

Abstract
Adoptive transfer of genetically T-cell receptor (TCR)–modified lymphocytes has been recently reported to cause objective cancer regression. However, a major limitation to this approach is the mispairing of the introduced chains with the endogenous TCR subunits, which leads to reduced TCR surface expression and, subsequently, to their lower biological activity. We here show that it is possible to improve TCR gene transfer by adding a single cysteine on each receptor chain to promote the formation of an additional interchain disulfide bond. We show that cysteine-modified receptors were more highly expressed on the surface of human lymphocytes compared with their wild-type counterparts and able to mediate higher levels of cytokine secretion and specific lysis when cocultured with specific tumor cell lines. Furthermore, cysteine-modified receptors retained their enhanced function in CD4+ lymphocytes. We also show that this approach can be employed to enhance the function of humanized and native murine receptors in human cells. Preferential pairing of cysteine-modified receptor chains accounts for these observations, which could have significant implications for the improvement of TCR gene therapy.
Introduction
Cellular adoptive immunotherapy has been shown to mediate the regression of large solid tumors in 50% of patients with metastatic melanoma (1). A limitation of this treatment is the need to isolate and expand tumor-reactive lymphocytes that preexist in the patient. Those requirements can be overcome by transducing genes encoding antitumor T-cell receptor (TCR), thus providing an alternative source of tumor-specific lymphocytes. We recently reported that this approach lead to objective cancer regression in two patients (2). However, one of the factors that might have influenced the response rate observed in this study (13%) could be the reduced level of TCR surface expression partly caused by the mispairing of the introduced chains with the endogenous TCR subunits. Therefore, there is a need to develop novel approaches that will result in exclusive or preferential pairing of exogenous TCR chains.
In that regard, we recently showed that TCR in which the human constant regions were replaced with murine ones functioned better in human cells, leading to an increased sensitivity to tumor cells (3). Although the possible antigenicity of murine constant regions expressed in human patients remains to be proven, we sought to develop additional strategies that could promote preferential pairing of the introduced TCR. Boulter et al. developed an approach to stabilize soluble TCR by mutating defined residues in the constant regions to cysteines to generate an additional disulfide bond between the TCR chains (4). Herein, we sought to examine whether this additional disulfide bond introduced into native TCR would function in the context of a living cell. Several TCRs that were accordingly mutated exhibited enhanced surface expression, tumor recognition, and more efficient pairing. These results could have significant implications for the improvement of TCR gene therapy in patients.
Materials and Methods
Patient peripheral blood mononuclear cells and cell lines
All peripheral blood mononuclear cells (PBMC) used in this study were from metastatic melanoma patients treated at the Surgery Branch, National Cancer Institute, NIH, Bethesda, MD. Jurkat/RT3-T3.5 is a surface TCR-negative mutant (ATCC TIB-153). Melanoma cell lines HLA-A2+/MART-1+ (526 and 624) and HLA-A2− (888 and 938) were previously described (5). All cells were cultured in RPMI 1640 with 10% fetal bovine serum (Biofluids, Rockville, MD). Lymphocytes were cultured in AIM-V medium (Invitrogen, Carlsbad, CA) with 5% human serum (Valley Biomedical, Winchester, VA) and 300 IU/mL interleukin 2.
Cysteine-modified TCR generation and cloning
We generated cysteine-modified α and β chains from different TCRs using a PCR megaprimer approach. We generated the first fragment by amplifying the chains with a 5′-specific primer and either the α-Cys48rev primer, TAGACCTCATGTCTAGCACGCATTTGTCTGTGATATACACATC or the β-Cys57rev primer, TGAGGGGCTGCGGGTCCGTGCAGACCCCACTGTGCAC-CTCC for the α and β chains, respectively (mutated bases are underlined). The product of this reaction was used as a megaprimer to amplify full-length TCR chain with a 3′-specific primer. The full-length chain was subcloned into the pGEM-4Z/64A vector as previously described (3).
Electroporation of peripheral blood lymphocytes
This technique has been extensively described in our previous reports (3, 6). Briefly, in vitro transcribed mRNA encoding α and β TCR chains generated using mMESSAGE-mMACHINE (Ambion, Austin, TX) was electroporated into OKT3-stimulated lymphocytes using an ElectroSquarePorator ECM-830 (BTX, San Diego, CA). The amount of in vitro transcribed mRNA for each chain was 1.5 μg per 106 PBMCs unless indicated otherwise.
