-onset AD, including both familial and sporadic and accounting for over 95% of total cases, is the ε4 allele of apolipoprotein E (apoE4) (Strittmatter and Roses 1996; Cedazo-Minguez and Cowburn 2001). The mechanism(s) underlying the significance of the apoE4 allele for AD pathogenesis remain to be elucidated. Both in vitro and more recently in vivo data strongly suggest that the ability of apoE to modify Aβ deposition may underlie the importance of apoE4 as an AD risk factor (Holtzman 2004). Since apoE is a lipid transport protein (Mahley 1988; Han 2004), we hypothesized that alterations in apoE-mediated lipid trafficking and metabolism must play a role in AD pathogenesis. Therefore, we (Han et al. 2001, 2002) have first determined alterations in the lipid profiles of pure gray and white matter from post-mortem brain of subjects with very mild AD (Morris 1993) employing a lipidomics approach by using an electrospray ionization mass spectrometry (ESI/MS)-based technology termed shotgun lipidomics (Han and Gross 2005a,b). It has been found that specific lipid changes are present in subjects at the earliest clinically recognizable stage of AD relative to cognitively normal, age-matched controls (see Han 2005 for recent review). Specifically, shotgun lipidomics reveals the substantial loss of sulfatide (Han et al. 2002) which is a class of specialized myelin glycosphing-olipids (Vos et al. 1994; Marcus et al. 2006). Moreover, shotgun lipidomics demonstrates a large content increase and molecular species compositional change in ceramide (Han et al. 2002) which is a class of central sphingolipid metabolites and is associated with cell death (Hannun and Luberto 2000).
In this short review, following a brief introduction of lipidomics research field and shotgun lipidomics technology, the importance of shotgun lipidomics in determining the degree of the cross presence of gray and white matter, which used to be a hidden and unpredictable variable for investigations using human brain tissue, will be discussed. Next, the specific and dramatic loss of sulfatide content in subjects with very mild AD as demonstrated by shotgun lipidomics is reviewed. Finally, a working model related to the potential mechanism(s) leading to sulfatide depletion is proposed.
Lipidomics and shotgun lipidomics
Lipidomics, defined as the large-scale study of the pathways and networks of cellular lipids, is an emerging and rapidly expanding research field (Han and Gross 2003; Lagarde et al. 2003). Interest in lipidomics has been fueled by the recognition that cellular lipids play many essential roles in cellular functions and that the metabolism of individual lipid molecular species or lipid classes is interwoven. To conduct research on lipid metabolism, it is often necessary to examine numerous molecular species and lipid classes to gain insights into factors which contribute to a given pathogenic state. Currently, lipidomics research has been focused on identifying alterations in lipid metabolic pathways and networks induced by a disease state, a gene mutation (knockout, or over-expression), a therapeutic treatment, or other perturbations. In the future, research in lipidomics will expand to include the dynamics of lipidomes, subcellular organizations among lipidomes, and interactions of lipids with lipids, proteins, and other cellular moieties.
Owing to the complexity of the lipidome, many modern technologies (including MS, NMR, and fluorescence spectroscopy) have been employed to identify, quantify, and characterize the chemical properties and function of each constituent lipid, and the metabolic nodes that they represent (Feng and Prestwich 2006; Mossoba et al. 2006). Among these analytical techniques, MS has played a leading role in lipid characterization, identification, and quantitation (Byrdwell 2003; Griffiths 2003; Han and Gross 2003, 2005a; Pulfer and Murphy 2003; Hsu and Turk 2005; Schiller et al. 2007). In particular, ESI/MS is the most prominent and has been the most successful in this endeavor (Griffiths 2003; Han and Gross 2003, 2005a,b; Pulfer and Murphy 2003; Ivanova et al. 2004; Welti and Wang 2004). Although lipidomics has only emerged as a distinct field within the past few years (Han and Gross 2003; Lagarde et al. 2003), several new discoveries and/or advances have already been made (Han and Gross 2005a; Serhan 2005; Walker et al. 2005; Wenk 2005; Han 2007; Welti et al. 2007).
One of the major new developments in current lipidomics practice is the multi-dimensional MS-based shotgun lipidomics which we have recently developed (Han and Gross 2001 2005a,b; Han et al. 2004). This platform has now evolved into a mature technology that includes a series of simple steps, such as multiplexed extractions, intrasource separation, identification of individual lipid molecular species using multi-dimensional MS and array analyses, and quantitation of the identified lipid molecular species using a two-step procedure in conjunction with data processing (Han and Gross 2005a; Cheng et al. 2007; Han 2007). Through lipid class-selective intrasource ionization and subsequent multi-dimensional MS analysis, shotgun lipidomics, at its current stage, enables us to fingerprint and quantitate the individual molecular species of most of the major and many of the minor lipid classes in cellular lipidomes, which collectively represent > 95% of the total lipid content and as many as 1000 molecular species, directly from their chloroform extracts. The classes of lipids include choline glycerophospholipid (GPCho), ethanolamine glycerophospholipid (GPEtn), phosphatidylinositol, phos-phatidylglycerol, phosphatidylserine, phosphatidic acid, sphingomyelin, galactosylceramide, glucosylceramide, sulfatide, free fatty acid, triacylglycerol, lysoGPCho, lysoGPEtn, lysophosphatidic acid, acylcarnitine, cholesterol and cholesterol esters, and ceramide (including dihydroceramide). Cardiolipin (Han et al. 2006) and sphingoid base-1-phosphate (Jiang and Han 2006) are the newest classes that have been added to the list of lipids identified and quantified by shotgun lipidomics.
