

Abstract
Bartonella henselae is a zoonotic pathogen and the causative agent of cat scratch disease and a variety of other disease manifestations in humans. Previous investigations have suggested that a limited subset of B. henselae isolates may be associated with human disease. In the present study, 182 human and feline B. henselae isolates from Europe, North America and Australia were analysed by multi-locus sequence typing (MLST) to detect any associations between sequence type (ST), host species and geographical distribution of the isolates. A total of 14 sequence types were detected, but over 66% (16/24) of the isolates recovered from human disease corresponded to a single genotype, ST1, and this type was detected in all three continents. In contrast, 27.2% (43/158) of the feline isolates corresponded to ST7, but this ST was not recovered from humans and was restricted to Europe. The difference in host association of STs 1 (human) and 7 (feline) was statistically significant (P≤0.001). eBURST analysis assigned the 14 STs to three clonal lineages, which contained two or more STs, and a singleton comprising ST7. These groups were broadly consistent with a neighbour-joining tree, although splits decomposition analysis was indicative of a history of recombination. These data indicate that B. henselae lineages differ in their virulence properties for humans and contribute to a better understanding of the population structure of B. henselae.
Introduction
Bartonella henselae is a fastidious bacterium associated with a broad spectrum of clinical disease manifestations in humans, including cat scratch disease (CSD) and bacillary angiomatosis (BA). CSD is characterized by subacute regional lymphadenopathy that usually occurs in immunocompetent individuals [1]. BA is a vasculoproliferative disorder which is predominantly encompassed in immunocompromised patients and often associated with chronic or relapsing bacteremia [2]. Cats represent the natural host and main reservoir for B. henselae. Infected animals develop relapsing bacteremia of several months duration without overt clinical symptoms [3].
Isolation of B. henselae is hampered by the fastidious nature of the organism. The sensitivity of cultural detection of B. henselae from tissues other than blood (e.g. lymph node biopsy specimen) is relatively low. The diagnosis of CSD and most other disease manifestations relies on detection of bacterial DNA in tissue specimens by PCR or serology [4], [5], [6]. Therefore, only few human-derived B. henselae isolates are available worldwide [7], [8], [9], [10]. In contrast, Bartonellae can be more easily isolated from the blood of infected cats. Several feline isolates have been collected during prevalence studies from different geographical regions [11], [12], [13], [14], [15]. Thus, feline isolates usually outnumber the human-derived isolates in investigations of the molecular epidemiology of B. henselae.
Previous studies have shown a considerable genetic heterogeneity among B. henselae isolates by using different DNA fingerprinting methods [10], [16], [17], [18]. The first suggestion that human-associated isolates represent a limited subset of the total B. henselae population came from a Dutch study of lymph nodes obtained from CSD patients, which revealed a higher prevalence of isolates displaying the 16S RNA-type I in tissue samples of CSD patients than among feline isolates obtained from the same geographic region [19]. Subsequent studies from Germany [4], [16], [20] and Australia [9] further supported the hypothesis that isolates responsible for human disease are not drawn randomly from the feline reservoir. Recent studies have shown that the delineation of B. henselae isolates into two genotypes based on the 16S rRNA sequence is not congruent with phylogenetic classifications using other genetic loci such as groEL, ftsZ and rpoB [18], [21]. In 2003, Iredell et al. developed a MLST scheme for B. henselae based on comparison of the nucleotide sequences of nine genetic loci [22]. Analysis of 37 feline and human B. henselae isolates from Australia by MLST revealed a considerable genetic diversity among feline isolates, while human isolates were more homogeneous [22].
We have recently validated the use of MLST for the definition of B. henselae strains by comparison with pulsed-field gel electrophoresis (PFGE) analysis [23]. MLST is a pangenomic approach that identifies very closely related bacterial isolates and allows the reconstruction of micro-evolutionary events [24]. In the present study, MLST was applied to a larger collection of feline and human B. henselae isolates from Europe, Australia and the USA in order to further investigate the association between ST, host species and geographical distribution. The clonal and phylogenetic relationships among the isolates was analysed using three different procedures: i) eBURST was used to define clonal lineages and reconstruct very recent events within each lineage [25], ii) a neighbour-joining tree was reconstructed based on concatenated MLST alleles, and iii) splits decomposition was used to detect inconsistent phylogenetic signals in the data indicative of recombination [26].
Results
Assignment of the B. henselae isolates to STs
From the 184 isolates studied, 182 isolates were assigned to 14 different STs. Two isolates could not be assigned to a ST because they contained different 16S RNA alleles; they will be described elsewhere. A new allele for rpoB was obtained from a feline isolate from Israel and designated as rpoB-allele 4. The nucleotide sequence of this allele has been deposited in Genbank (Accession No. EU289215) and the MLST web site (in preparation). Six STs were encountered for the first time in this study and designated as ST9 to ST14 in order of detection. The allelic profile, frequency of isolation and geographical distribution of STs, as well as a reference strain for each ST have been presented in Table 1. The average sequence divergence between all pairwise allelic comparisons was 0.5, 0.6, 0.5, 0.4, 0.3, 0.2, 1 and 0.3 percent for the rrs, batR, ftsZ, gltA, groEL, nlpD, ribC, and rpoB locus, respectively.