Antibodies and MHC multimers
Human Vβ12 antibody phycoery-thrin-conjugated was supplied by Immunotech (Westbrook, ME). p53264-272/HLA-A2 pentamer was supplied by ProImmune (Oxford, United Kingdom), and MART-127L/HLA-A2 tetramer was supplied by Beckman Coulter (San Jose, CA).
Cytokine release assays
Peripheral blood lymphocytes (PBL) cultures were tested for reactivity in cytokine release assays using commercially available ELISA kits (Endogen, Cambridge, MA).
Statistical analysis
Results of cytokine secretion shown as mean ± SD were compared using Student’s t test. The P obtained is indicated in the figure legends.
51Cr release assay
The ability of the electroporated PBLs to lyse targets was measured using a 51Cr release assay as described (5).
CD4/CD8 separation
CD4+ and CD8+ populations were separated using commercially available kits (Dynal Biotech, Brown Deer, WI and Miltenyi Biotech, Auburn, CA).
Results and Discussion
Engineering of TCR with an additional disulfide bond mediates enhanced expression and biological activity in human PBLs
We designed a mutant NY-ESO-1–specific TCR (1G4) in which we replaced the Thr48 on the α chain and Ser57 on the β chain with cysteines to facilitate the creation of an additional disulfide bond between the TCR constant regions (ref. 4; Fig. 1A). Boulter et al. originally showed the successful stabilization of the soluble form of this TCR by the addition of the same nonnative interchain disulfide bond, later confirmed by crystallographic analysis (4). We electroporated OKT3-stimulated human PBLs with mRNA encoding the TCR α and β chains from 1G4 or its cysteine-modified form (1G4-Cys) and cocultured those cells with different NY-ESO+ cell lines. RNA electroporation enables the use of normalized levels of mRNA and is, unlike other common gene transfer methods, not dependent on retroviral vector titer, insertion sites, or the influence of diverse transcription mechanisms (3, 6). We observed an increased secretion of IFN-γ mediated by 1G4-Cys compared with the wild-type receptor 1G4 (e.g., 2,639 versus 942 pg/mL for 624.38, respectively, in Fig. 1B). It is interesting to note that the validity of observations first shown in soluble molecules (4) could be extended to cellular systems, paving the way for novel immunologic approaches for the study of TCR structure and function.
Figure 4.
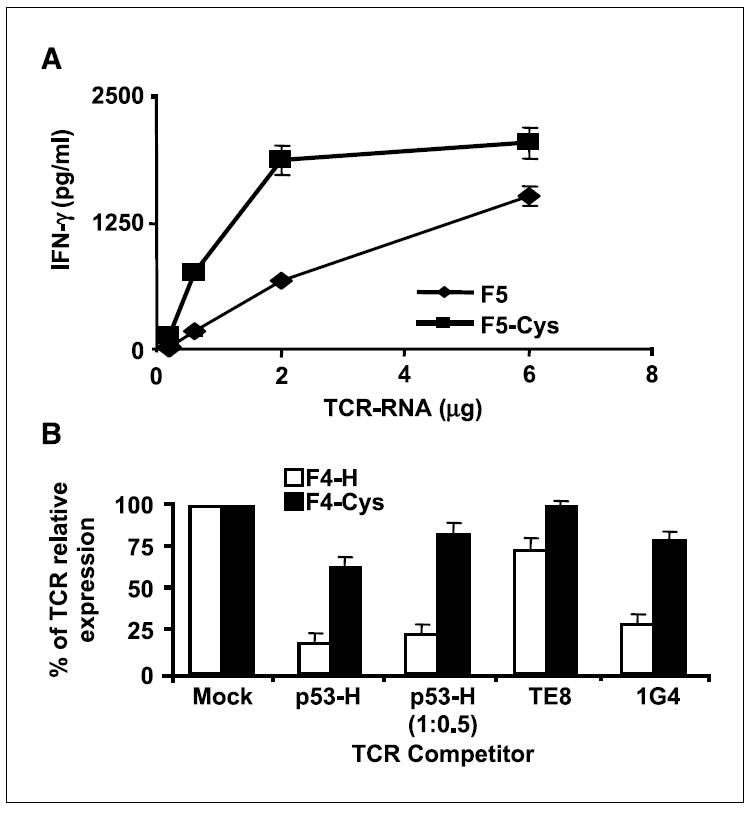
Efficient and preferential pairing of cysteine-modified TCRs. A, TCR titration. OKT3-stimulated PBLs, electroporated with different amount of mRNA (0.2, 0.6, 2, and 6 μg total) encoding either the wild-type receptor F5 or its cysteine-modified version F5-Cys, were cocultured with the melanoma tumor 526. Twenty-four hours after the beginning of the coculture, the concentration of IFN-γ secreted in the medium was measured using an ELISA procedure. B, TCR competition assay. TCR-deficient Jurkat/RT3-T3.5 cells were electroporated with 1 μg of each chain of F4 (white) or F4-Cys (black) along with 1 μg of each chain of the competitor TCR: 1G4, TE-8 (an NY-ESO-1 class I–restricted TCR), and p53-H [for p53-H, we also used an additional amount (i.e., 0.5 μg) indicated as p53-H (1:0.5)]. Twenty-four hours after electroporation, the cells were stained with MART-1 tetramer, and the percentage of MART-1 TCR relative expression was calculated by dividing percentage of tetramer-positive cells of a given sample by that of the control sample (without competitor TCR). All the differences were found to be statistically significant based on Student’s t test (P < 0.001).