Shotgun lipidomics unambiguously identifies the degree of the cross existing of gray and white matter: one of its major advantages for neurolipidomics
One of the complexities of the brain is reflected by the presence of multiple cell types (e.g., neurons, astrocytes, oligodendrocytes, microglia, etc). Neurons are enriched in gray matter whereas oligodendrocytes are mainly present in white matter. One complicating factor present in any study using human brain samples is the varying degree of the cross presence of gray and white matter. Differences in the degree of this co-existing due to sampling represent an unpredictable variable which may overshadow real differences between the samples from diseased and normal states. Previous analytical methods have not been able to effectively identify such cross contamination which has subsequently been neglected by the majority of studies using human brain tissue. It should be particularly emphasized that the degree of this type of co-existing is very critical for the determination of alterations in neurolipidomes (and other metabolomes) as well as other measurements, including changes in gene expression and protein levels induced by disease states.
In early studies using shotgun lipidomics, we demonstrated the presence of very distinct lipid profiles of GPEtn molecular species in gray matter and white matter samples from human brain (Fig. 1) (Han et al. 2001). This finding has established a solid foundation for lipidomics to identify alterations in lipids, metabolites, and proteins induced by neurodegenerative diseases, to investigate the biochemical mechanisms underlying these disorders, and to discover their associated biomarkers. In addition to its many other advantages, shotgun lipidomics, at present, is the only analytical approach that enables us to successfully and efficiently determine the degree of the cross presence of gray and white matter based upon the peak intensity ratio of ions at m/z 726.4 (18:1–18:1 plasmenylethanolamine (pGPEtn)) and 790.4 (18:0–22:6 GPEtn) (Fig. 1).
Fig. 1.
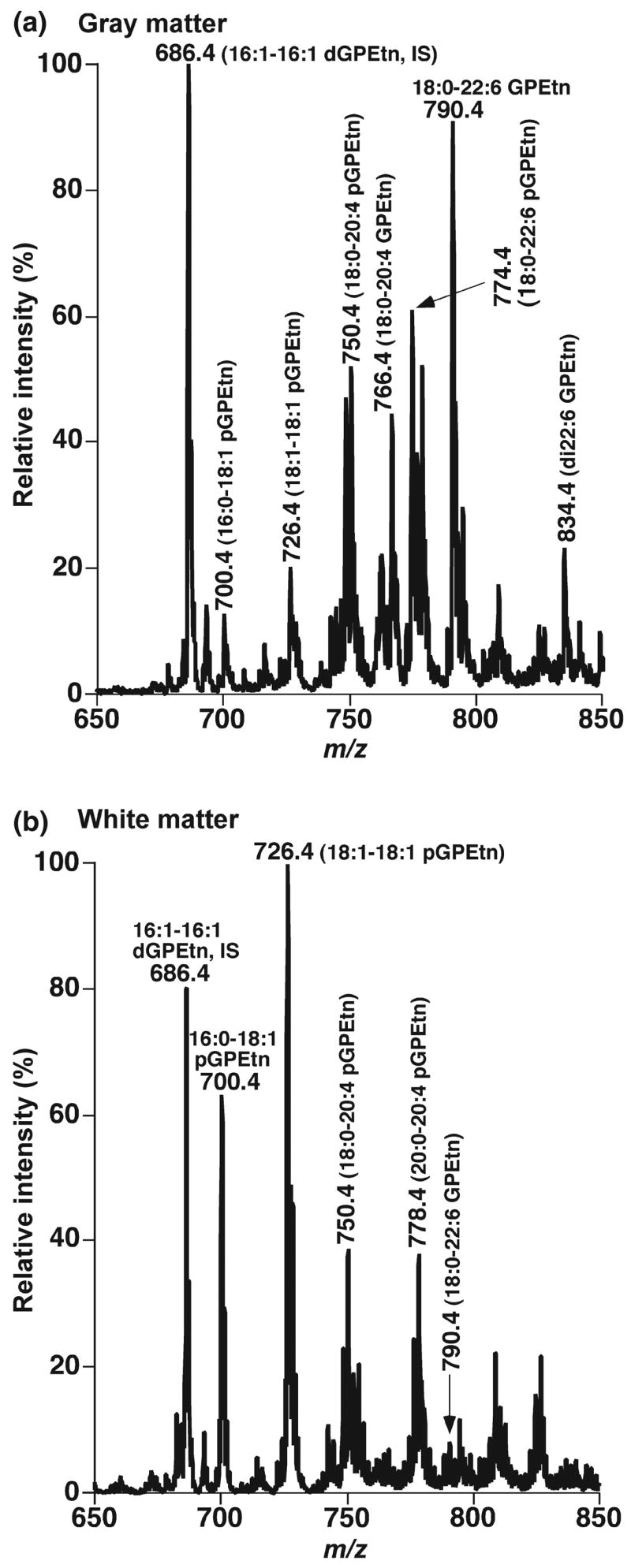
Distinct profiles of ethanolamine glycerophospholipid molecular species in lipid extracts of cognitively normal human occipital gray and white matter. Brain samples were obtained from the brain bank of the Washington University ADRC Neuropathology/Tissue Resource Core and brain lipids were extracted by a modified procedure of Bligh-Dye as previously described (Bligh and Dyer 1959). Negative-ion ESI mass spectra of lipid extracts of occipital gray matter (Panel a) and white matter (Panel b) were acquired in the presence of a small amount of LiOH as previously described (Han and Gross 2005a). Individual molecular species corresponding to each ion peak were identified using multi-dimensional MS analysis as previously described (Han and Gross 2005b). Plasmenylethanolamine and phosphatidylethanolamine are abbreviated as ‘pGPEtn’ and ‘GPEtn,’ respectively. ‘IS’ denotes internal standard (Han, Unpublished data).