Table 1. Allelic profile, frequency of isolation, and geographic distribution of STs.
| ST | Reference strain | Host a | Country of origin b | rrs | batR | gltA | ftsZ | groEL | nlpD | ribC | rpoB | Frequency (n) | Distribution c |
| 1 | Houston-1 | H | USA | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 43 | AM, AU, EU |
| 2 | JR2 | H | AU | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | AU |
| 3 | HC62 | F | AU | 1 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | AU |
| 4 | HC35 | F | AU | 2 | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 11 | AU, EU |
| 5 | CA-1 | H | USA | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 38 | AM, AU, EU |
| 6 | Urlly8 (Marseille) | H | FR | 2 | 3 | 2 | 2 | 2 | 1 | 1 | 2 | 28 | AM, AU, EU |
| 7 | Berlin-2 | F | GE | 2 | 4 | 2 | 3 | 1 | 2 | 2 | 1 | 43 | EU |
| 8 | I107 | F | FR | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 7 | EU |
| 9 | Ber-K143 | F | GE | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 4 | AM, EU |
| 10 | G449 | F | UK | 2 | 3 | 2 | 1 | 2 | 1 | 1 | 2 | 2 | EU |
| 11 | I112 | F | FR | 2 | 3 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | EU |
| 12 | Is-959 | F | IS | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 4 | 1 | EU |
| 13 | C27 | F | CZ | 2 | 3 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | EU |
| 14 | FR96/BK36 | F | GE | 1 | 2 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | EU |
Reference strain represents the first isolate described or obtained with the allelic profile of a distinct ST.
F, feline; H, human
USA, United States of America; AU, Australia; FR, France; GE, Germany; UK, United Kingdom; IS, Israel; CZ, Czech Republic
Geographical distribution; EU, Europe (including Israel); AM, America; AU, Australia
The Urlly8 (Marseille) isolate displayed the rpoB-allele 2 and was assigned to ST6 in the present study (Table 1). These results are in accordance with data by Renesto et al. [27], Iredell et al. [22], and our previous results [23], but differ from the data presented by Lindroos et al. [28], who found an extra single nucleotide polymorphism in the rpoB allele of the Urlly8 isolate and assigned it to a new ST. The CA-1 isolate displayed the rrs- and groEL-alleles 2 in our study, corresponding to ST5. This is in accordance with previous results of different groups [18], [21], [29], but again differs from the data presented by Lindroos et al. [28], who obtained the rrs- and groEL-alleles 1 for the CA-1 isolate and assigned it to ST1.
Geographical distribution and frequency of STs
ST1, ST5, ST6, and ST7 were the most common STs, representing 23.6%, 20.9%, 15.4%, and 23.6% of the isolates, respectively. ST1, ST5 and ST6 were isolated in Europe, America and Australia, while ST7 was only distributed in Europe. ST4 and ST9 were distributed in two continents only, being absent from the USA and Australian samples respectively. The less common STs, noted in 1-7 isolates, were found in one continent only. Figure 1 shows the distribution of STs in different continents and among European countries that were represented by more than 10 isolates. The distribution of the major STs in different continents was found to be significantly non-random by chi-square test for STs 1 and 7 (p<0.00001 each), and ST6 (p = 0.039). In contrast, ST5 was evenly distributed among the three continents (p>0.1). The distribution of STs varied also considerably between different countries within Europe. The dominance of ST1 in Italy and Israel, but near absence in France, UK and Germany was particularly striking. To examine this further, we divided the European isolates in two subgroups: i) Mediterranean isolates including all isolates from Israel, Italy, and the Urlly8 (Marseille) isolate, and ii) North-western European isolates (NW-Europe) including all other European isolates. The relative frequency of major STs in different geographic regions was again evaluated by chi-square test (Table 2). This revealed a highly non-random distribution for STs 1 and 7 in Europe, these being over-represented in the Mediterranean and NW Europe, respectively.
Figure 1. Geographical distribution of B. henselae STs in different continents.

The lower panel shows the ST distribution in European countries that were represented by at least 10 isolates.