Figure 1.
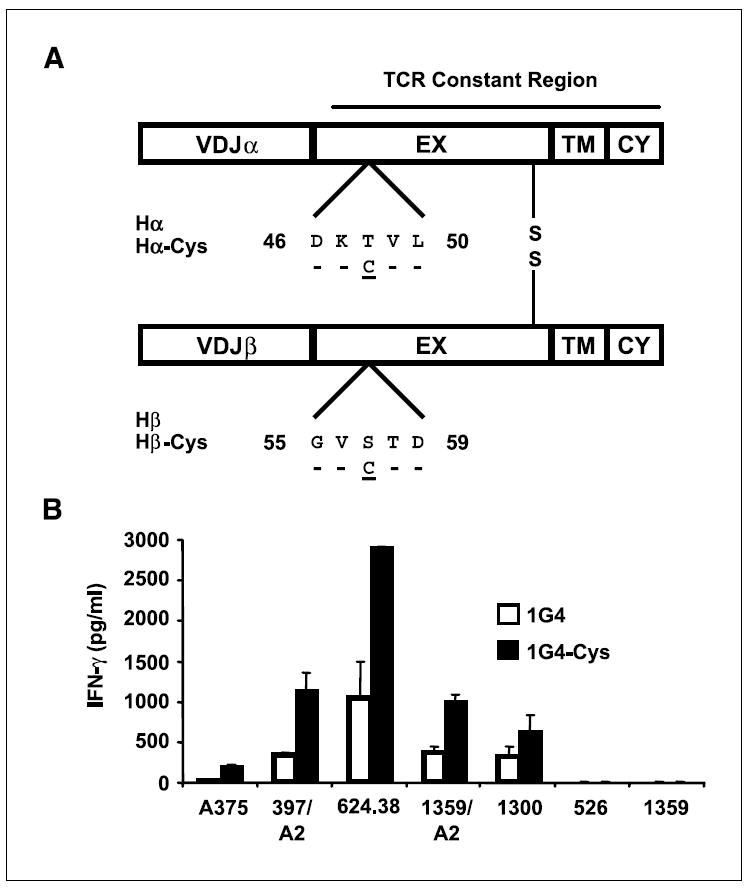
TCR modification and function. A, schematic representation of the cysteine-modified TCR chains. The numbers indicate amino acid position according to the Immunogenetics database (18). EX, extracellular region; TM, transmembrane region; CY, cytoplasmic regions. Hα or Hβ, sequence of the wild-type TCR constant region; Hα-Cys or Hβ-Cys show where the cysteine residue was inserted. B, human PBLs were electroporated with 1G4 or 1G4-Cys and cocultured with HLA-A2+/NY-ESO-1+ cell lines (A375, 397/A2, 624.38, 1359/A2, and 1300) and with the control cell lines 526 (HLA-A2+/NY-ESO-1−) and 1359 (HLA-A2−/NY-ESO-1+). Twenty-four hours after the beginning of the coculture, the concentration of cytokine secreted in the medium (IFN-γ), normalized to the amount of mRNA used, was measured using an ELISA procedure. The difference between 1G4 and 1G4-Cys, based on seven independent experiments, was found to be statistically significant (P = 0.01).