Specifically, ESI/MS analysis of CHCl3 extracts of cortex gray matter from postmortem subjects has demonstrated multiple predominant deprotonated ion peaks corresponding to GPEtn molecular species (Fig. 1a) (Han et al. 2001). It has been identified that over 80 mol% of GPEtn and 55–60% of pGPEtn molecular species contain four or more double bonds at the sn-2 position in gray matter (Han et al. 2001). In contrast, ESI/MS analysis of lipid extracts of white matter from different brain regions from postmortem subjects has revealed the presence of one predominant peak at m/z 726.4 and that pGPEtn represents over 85 mol% of the total GPEtn (Fig. 1b). Shotgun lipidomics analyses have also shown that over 50 mol% of GPEtn molecular species containing one double bond at the sn-2 position are present in white matter in comparison to < 15 mol% of those containing one double bond at the sn-2 position in gray matter (Han et al. 2001).
The distinct molecular species profiles of GPEtn in brain gray matter and white matter likely reflect the compartmentalization and distinct functions of these fundamental brain tissues. Myelin-dominated white matter requires plasmalogen to provide a sufficiently compact sheath for efficient axonal function, since plasmalogen possesses a compact smembrane conformation in comparison to its diacyl counterpart (Han and Gross 1990). Thus, it is not surprising that pGPEtn molecular species in white matter contain less unsaturated acyl constituents at the sn-2 position. In contrast, pGPEtn molecular species in gray matter contains abundant polyunsaturated acyl chains (e.g., 20:4 and 22:6 FA). It is well known that 20:4 and 22:6 fatty acids are essential components for signal transduction. Moreover, polyunsaturated acyl chain-containing pGPEtn can efficiently facilitate membrane fusion between synaptic vesicles and the neuronal plasma membrane (Glaser and Gross 1994, 1995; Han et al. 1998) which is an essential process associated with neurotransmitter release at synaptic terminals. Accordingly, the distinct molecular species profiles of GPEtn between brain gray matter and white matter not only represent an important criterion by which one is able to determine the degree of the cross presence of gray matter and white matter, but also serves as an example of the power of shotgun lipidomics to reveal biological relationships between the structure and function of lipids.
Shotgun lipidomics identifies the specific depletion of sulfatides at the earliest clinically recognizable stage of Alzheimers’s disease
We have determined alterations in the brain lipid profiles of subjects with very mild AD (Morris 1993) by using shotgun lipidomics in ‘pure’ gray and white matter (Han et al. 2001, 2002). A dramatic depletion of sulfatide content (up to 90%) in gray matter and approximately 50% sulfatide content in white matter of all examined cerebral regions were demonstrated (Han et al. 2002). Fig. 2 displays some representative examples of the expanded mass spectra showing the depleted molecular ions of both hydroxy- and non-hydroxysulfatides from three AD cases in comparison to a control case. Fig. 3 exemplifies the summarized results of the altered levels of total sulfatide in temporal gray and white matter from subjects with diverse severities of AD dementia. Notably, sulfatide loss occurs in all examined brain regions including both cerebrum (e.g., frontal, parietal, and temporal regions) and cerebellum.
Fig. 2.

Loss of sulfatide molecular species in lipid extracts of temporal gray matter from subjects with very mild AD relative to control. Brain samples were obtained from the brain bank of the Washington University ADRC Neuropathology/Tissue Resource Core and lipids were extracted by a modified method of Bligh-Dyer as previously described (Bligh and Dyer 1959). The mass region showing sulfatide molecular ions was displayed, which was expanded from negative-ion zESI mass spectra of lipid extracts from temporal gray matter of cognitively normal control (Panel a), or of subjects with very mild AD (Panels b–d). Negative-ion ESI mass spectra were acquired in the absence of LiOH after direct infusion of diluted lipid solution as previously described (Han and Gross 2005a). All mass spectra are displayed after being normalized to the internal standard for analysis of phosphatidylinositol (denoted as GPIns) molecular species. GPSer stands for phosphatidylserine (Han, unpublished data).
Fig. 3.

Correlation of sulfatide content in lipid extracts of temporal gray and white matter with the severity stages of AD dementia. The total content of sulfatide molecular species in chloroform extracts of temporal gray matter (Left panel) and white matter (Right panel) was quantitated using shotgun lipidomics as previous described (Han et al. 2002). The data were normalized to the protein content of each tissue sample and are presented as the means ± SEM from six separate subjects per group. Some error bars are within the symbols.
We also determined alterations of the contents in other lipid classes, including GPCho, diacyl GPEtn, pGPEtn, phosphatidylinositol, phosphatidylserine, sphingomyelin, cerebrosides, ceramide, free fatty acid, and cholesterol by shotgun lipidomics analyses of both gray and white matter samples of multiple brain regions from very mild AD subjects in comparison to age-matched, cognitively normal controls. We did not find significant alterations in enzymatic activities and the contents of these lipid classes except pGPEtn and ceramide (Han et al. 2001, 2002). These results are consistent with the previous findings that no apparent losses of neurons and synapses occur at the very mild stages of AD (Morris 1993; Berg et al. 1998; Morris et al. 2001). The substantial increases in ceramide content present in subjects with very mild AD may be related to the dramatic depletion of sulfatide content, but other explanations include abnormal de novo ceramide synthesis or accelerated hydrolysis of sphingomyelin. Therefore, these results suggest that the loss of sulfatide content in very mild AD is lipid class-specific and not due to the general neuronal loss associated with AD from which losses of most lipid classes would occur.