Table 2. Correlation between ST and geographic origin among 182 B. henselae isolates analysed.
| ST | Australia | North America | NW-Europe a | Mediterranean b | P c |
| 1 | 13 | 17 | 1 | 12 | <0.00001 |
| 2 | 1 | 0 | 0 | 0 | 0.165 d |
| 3 | 1 | 0 | 0 | 0 | 0.165 d |
| 4 | 4 | 0 | 7 | 0 | 0.090 d |
| 5 | 3 | 10 | 20 | 5 | 0.203 |
| 6 | 8 | 1 | 16 | 3 | 0.085 |
| 7 | 0 | 0 | 42 | 1 | <0.00001 |
| 8 | 0 | 0 | 6 | 1 | 0.281 d |
| 9 | 0 | 3 | 1 | 0 | 0.020 d |
| 10 | 0 | 0 | 2 | 0 | 0.630 d |
| 11 | 0 | 0 | 1 | 0 | 0.835 d |
| 12 | 0 | 0 | 0 | 1 | 0.073 d |
| 13 | 0 | 0 | 1 | 0 | 0.835 d |
| 14 | 0 | 0 | 1 | 0 | 0.835 d |
| Total | 30 | 31 | 98 | 23 |
North-western Europe including Denmark, Sweden, UK, the Netherlands, Germany, Czech Republic, France (isolates from Paris and Strasbourg), and Switzerland
Mediterranean region including Italy, Marseille (Urlly8 isolate), and Israel
As determined by chi square test
p values have a relative low reliability because of the small number of isolates in this ST
Relationship between ST and host species
The feline isolates (n = 158) were assigned to 13 STs. The human-derived isolates were assigned to 4 STs, including ST1 (16 isolates), ST5 (5 isolates), ST6 (2 isolates), and ST2 (1 isolate). Interestingly, ST7 was not encountered among the human isolates, although it was displayed by 43/121 (35.5%) of the feline isolates from Europe. Figure 2 shows the frequency of feline and human isolates within each ST, along with geographical source. The relative frequency of feline and human isolates within each ST was compared with their frequency in the whole panel by using Fisher's exact test. Human-derived isolates were over proportionally allocated to ST1 and under proportionally associated with ST7 (p≤0.001). The other major STs did not show a disproportional distribution pattern (Table 3).
Figure 2. Frequency of feline and human B. henselae isolates within each ST in correlation with the geographic origin of the isolates.

Table 3. Correlation between ST and host species in 182 B. henselae isolates analysed.
| ST | Feline (%) a | Human (%) b | Total (%) c | P d |
| 1 | 27 (17.1) | 16 (66.7) | 43 (23.6) | <0.00001 |
| 2 | 0 | 1 (4.2) | 1 (0.5) | 0.132 |
| 3 | 1 (0.6) | 0 | 1 (0.5) | 1 |
| 4 | 11 (7.0) | 0 | 11 (6) | 0.364 |
| 5 | 33 (20.9) | 5 (20.8) | 38 (20.9) | 1 |
| 6 | 26 (16.5) | 2 (8.3) | 28 (15.4) | 0.542 |
| 7 | 43 (27.2) | 0 | 43 (23.6) | 0.001 |
| 8 | 7 (4.4) | 0 | 7 (3.9) | 0.597 |
| 9 | 4 (2.5) | 0 | 4 (2.2) | 1 |
| 10 | 2 (1.3) | 0 | 2 (1.1) | 1 |
| 11 | 1 (0.6) | 0 | 1 (0.5) | 1 |
| 12 | 1 (0.6) | 0 | 1 (0.5) | 1 |
| 13 | 1 (0.6) | 0 | 1 (0.5) | 1 |
| 14 | 1 (0.6) | 0 | 1 (0.5) | 1 |
| Total | 158 (100) | 24 (100) | 182 (100) |
Frequency of each ST among feline isolates
Frequency of each ST among human isolates
Frequency of an individual ST among all isolates
As determined by Fisher's exact test
Evaluation of eno for the MLST scheme
The original MLST scheme proposed by Iredell et al. [22] contained eno, however, no allelic variability was found in the latter study or in subsequent studies [28]. We therefore decided to determine the eno sequence in a selected panel of isolates to evaluate its appropriateness for the B. henselae MLST scheme. Fifty isolates that represented every ST and corresponded with their frequency of isolation were analysed. We did not find any allelic diversity among these isolates. Since the selected 50 isolates represent the most heterogeneous B. henselae strain collection analysed so far, we conclude that this region of the eno gene is not an appropriate target for the B. henselae MLST scheme.
Phylogenetic analysis
The relationships between the STs was first examined using eBURST (Figure 3), which uses allele profiles rather than sequences and does not attempt to reconstruct the relationships between the different clonal lineages. The majority of the isolates (107/182; 58.8%) corresponded to nine STs, which formed a single clonal complex, Group 1, with ST5 as primary founder. A clonal complex contains STs that have 7 out of 8 alleles in common and a primary founder is the ST with the largest number of single locus variants (SLVs) within a clonal complex. ST5 corresponded to 38/182 (20.8%) of the isolates, which is consistent with its positioning as the founder of STs 9, 4, 12 and 2. The links connecting the other STs in this complex, STs 1, 3, 8 and 14, are less certain, and may have been obscured by recombination. Two doublets were also identified (ST6-ST10, Group 2, and ST13-ST11, Group 3), whilst ST7 differed in two or more genes from every other isolate and was therefore assigned as a singleton.