To determine the specificity of the altered sulfatide levels in AD among other neurodegenerative diseases, we have also examined the contents of sulfatide in postmortem brain samples from subjects with non-AD-related Parkinson’s disease (PD) and non-PD-related dementia with Lewy bodies (Cheng et al. 2003), as well as multiple sclerosis and frontotemporal dementia (Han, unpublished data). These diseases commonly occur concomitantly with AD and are complicating factors for the early diagnosis of AD. In contrast to AD cases, the sulfatide levels of all examined brain regions in both gray and white matter were dramatically elevated from PD subjects and modestly depleted in multiple sclerosis compared to cognitively normal controls, whereas the sulfatide contents in both gray matter and white matter in dementia with Lewy bodies and frontotemporal dementia were similar to those observed in controls. These results suggest that sulfatide depletion in subjects with very mild AD is specific among the examined neurodegenerative diseases.
It should be specifically pointed out that, from the studies using shotgun lipidomics, we have also found another intriguing phenomenon which is a sharp loss of sulfatide and a maximal content of ceramide present at the earliest clinically recognizable stage of AD. At more severe stages of AD dementia, minimal additional losses of sulfatide content occur relative to that at the very mild stage of AD and a decreased rate of ceramide accumulation is manifest in comparison to that at the earliest onset of AD pathogenesis. These observations suggest the existence of a preclinical stage of AD during which sulfatide levels would begin to decrease and the content of ceramide may be transiently higher than that present at the very mild stage of AD. This suggestion is consistent with autopsy studies which show that approximately 30% of cognitively normal subjects who died in their mid 70s have a marked AD pathology (i.e., the presence of large amounts of plaques and tangles), but do not yet have the substantial neuronal cell loss that is present in subjects who died with very mild AD (Price and Morris 1999; Morris et al. 2001). Therefore, this finding strongly supports the notion that extensive AD pathology (i.e., plaques and tangles) likely develops over a 10–20-year period prior to any cognitive impairment or neuronal cell death (Price and Morris 1999) and indicates that alterations in sulfatide and ceramide levels are among the earliest events of AD pathogenesis.
Potential mechanisms responsible for sulfatide depletion at the earliest clinically recognizable stage of AD: What we have learned from lipidomics
During our studies to elucidate the mechanism(s) underlying the sulfatide depletion in subjects with very mild AD, we uncovered a novel role of apoE in both the CNS and the peripheral nervous system which is modulation of sulfatide content (Han et al. 2003a; Cheng et al. 2007). For example, sulfatide levels are increased in both the CNS and the peripheral nervous system in apoE knockout mice relative to their wild-type littermates. Furthermore, modulation of sulfatide content in brain depends on specific apoE isoforms, since substantially lower sulfatide content is manifest in human apoE4 transgenic mice than that in human apoE3 transgenic mice (Han et al. 2003a).
These results have led us to ask several specific questions. For instance, is sulfatide associated with apoE-containing lipoprotein particles and is this association specific to these particles? If sulfatide is only present in apoE-associated lipoproteins, how do these particles acquire sulfatide since apoE-containing lipoprotein particles are mainly secreted from astrocytes (DeMattos et al. 2001; Koch et al. 2001) whereas sulfatide is synthesized de novo in oligodendrocytes and is predominantly present in myelin sheath (Svennerholm et al. 1992; Vos et al. 1994)? Are the sulfatide levels varied between the lipoproteins containing different apoE isoforms? What are the factors that influence the metabolism of the apoE-associated lipoprotein particles? What are the functions of sulfatide that is transported by apoE-associated lipoproteins in the nervous system? Thus, the results revealed by shotgun lipidomics have led us to test multiple hypotheses.
Interestingly, it has been found that sulfatide precisely co-localizes with high density lipoprotein-like lipoproteins present in human cerebrospinal fluid (CSF) (Han et al. 2003a). Since there are two predominant apolipoprotein subtypes in the CNS, i.e., apoE and apoJ (DeMattos et al. 2001; Koch et al. 2001), further experiments have been performed to identify the specific carrier(s) of sulfatide in human CSF by immunoprecipitation followed by shotgun lipidomics analysis. It has been demonstrated that sulfatide is specifically associated with apoE-containing HDL-like lipoproteins in human CSF (Han et al. 2003a). These results not only indicate that sulfatide is transported by apoE-associated lipoproteins in brain interstitial fluid and CSF, but also imply that sulfatide is metabolized through endocytotic recycling of apoE-associated particles via the low density lipoprotein (LDL) receptor or its family members (Krieger and Herz 1994) which are abundantly expressed in neurons (Herz 2001).
Moreover, the results from these studies also lead us to establish a working model for understanding sulfatide metabolism and elucidating potential mechanism(s) responsible for sulfatide depletion in subjects with very mild AD (Fig. 4). In this model, apoE-associated lipoprotein particles are released from astrocytes and acquire sulfatide from the myelin sheath likely through a ‘kiss-and-run’ mechanism. Then the sulfatide-containing apoE-associated lipoproteins can be metabolized through an endocytotic pathway by neuronal cells possessing LDL receptors or its family members, such as the LDL receptor-related protein. Alternatively, these sulfatide-enriched apoE-associated lipoproteins can be transported to destinations in the peripheral system through the CSF.
Fig. 4.
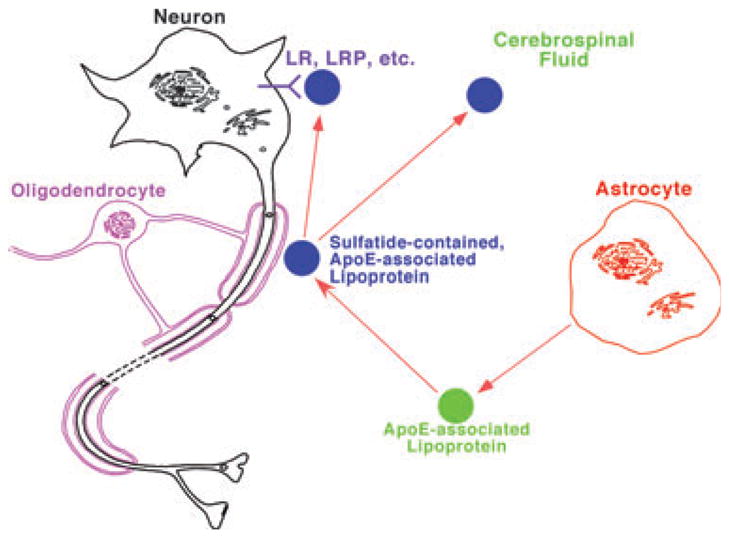
A proposed model for the metabolism of apolipoprotein E-associated lipoproteins which mediate sulfatide homeostasis. In the model, apoE-associated lipoproteins are released from astrocytes, acquire sulfatide from myelin sheath, and then either metabolized through endocytotic pathway through LDL receptors or its family members, such as the LDL receptor-related protein or transported to cerebrospinal fluid. Therefore, any factors that modulate apoE-associated lipoprotein metabolism can alter sulfatide levels in the nervous system.
This model is supported by all of our currently available experimental data. For example, sulfatide is present in CSF (Han et al. 2003b); apoE4-associated lipoprotein articles contain a significantly higher sulfatide content in CSF than that in their apoE3-associated counterparts (Han et al. 2003a), thereby accounting for the lower sulfatide levels in apoE4 transgenic mouse brain tissue relative to that in apoE3 mice (Han et al. 2003a); and apoE deficiency leads to the accumulation of sulfatide in the brain (Han et al. 2003a) and in the ganglia (Cheng et al. 2007). This model not only suggests that apoE, as a risk factor for AD, is involved in the sulfatide loss in AD patients, but also accounts for the abnormal sulfatide metabolism of AD pathogenesis. Moreover, other reported AD risk factors, such as the LDL receptor, LDL receptor-related protein (Van Uden et al. 2000; Abdulkarim and Hameed 2006), and heparan sulfate proteoglycans [which have been proposed to be associated with lipoprotein metabolism (Mulder and Terwel 1998)] are linked together with this model.
Therefore, it can be speculated that any factors that alter this apoE-mediated sulfatide metabolic network will change the levels of sulfatide in the CNS. For example, lower sulfatide contents have been determined by shotgun lipidomics from the white matter-containing brain samples of mice which over-express LDL receptor in comparison to those of wild type mice (Han, unpublished data). It has also been found that altered cholesterol metabolism through either increased dietary intake of cholesterol or pharmacologic inhibition of cholesterol synthesis affects the secretion of apoE from astrocytes, thereby disturbing sulfatide levels in the brain (Han, unpublished data). It has previously been documented that the expression levels of apoE, LDL receptor and the LDL receptor-related protein are higher in AD patients than those in control (LaFerla et al. 1997; Ulery and Strickland 2000; Herz and Strickland 2001; Laws et al. 2002). It can be anticipated based upon our working model that increased expression of these apolipoproteins and receptors can lead to accelerated sulfatide metabolism, thereby resulting in accelerated sulfatide depletion in AD patients. It can be speculated that this accelerated sulfatide depletion would occur at the relatively earlier stages in apoE4 carriers than other apoE isoform carriers since apoE4 lipoproteins carry more sulfatide content than their apoE3 counterparts in the CNS (Han et al. 2003a), thereby accelerating the processing of sulfatide metabolism.
It should be specifically pointed out that we have recognized the importance of sulfatide in the immune system and in anti-inflammatory responses and have discussed the advantages of apoE4 in younger aged carriers (Han 2005). This argument is consistent with the ability of apoE4 lipoproteins to carry more sulfatide content than their apoE3 counterparts in the CNS (Han et al. 2003a) and the fact that AD appears as an age-related issue in modern societies. Therefore, the frequency of apoE4 is higher in certain geographical areas where protection against infectious disease remains a priority such as in the African subcontinent and in certain other isolated populations (e.g., in Papua New Guinea). Clearly, apoE-associated sulfatide and their roles in neurobiology, inflammation, and infectious disease require further exploration.