Figure 3. Phylogenetic relationship between different B. henselae STs as determined by eBURST.

A clonal complex contains STs that have 7 out of 8 alleles in common. ST7 is assigned as a singleton since it differed in 3–7 alleles from all other STs. The size of the circles relates to the frequency of the corresponding ST, and illustrates that the assigned primary founder of the major clonal complex (ST5) is a common clone.
Phylogenetic analysis of the data was carried out by reconstructing a neighbour-joining tree based on concatenated sequences as implemented in MEGA4 [30] (Figure 4). Although the bootstrap support on this tree was generally poor, owing to a paucity of informative sites and possibly a history of recombination, 75% of 1,000 bootstrap trees supported the delineation of Group 1 from the other STs. This is consistent with the hypothesis that it represents a real division within the B. henselae population.
Figure 4. Neighbour-joining tree of the concatenated sequences of B. henselae STs as reconstructed by MEGA4.
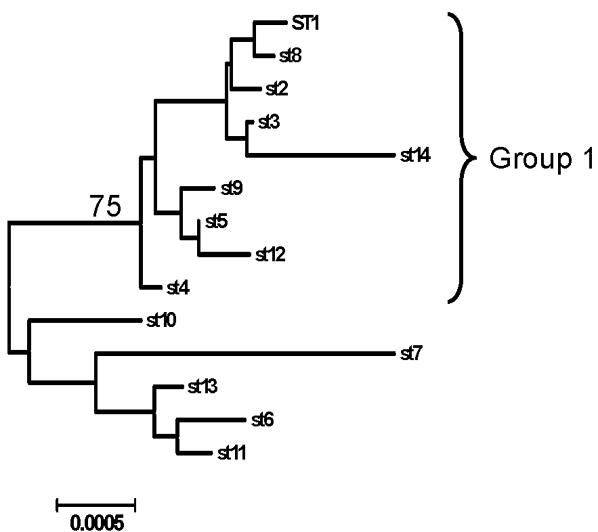
1,000 bootstrap replicates were used to examine the confidence in the tree. The only node to score above 60 is the one leading to Group 1 (75%, as indicated), indicating that the delineation of Group 1 represents a real division in the B. henselae population.
Iredell et al [22] noted evidence for recombination from their MLST data, and to explore this issue further we used splits decomposition analysis as implemented in Splitstree4 [26]. The approach examines the degree to which the data correspond to a bifurcating tree, which would indicate limited recombination, or alternatively a network structure, which would be consistent with more frequent recombination. Figure 5 shows that the approach resulted in extensive reticulation between the STs, which is consistent with a history of recombination. Furthermore, the phi test, as implemented in Splitstree4, revealed significant evidence for recombination (P<0.004). Splits decomposition analysis also confirmed the delineation of Group 1, and placed ST7 as a distinct genotype more closely related to Groups 2 and 3 than to Group 1.
Figure 5. Splits decomposition was used to detect evidence for a past history of recombination in the sequences.
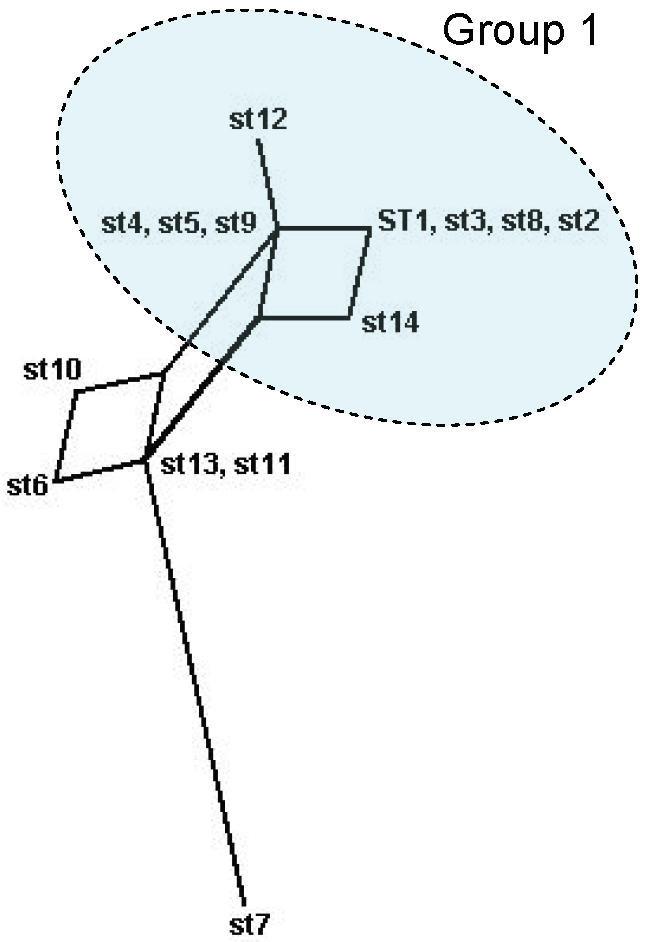
The extensive reticulation suggests that recombination has occurred relatively frequently. However, Group 1 remains distinct (as indicated by the filled oval).
Discussion
In this study, a collection of 182 B. henselae isolates from 12 countries and three continents was analysed by MLST to elucidate i) the relationship between ST and host species, ii) the geographical distribution of STs, and iii) the phylogenetic relationship among different STs. To our knowledge, this is the largest B. henselae collection that has been analysed by MLST or other molecular typing techniques hitherto. We have tried to minimise sampling artefacts by making every effort to include human isolates from all geographic regions that were represented by feline isolates. However, human isolates were not available from some regions or were outnumbered by feline isolates in other areas. In addition, as the isolates were collected by different investigators in different settings and during a long period of time (approximately 15 years), we can not exclude temporal or seasonal variations or a bias caused by the population (stray versus pet) or breed of the cats examined. Therefore, the panel is still not truly representative of the natural population of B. henselae.
Fourteen STs were encountered among 182 B. henselae isolates. ST1, ST5, ST6 and ST7 represented major STs and accounted for 83.5% of the isolates. The geographical distribution of STs was not homogeneous. ST1, ST5 and ST6 were found in three continents, suggesting that they may be distributed world wide. In contrast, ST7 was detected only in Europe, suggesting that its distribution may be restricted to Europe. The differences in distribution of STs 1, 6 and 7 on three continents were statistically significant. The distribution of STs varied also between different European countries. ST7 was more prevalent in North and West Europe (UK, Sweden, Denmark, Germany, the Netherlands, France), whereas ST1 was more frequently obtained from the Mediterranean region (Italy, Israel).
We found a significant correlation between distinct STs and human disease. ST1 was statistically significantly associated with human infection, suggesting that it represents a hypervirulent strain. This finding is in accordance with data by Iredell et al. [22], who found a significant association of ST1 with CSD in Australia. They contradict the results by Lindroos et al. [28], who did not find a disproportional association between a distinct ST and human-derived isolates. This discrepancy might be due to differences in size and composition of the panels of isolates. In the latter study, the panel was smaller (n = 38), composed to 60.5% of ST1, and did not contain matched human and feline isolates from the same geographic regions.
ST7 was underrepresented among the human isolates in our study, suggesting that ST7 may be less virulent for humans. However, we can not completely rule out the possibility that the absence of ST7 among the human isolates could be due to a bias in composition of our panel, which contained more feline than human isolates from countries with a higher prevalence of ST7 (e.g. Germany, UK, and France). Further studies with more human-derived isolates from Europe would help to evaluate this hypothesis.
eBURST and phylogenetic analyses were broadly consistent and revealed a major division within the population of B. henselae. Of the four predominant genotypes, ST1 and ST5 are related and belong to the major clade, Group 1. ST6 belongs to the minor clade, Group 2, and ST7 is a distinct genotype, probably more closely related to Groups 2 and 3 than to Group 1. Our analysis also supports previous studies which have suggested a history of recombination between the isolates. This is further supported by the “straggly” shape of the major clonal complex as revealed by eBURST. Recent simulation studies have shown that such a structure is indicative of frequent recombination [31].
In summary, our data indicate that different STs of B. henselae may vary with regard to virulence for humans. It can be hypothesized that ST1 might possess additional virulence factors, which could encode for a more effective transmission from cats to humans, or a better survival of the pathogen in the human host. It can also be speculated that ST7 may lack one or more virulence determinants, and lower transmission potential may possibly account for the restriction of this genotype to Europe. Future studies using comparative genomic or proteomic approaches could help to identify and characterize these factors. The MLST approach has been previously used for tracking hypervirulent or antibiotic resistant lineages in other bacterial pathogens, e.g. Neisseria meningitidis, Streptococcus pneumoniae or Staphylococcus aureus [24], [25]. MLST data are unambiguous and can be easily transferred electronically between laboratories. Furthermore, MLST can be applied directly to clinical specimens and is therefore not strictly dependent on culture. It can be expected that more MLST data will become available in future, and the establishment of a B. henselae site on www.mlst.net should greatly facilitate this.