It should also be emphasized that our working model does not exclude the possibility that factors which affect apoE-mediated sulfatide metabolism may also alter the intra-cellular metabolic pathways of sulfatide in particular and of the sphingolipidome in general. As pointed out in the ‘Lipidomics and shotgun lipidomics’ section, the metabolism of lipid classes and individual lipid molecular species in a cellular lipidome are interwoven. The factors that influence apoE-mediated sulfatide metabolism could directly or indirectly affect the entire sphingolipidome, thereby causing dramatic changes in ceramide levels present during AD pathogenesis, which directly result in the dramatic increases in ceramide levels at the very mild stage of AD. In our future studies, we will pursue both possibilities by using shotgun lipidomics.
Conclusions
In this review, we have discussed the powerful role of shotgun lipidomics in the study of AD and summarized the results from our recent studies on AD. A working model which leads us to understand the potential biochemical mechanism(s) responsible for sulfatide depletion in subjects with very mild AD has been proposed and discussed. This model is not only consistent with the current experimental results available, but also provides future directions for the elucidation of the biochemical mechanism(s) underlying AD pathogenesis.
Acknowledgments
This work was supported by National Institute on Aging Grant R01 AG23168 and the Neurosciences Education and Research Foundation. The author is grateful to Dr Christopher M. Jenkins for his comments, and Dr Kui Yang and Ms Hua Cheng for their technical help.
Abbreviation used
- AD
Alzheimer’s disease
- apoE
apolipoprotein E
- CSF
cerebrospinal fluid
- GPCho
glycerophosphocholine(s)
- GPEtn
glycerophosphoethanolamine(s)
- LDL
low density lipoprotein
- MS
mass spectrometry
- pGPEtn
alkenyl-acyl GPEtn (i.e., plasmenylethanolamine)
References
- Abdulkarim Y, Hameed Z. Is the LDL receptor involved in cortical amyloid protein clearance? Neurochem Res. 2006;31:839–847. doi: 10.1007/s11064-006-9084-0. [DOI] [PubMed] [Google Scholar]
- Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J Neural Transm Suppl. 2000;59:133–154. doi: 10.1007/978-3-7091-6781-6_16. [DOI] [PubMed] [Google Scholar]
- Armstrong RA, Winsper SJ, Blair JA. Aluminium and Alzheimer’s disease: review of possible pathogenic mechanisms. Dementia. 1996;7:1–9. doi: 10.1159/000106845. [DOI] [PubMed] [Google Scholar]
- Berg L, McKeel DW, Jr, Miller JP, et al. Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55:326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Byrdwell WC. APCI-MS lipid analysis. Oily Press Lipid Library. 2003;16:171–253. [Google Scholar]
- Cedazo-Minguez A, Cowburn RF. Apolipoprotein E: a major piece in the Alzheimer’s disease puzzle. J Cell Mol Med. 2001;5:254–266. doi: 10.1111/j.1582-4934.2001.tb00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Xu J, McKeel DW, Jr, Han X. Specificity and potential mechanism of sulfatide deficiency in Alzheimer’s disease: An electrospray ionization mass spectrometric study. Cell Mol Biol. 2003;49:809–818. [PubMed] [Google Scholar]
- Cheng H, Jiang X, Han X. Alterations in lipid homeostasis of mouse dorsal root ganglia induced by apolipoprotein E deficiency: A shotgun lipidomics study. J Neurochem. 2007;101:57–76. doi: 10.1111/j.1471-4159.2006.04342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Cole G. Alzheimer disease. JAMA. 2002;287:2335–2338. doi: 10.1001/jama.287.18.2335. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Brendza RP, Heuser JE, Kierson M, Cirrito JR, Fryer J, Sullivan PM, Fagan AM, Han X, Holtzman DM. Purification and characterization of astrocyte-secreted apolipoprotein E and J-containing lipoproteins from wild-type and human apoE transgenic mice. Neurochem Int. 2001;39:415–425. doi: 10.1016/s0197-0186(01)00049-3. [DOI] [PubMed] [Google Scholar]
- Feng L, Prestwich GD, editors. Functional Lipidomics. CRC Press, Taylor & Francis Group; Boca Raton, FL, USA: 2006. [Google Scholar]
- Glaser PE, Gross RW. Plasmenylethanolamine facilitates rapid membrane fusion: a stopped-flow kinetic investigation correlating the propensity of a major plasma membrane constituent to adopt an HII phase with its ability to promote membrane fusion. Biochemistry. 1994;33:5805–5812. doi: 10.1021/bi00185a019. [DOI] [PubMed] [Google Scholar]
- Glaser PE, Gross RW. Rapid plasmenylethanolamine-selective fusion of membrane bilayers catalyzed by an isoform of glyceraldehyde-3-phosphate dehydrogenase: discrimination between glycolytic and fusogenic roles of individual isoforms. Biochemistry. 1995;34:12193–12203. doi: 10.1021/bi00038a013. [DOI] [PubMed] [Google Scholar]
- Griffiths WJ. Tandem mass spectrometry in the study of fatty acids, bile acids, and steroids. Mass Spectrom Rev. 2003;22:81–152. doi: 10.1002/mas.10046. [DOI] [PubMed] [Google Scholar]
- Han X. The role of apolipoprotein E in lipid metabolism in the central nervous system. Cell Mol Life Sci. 2004;61:1896–1906. doi: 10.1007/s00018-004-4009-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X. Lipid alterations in the earliest clinically recognizable stage of Alzheimer’s disease: Implication of the role of lipids in the pathogenesis of Alzheimer’s disease. Curr Alz Res. 2005;2:65–77. doi: 10.2174/1567205052772786. [DOI] [PubMed] [Google Scholar]
- Han X. Neurolipidomics: challenges and developments. Front Biosci. 2007;12:2601–2615. doi: 10.2741/2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Gross RW. Plasmenylcholine and phosphatidyl-choline membrane bilayers possess distinct conformational motifs. Biochemistry. 1990;29:4992–4996. doi: 10.1021/bi00472a032. [DOI] [PubMed] [Google Scholar]
- Han X, Gross RW. Quantitative analysis and molecular species fingerprinting of triacylglyceride molecular species directly from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal Biochem. 2001;295:88–100. doi: 10.1006/abio.2001.5178. [DOI] [PubMed] [Google Scholar]
- Han X, Gross RW. Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry: a bridge to lipidomics. J Lipid Res. 2003;44:1071–1079. doi: 10.1194/jlr.R300004-JLR200. [DOI] [PubMed] [Google Scholar]
- Han X, Gross RW. Shotgun lipidomics: Electrospray ionization mass spectrometric analysis and quantitation of the cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev. 2005a;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- Han X, Gross RW. Shotgun lipidomics: multi-dimensional mass spectrometric analysis of cellular lipidomes. Expert Rev Proteomics. 2005b;2:253–264. doi: 10.1586/14789450.2.2.253. [DOI] [PubMed] [Google Scholar]
- Han X, Ramanadham S, Turk J, Gross RW. Reconstitution of membrane fusion between pancreatic islet secretory granules and plasma membranes: catalysis by a protein constituent recognized by monoclonal antibodies directed against glyceraldehyde-3-phosphate dehydrogenase. Biochim Biophys Acta. 1998;1414:95–107. doi: 10.1016/s0005-2736(98)00154-0. [DOI] [PubMed] [Google Scholar]
- Han X, Holtzman DM, McKeel DW., Jr Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J Neurochem. 2001;77:1168–1180. doi: 10.1046/j.1471-4159.2001.00332.x. [DOI] [PubMed] [Google Scholar]
- Han X, Holtzman DM, McKeel DW, Jr, Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J Neurochem. 2002;82:809–818. doi: 10.1046/j.1471-4159.2002.00997.x. [DOI] [PubMed] [Google Scholar]
- Han X, Cheng H, Fryer JD, Fagan AM, Holtzman DM. Novel role for apolipoprotein E in the central nervous system: Modulation of sulfatide content. J Biol Chem. 2003a;278:8043–8051. doi: 10.1074/jbc.M212340200. [DOI] [PubMed] [Google Scholar]
- Han X, Fagan AM, Cheng H, Morris JC, Xiong C, Holtzman DM. Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. [see comment][erratum appears in Ann Neurol 2003 Nov;54(5):693] Ann Neurol. 2003b;54:115–119. doi: 10.1002/ana.10618. [DOI] [PubMed] [Google Scholar]
- Han X, Yang J, Cheng H, Ye H, Gross RW. Towards fingerprinting cellular lipidomes directly from biological samples by two-dimensional electrospray ionization mass spectrometry. Anal Biochem. 2004;330:317–331. doi: 10.1016/j.ab.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Han X, Yang K, Yang J, Cheng H, Gross RW. Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J Lipid Res. 2006;47:864–879. doi: 10.1194/jlr.D500044-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun YA, Luberto C. Ceramide in the eukaryotic stress response. Trends Cell Biol. 2000;10:73–80. doi: 10.1016/s0962-8924(99)01694-3. [DOI] [PubMed] [Google Scholar]
- Hauptmann S, Keil U, Scherping I, Bonert A, Eckert A, Mueller WE. Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp Gerontol. 2006;41:668–673. doi: 10.1016/j.exger.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Herz J. The LDL receptor gene family: (un)expected signal transducers in the brain. Neuron. 2001;29:571–581. doi: 10.1016/s0896-6273(01)00234-3. [DOI] [PubMed] [Google Scholar]
- Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM. In vivo effects of ApoE and clusterin on amyloid-beta metabolism and neuropathology. J Mol Neurosci. 2004;23:247–254. doi: 10.1385/JMN:23:3:247. [DOI] [PubMed] [Google Scholar]
- Hsu F-F, Turk J. Electrospray ionization with low-energy collisionally activated dissociation tandem mass spectrometry of complex lipids: structural characterization and mechanism of fragmentation. In: Byrdwell WC, editor. Modern Methods for Lipid Analysis by Liquid Chromatography/Mass Spectrometry and Related Techniques. AOCS Press; Champaign, IL, USA: 2005. pp. 61–178. [Google Scholar]
- Ivanova PT, Milne SB, Forrester JS, Brown HA. LIPID arrays: New tools in the understanding of membrane dynamics and lipid signaling. Mol Interv. 2004;4:86–96. doi: 10.1124/mi.4.2.6. [DOI] [PubMed] [Google Scholar]
- Jiang X, Han X. Characterization and direct quantitation of sphingoid base-1-phosphates from lipid extracts: A shotgun lipidomics approach. J Lipid Res. 2006;47:1865–1873. doi: 10.1194/jlr.D600012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Donarski N, Goetze K, Kreckel M, Stuerenburg HJ, Buhmann C, Beisiegel U. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 2001;42:1143–1151. [PubMed] [Google Scholar]
- Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP) Annu Rev Biochem. 1994;63:601–637. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Troncoso JC, Strickland DK, Kawas CH, Jay G. Neuronal cell death in Alzheimer’s disease correlates with apoE uptake and intracellular Abeta stabilization. J Clin Invest. 1997;100:310–320. doi: 10.1172/JCI119536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarde M, Geloen A, Record M, Vance D, Spener F. Lipidomics is emerging. Biochim Biophys Acta. 2003;1634:61. doi: 10.1016/j.bbalip.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Laws SM, Hone E, Taddei K, Harper C, Dean B, McClean C, Masters C, Lautenschlager N, Gandy SE, Martins RN. Variation at the APOE -491 promoter locus is associated with altered brain levels of apolipoprotein E. Mol Psychiatry. 2002;7:886–890. doi: 10.1038/sj.mp.4001097. [DOI] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, Dupree JL. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 2006;53:372–381. doi: 10.1002/glia.20292. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Will caloric restriction and folate protect against AD and PD? Neurology. 2003;60:690–695. doi: 10.1212/01.wnl.0000042785.02850.11. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Inflammatory processes in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:741–749. doi: 10.1016/S0278-5846(03)00124-6. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Ihara Y. Alzheimer’s disease: beta-Amyloid protein and tau. J Neurosci Res. 2002;70:392–401. doi: 10.1002/jnr.10355. [DOI] [PubMed] [Google Scholar]
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH, Berg L. Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001;58:397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- Mossoba MM, Kramer JKG, Brenna JT, McDonald RE, editors. Lipid analysis and lipidomics: New techniques and applications. AOCS Press; Champaign, IL, USA: 2006. [Google Scholar]
- Mulder M, Terwel D. Possible link between lipid metabolism and cerebral amyloid angiopathy in Alzheimer’s disease: A role for high-density lipoproteins? Haemostasis. 1998;28:174–194. doi: 10.1159/000022429. [DOI] [PubMed] [Google Scholar]
- Onyango IG, Khan SM. Oxidative stress, mitochondrial dysfunction, and stress signaling in Alzheimer’s disease. Curr Alz Res. 2006;3:339–349. doi: 10.2174/156720506778249489. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and ‘preclinical’ Alzheimer’s disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Pulfer M, Murphy RC. Electrospray mass spectrometry of phospholipids. Mass Spectrom Rev. 2003;22:332–364. doi: 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- Schiller J, Suss R, Fuchs B, Muller M, Zschornig O, Arnold K. MALDI-TOF MS in lipidomics. Front Biosci. 2007;12:2568–2579. doi: 10.2741/2255. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140:627–638. doi: 10.7326/0003-4819-140-8-200404200-00047. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Mediator lipidomics. Prostaglandins Other Lipid Mediat. 2005;77:4–14. doi: 10.1016/j.prostaglandins.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Sivaprakasam K. Towards a unifying hypothesis of Alzheimer’s disease: cholinergic system linked to plaques, tangles and neuroinflammation. Curr Med Chem. 2006;13:2179–2188. doi: 10.2174/092986706777935203. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci. 1996;19:53–77. doi: 10.1146/annurev.ne.19.030196.000413. [DOI] [PubMed] [Google Scholar]
- Svennerholm L, Bostrom K, Fredman P, Jungbjer B, Mansson JE, Rynmark BM. Membrane lipids of human peripheral nerve and spinal cord. Biochim Biophys Acta. 1992;1128:1–7. doi: 10.1016/0005-2760(92)90250-y. [DOI] [PubMed] [Google Scholar]
- Tabet N. Acetyl cholinesterase inhibitors for Alzheimer’s disease: anti-inflammatories in acetylcholine clothing! Age Ageing. 2006;35:336–338. doi: 10.1093/ageing/afl027. [DOI] [PubMed] [Google Scholar]
- Ulery PG, Strickland DK. LRP in Alzheimer’s disease: friend or foe? J Clin Invest. 2000;106:1077–1079. doi: 10.1172/JCI11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Uden E, Kang DE, Koo EH, Masliah E. LDL receptor-related protein (LRP) in Alzheimer’s disease: towards a unified theory of pathogenesis. Microsc Res Tech. 2000;50:268–272. doi: 10.1002/1097-0029(20000815)50:4<268::AID-JEMT3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Vos JP, Lopes-Cardozo M, Gadella BM. Metabolic and functional aspects of sulfogalactolipids. Biochim Biophys Acta. 1994;1211:125–149. doi: 10.1016/0005-2760(94)90262-3. [DOI] [PubMed] [Google Scholar]
- Walker JM, Krey JF, Chen JS, Vefring E, Jahnsen JA, Bradshaw H, Huang SM. Targeted lipidomics: fatty acid amides and pain modulation. Prostaglandins Other Lipid Mediat. 2005;77:35–45. doi: 10.1016/j.prostaglandins.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Welti R, Wang X. Lipid species profiling: a high-throughput approach to identify lipid compositional changes and determine the function of genes involved in lipid metabolism and signaling. Curr Opin Plant Biol. 2004;7:337–344. doi: 10.1016/j.pbi.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Welti R, Shah J, Li W, Li M, Chen J, Burke JJ, Fauconnier ML, Chapman K, Chye ML, Wang X. Plant lipidomics: discerning biological function by profiling plant complex lipids using mass spectrometry. Front Biosci. 2007;12:2494–2506. doi: 10.2741/2250. [DOI] [PubMed] [Google Scholar]
- Wenk MR. The emerging field of lipidomics. Nat Rev Drug Discov. 2005;4:594–610. doi: 10.1038/nrd1776. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Yankner BA. New clues to Alzheimer’s disease: unraveling the roles of amyloid and tau. Nat Med. 1996;2:850–852. doi: 10.1038/nm0896-850. [DOI] [PubMed] [Google Scholar]