Materials and Methods
Bartonella isolates
One-hundred and eighty four B. henselae isolates collected by different investigators in several European countries, Australia, and the USA were analysed. One-hundred and sixty isolates were isolated from feline blood, and 24 isolates were obtained from human tissue specimens, including lymph node, cutaneous BA lesion, and blood. Table 4 summarises the epidemiological data of the isolates studied. Bacteria were stored at −20°C or −80°C until use. The isolates were grown on Columbia blood agar with 5% sheep blood (Becton Dickenson) at 37°C in 5% CO2 for 7–14 d, and passaged once on agar prior to isolation of bacterial DNA.
Table 4. Geographic origin, host species and clinical source of B. henselae isolates.
| Country | Cat | Human | Total | Isolate obtained from/reference | ||
| CSD | bacteremia, BAa | unknown | ||||
| Australia | 17 | 13 | 30 | J. Iredell [22] | ||
| Czech Republic | 2 | 2 | O. Melter [32] | |||
| Denmark | 3 | 3 | R. Birtles | |||
| France | 16 | 1 | 17 | H-J. Boulouis, P-E. Fournier, Y. Piémont [12], [33] | ||
| Germany | 49 | 1 | 50 | our group, A. Sander, D. Schimmel [13], [20], [34] | ||
| Israel | 9 | 3 | 12 | M. Giladi | ||
| Italy | 11 | 11 | M. Fabbi [35] | |||
| Netherlands | 5 | 5 | A. Bergmans [14] | |||
| Sweden | 2 | 2 | E. Hjelm | |||
| Switzerland | 1 | 1 | P-E. Fournier [29] | |||
| UK | 20 | 20 | R. Birtles [36] | |||
| USA | 25 | 1 | 3 | 2 | 31 | B. Anderson, L. Guptill [7], [8], [37] |
| Total | 158 | 18 | 4 | 182 | ||
BA, bacillary angiomatosis
MLST
Nucleotide sequence data were collected from all B. henselae isolates for approximately 320–500 bp fragments of eight genetic loci (16S rRNA [rrs], batR, gltA, groEL, ftsZ, nlpD, ribC, and rpoB) as described previously [22], [23]. In addition, the partial sequence of eno was determined for 50 isolates [22]. All sequences were determined for both strands and the results were confirmed by repeats when necessary. The reliability of the sequence data was controlled by subjecting 20 randomly selected isolates in a blinded manner as “quality control strains” to MLST analysis. The results of the quality control strains were compared with the data obtained from the “original isolates”. The MLST results were 100% consistent for each pair of quality control strain-original isolate.
Analysis of MLST Data
The nucleotide sequences were analysed with the DNASTAR Lasergene software package 7 (DNASTAR, Madison, USA). Alleles and STs were assigned in accordance with the published data [22], [23]. New alleles were confirmed by repeats and the sequence was deposited in GenBank (see below). New allelic combinations that were encountered for the first time in this study were assigned to new STs in order of detection.
Phylogenetic analysis
The definition of clonal complexes and the examination of relationships between STs within clonal complexes were carried out by using eBURST (http://eburst.mlst.net). A neighbour-joining tree was reconstructed from the concatenated MLST alleles using the kimura-2-parameter distance measures as implemented in MEGA4 [30]. Splits decomposition analysis and the phi test were carried out using the default settings in Splitstree4 [26].
Statistical analysis
Chi square test was used to compare the geographical distribution patterns of major STs. Two-tailed Fisher's exact test was used to compare the frequency of feline and human-derived isolates within a ST with the frequency within the whole panel. P values of <0.05 were considered significant.
Nucleotide sequence accession number
A new rpoB allele was encountered from the isolate Is-959 and was designated as rpoB-allele 4. This allele contains a single nucleotide variation (G instead of A) at position 711758 of the B. henselae Houston-1 chromosome (Accession No. BX897699.1). The rpoB-allele 4 sequence has been deposited in GenBank under the Accession No. EU289215. The data were also deposited at http://www.mlst.net (in preparation).
Acknowledgments
We thank Drs. Burt Anderson, Anneke Bergmans, Richard Birtles, Massimo Fabbi, Pierre-Edouard Fournier, John Iredell, Lynn Guptill, Eva Hjelm, Oto Melter, Yves Piémont, Anna Sander, and Dietrich Schimmel for providing B. henselae isolates.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This project was supported by a grant from the Deutsche Forschungsgemeinschaft to MA.
References
- 1.Margileth AM. Cat scratch disease. Adv Pediatr Infect Dis. 1993;8:1–21. [PubMed] [Google Scholar]
- 2.Koehler JE, Quinn FD, Berger TG, LeBoit PE, Tappero JW. Isolation of Rochalimaea species from cutaneous and osseous lesions of bacillary angiomatosis. N Engl J Med. 1992;327:1625–1631. doi: 10.1056/NEJM199212033272303. [DOI] [PubMed] [Google Scholar]
- 3.Guptill L, Slater L, Wu CC, Lin TL, Glickman LT, et al. Experimental infection of young specific pathogen-free cats with Bartonella henselae. J Infect Dis. 1997;176:206–216. doi: 10.1086/514026. [DOI] [PubMed] [Google Scholar]
- 4.Sander A, Posselt M, Bohm N, Ruess M, Altwegg M. Detection of Bartonella henselae DNA by two different PCR assays and determination of the genotypes of strains involved in histologically defined cat scratch disease. J Clin Microbiol. 1999;37:993–997. doi: 10.1128/jcm.37.4.993-997.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avidor B, Varon M, Marmor S, Lifschitz-Mercer B, Kletter Y, et al. DNA amplification for the diagnosis of cat-scratch disease in small-quantity clinical specimens. Am J Clin Pathol. 2001;115:900–909. doi: 10.1309/Y468-82G5-ACHW-YRMV. [DOI] [PubMed] [Google Scholar]
- 6.Arvand M, Schäd SG. Isolation of Bartonella henselae DNA from the peripheral blood of a patient with cat scratch disease up to 4 months after the cat scratch injury. J Clin Microbiol. 2006;44:2288–2290. doi: 10.1128/JCM.00239-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slater LN, Welch DF, Hensel D, Coody DW. A newly recognized fastidious gram-negative pathogen as a cause of fever and bacteremia. N Engl J Med. 1990;323:1587–1593. doi: 10.1056/NEJM199012063232303. [DOI] [PubMed] [Google Scholar]
- 8.Dolan MJ, Wong MT, Regnery RL, Jorgensen JH, Garcia M, et al. Syndrome of Rochalimaea henselae adenitis suggesting cat scratch disease. Ann Intern Med. 1993;118:331–336. doi: 10.7326/0003-4819-118-5-199303010-00002. [DOI] [PubMed] [Google Scholar]
- 9.Dillon B, Valenzuela J, Don R, Blanckenberg D, Wigney DI, et al. Limited Diversity among Human Isolates of Bartonella henselae. J Clin Microbiol. 2002;40:4691–4699. doi: 10.1128/JCM.40.12.4691-4699.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drancourt M, Birtles R, Chaumentin G, Vandenesch F, Etienne J, et al. New serotype of Bartonella henselae in endocarditis and cat-scratch disease. Lancet. 1996;347:441–443. doi: 10.1016/s0140-6736(96)90012-4. [DOI] [PubMed] [Google Scholar]
- 11.Chomel BB, Abbott RC, Kasten RW, Floyd-Hawkins KA, Kass PH, et al. Bartonella henselae prevalence in domestic cats in California: risk factors and association between bacteremia and antibody titers. J Clin Microbiol. 1995;33:2445–2450. doi: 10.1128/jcm.33.9.2445-2450.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heller R, Artois M, Xemar V, De Briel D, Gehin H, et al. Prevalence of Bartonella henselae and Bartonella clarridgeiae in stray cats. J Clin Microbiol. 1997;35:1327–1331. doi: 10.1128/jcm.35.6.1327-1331.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sander A, Buhler C, Pelz K, von Cramm E, Bredt W. Detection and identification of two Bartonella henselae variants in domestic cats in Germany. J Clin Microbiol. 1997;35:584–587. doi: 10.1128/jcm.35.3.584-587.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bergmans AM, de Jong CM, van Amerongen G, Schot CS, Schouls LM. Prevalence of Bartonella species in domestic cats in The Netherlands. J Clin Microbiol. 1997;35:2256–2261. doi: 10.1128/jcm.35.9.2256-2261.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Branley J, Wolfson C, Waters P, Gottlieb T, Bradbury R. Prevalence of Bartonella henselae bacteremia, the causative agent of cat scratch disease, in an Australian cat population. Pathology. 1996;28:262–265. doi: 10.1080/00313029600169124. [DOI] [PubMed] [Google Scholar]
- 16.Sander A, Ruess M, Bereswill S, Schuppler M, Steinbrueckner B. Comparison of different DNA fingerprinting techniques for molecular typing of Bartonella henselae isolates. J Clin Microbiol. 1998;36:2973–2981. doi: 10.1128/jcm.36.10.2973-2981.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez-Barradas MC, Hamill RJ, Houston ED, Georghiou PR, Clarridge JE, et al. Genomic fingerprinting of Bartonella species by repetitive element PCR for distinguishing species and isolates. J Clin Microbiol. 1995;33:1089–1093. doi: 10.1128/jcm.33.5.1089-1093.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeaiter Z, Fournier PE, Raoult D. Genomic variation of Bartonella henselae strains detected in lymph nodes of patients with cat scratch disease. J Clin Microbiol. 2002;40:1023–1030. doi: 10.1128/JCM.40.3.1023-1030.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergmans AM, Schellekens JF, van Embden JD, Schouls LM. Predominance of two Bartonella henselae variants among cat-scratch disease patients in the Netherlands. J Clin Microbiol. 1996;34:254–260. doi: 10.1128/jcm.34.2.254-260.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arvand M, Klose AJ, Schwartz-Porsche D, Hahn H, Wendt C. Genetic variability and prevalence of Bartonella henselae in cats in Berlin, Germany, and analysis of its genetic relatedness to a strain from Berlin that is pathogenic for humans. J Clin Microbiol. 2001;39:743–746. doi: 10.1128/JCM.39.2.743-746.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehrenborg C, Wesslen L, Jakobson A, Friman G, Holmberg M. Sequence variation in the ftsZ gene of Bartonella henselae isolates and clinical samples. J Clin Microbiol. 2000;38:682–687. doi: 10.1128/jcm.38.2.682-687.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iredell J, Blanckenberg D, Arvand M, Grauling S, Feil EJ, et al. Characterization of the natural population of Bartonella henselae by multilocus sequence typing. J Clin Microbiol. 2003;41:5071–5079. doi: 10.1128/JCM.41.11.5071-5079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arvand M, Viezens J. Evaluation of pulsed-field gel electrophoresis and multi-locus sequence typing for the analysis of clonal relatedness among Bartonella henselae isolates. Int J Med Microbiol. 2007;297:255–262. doi: 10.1016/j.ijmm.2007.02.001. Epub 2007 Mar 2030. [DOI] [PubMed] [Google Scholar]
- 24.Feil EJ, Enright MC. Analyses of clonality and the evolution of bacterial pathogens. Curr Opin Microbiol. 2004;7:308–313. doi: 10.1016/j.mib.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. Epub 2005 Oct 2012. [DOI] [PubMed] [Google Scholar]
- 27.Renesto P, Gouvernet J, Drancourt M, Roux V, Raoult D. Use of rpoB gene analysis for detection and identification of Bartonella species. J Clin Microbiol. 2001;39:430–437. doi: 10.1128/JCM.39.2.430-437.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindroos H, Vinnere O, Mira A, Repsilber D, Naslund K, et al. Genome Rearrangements, Deletions, and Amplifications in the Natural Population of Bartonella henselae. J Bacteriol. 2006;188:7426–7439. doi: 10.1128/JB.00472-06. Epub 2006 Aug 7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.La Scola B, Liang Z, Zeaiter Z, Houpikian P, Grimont PA, et al. Genotypic characteristics of two serotypes of Bartonella henselae. J Clin Microbiol. 2002;40:2002–2008. doi: 10.1128/JCM.40.6.2002-2008.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. Epub 2007 May 1597. [DOI] [PubMed] [Google Scholar]
- 31.Turner KM, Hanage WP, Fraser C, Connor TR, Spratt BG. Assessing the reliability of eBURST using simulated populations with known ancestry. BMC Microbiol. 2007;7:30. doi: 10.1186/1471-2180-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melter O, Hercik K, Weyant RS, Janecek J, Nemec A, et al. Detection and characterization of feline Bartonella henselae in the Czech Republic. Vet Microbiol. 2003;93:261–273. doi: 10.1016/s0378-1135(03)00032-4. [DOI] [PubMed] [Google Scholar]
- 33.Gurfield AN, Boulouis HJ, Chomel BB, Kasten RW, Heller R, et al. Epidemiology of Bartonella infection in domestic cats in France. Vet Microbiol. 2001;80:185–198. doi: 10.1016/s0378-1135(01)00304-2. [DOI] [PubMed] [Google Scholar]
- 34.Arvand M, Wendt C, Regnath T, Ullrich R, Hahn H. Characterization of Bartonella henselae isolated from bacillary angiomatosis lesions in a human immunodeficiency virus-infected patient in Germany. Clin Infect Dis. 1998;26:1296–1299. doi: 10.1086/516348. [DOI] [PubMed] [Google Scholar]
- 35.Fabbi M, De Giuli L, Tranquillo M, Bragoni R, Casiraghi M, et al. Prevalence of Bartonella henselae in Italian stray cats: evaluation of serology to assess the risk of transmission of Bartonella to humans. J Clin Microbiol. 2004;42:264–268. doi: 10.1128/JCM.42.1.264-268.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birtles RJ, Laycock G, Kenny MJ, Shaw SE, Day MJ. Prevalence of Bartonella species causing bacteraemia in domesticated and companion animals in the United Kingdom. Vet Rec. 2002;151:225–229. doi: 10.1136/vr.151.8.225. [DOI] [PubMed] [Google Scholar]
- 37.Guptill L, Wu CC, HogenEsch H, Slater LN, Glickman N, et al. Prevalence, risk factors, and genetic diversity of Bartonella henselae infections in pet cats in four regions of the United States. J Clin Microbiol. 2004;42:652–659. doi: 10.1128/JCM.42.2.652-659.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]