The TCR recently showed to mediate cancer regression in two patients following infusion of genetically modified lymphocytes was an anti-MART-1–specific TCR (termed F4) that was derived from a persistent tumor-infiltrating lymphocyte clone isolated from a melanoma patient who experienced cancer regression following adoptive cell therapy (patient 9 in ref. 1). We next sought to determine if the cysteine modification could improve this TCR. To compare relative levels of TCR surface expression of F4, we electroporated OKT3-stimulated PBLs with mRNA for each TCR chain (wild type and mutant) and, 24 h after electroporation, stained the cells with MART-1-tetramer. Cells that expressed the cysteine-modified TCR F4-Cys had a higher proportion of tetramer-positive cells (70%) and had a greater mean fluorescence intensity (MFI = 151) than the lymphocytes that expressed the wild-type F4 (35%; MFI = 85; Fig. 2A).
Figure 2.

Expression and function of the cysteine-modified anti-MART-1 TCR F4. A and B, comparison of the surface expression of the wild-type F4 and its cysteine-modified F4-Cys. OKT3-stimulated PBLs were electroporated with mRNA encoding different combinations of F4-TCR chains: F4, F4-Cys, the α chain of the wild-type F4 + the β chain of F4-Cys (noted as F4 α/β Cys), or the opposite noted as F4 α Cys /β. Twenty-four hours after electroporation, we assessed MART-1 tetramer (A) and Vβ12 (B) binding. Percentage of positive cells and the relative MFI (in brackets). The difference of TCR surface expression, based on 13 independent experiments done with 10 different donors, was found to be statistically significant (P < 0.001). C, recognition of tumor lines. Human PBLs were electroporated with the F4 (white), F4-Cys (black), the α chain of the wild-type F4 + the β chain of F4-Cys (noted as F4 α/β Cys; horizontal hatching), or the opposite noted as F4 α Cys/β (diagonal hatching). The electroporated cells (1 × 105) were cocultured with the HLA-A2+ 526 and 624 and the HLA-A2− 888 and 938 melanoma cell lines (1 × 105). Twenty-four hours after the beginning of the coculture, the concentration of IFN-γ and GM-CSF secreted in the medium were measured using an ELISA procedure. This was observed in independent experiments for 10 different donors, and the difference between F4 and F4-Cys was found to be statistically significant (P < 0.001). D, specific killing of tumor cell lines. CD8+ purified human PBLs expressing F4 (◆), F4-Cys (■), or mock electroporated (○) were cocultured for 3 h with the indicated tumor cell lines previously labeled with 51Cr. Specific lysis was measured at the effector/target (E/T) ratios indicated: (specific release − spontaneous release) / (total release − spontaneous release). We used the following melanoma cell lines: HLA-A2+ (526 and 624) and HLA-A2− (888 and 938) as control lines.
Both F4 and F4-Cys showed similar levels of Vβ12 staining (Fig. 2B), which suggested that the enhanced tetramer staining was likely the result of better pairing of the cysteine-modified chains rather than enhanced surface expression. In parallel, we electroporated PBLs with different F4 chain combinations [i.e., the F4 wild-type α chain with the cysteine-modified β (noted as F4-α/βCys), or the opposite (F4-αCys/β)]. PBLs expressing both F4 combinations α/βCys and αCys/β stained with MART-1 tetramer (23% and 25%, respectively). This shows that the αCys or βCys can pair with wild-type β or α chain, respectively, but that cysteines on both chains are simultaneously needed to achieve enhanced TCR expression.
We determined if the enhanced surface expression of F4-Cys was correlated with a higher biological activity by coculturing PBLs expressing F4 or F4-Cys with different human melanoma tumor lines. HLA-A2+ melanoma tumors (526 and 624) specifically stimulated MART-1 TCR-expressing T cells to secrete cytokines IFN-γ and granulocyte macrophage colony-stimulating factor (GM-CSF; Fig. 2C). F4-Cys was able to trigger up to 5-fold more cytokine secretion than F4. No significant cytokine secretion was noted in cocultures with control HLA-A2− melanoma lines (888 and 938). In addition, we observed TCR down-regulation in cells expressing either F4 or F4-Cys following overnight coculture with cells pulsed with the MART-1 epitope (data not shown), which shows that TCR internalization is not influenced by the cysteine modifications.
Additionally, cell-mediated cytotoxicity of PBLs expressing either F4 or F4-Cys was compared in a 3-h 51Cr release assay. CD8+ PBLs were electroporated with mRNA encoding both anti-MART-1 TCRs and cocultured with 51Cr-labeled tumors. Although both TCRs were able to mediate specific lysis of HLA-A2+ melanoma lines (Fig. 2D), the lymphocytes expressing F4-Cys showed higher lysis compared with the wild type (e.g., 25.4 % versus 11.8 % of specific lysis for the 624 target cell line, at 25:1 E/T ratio, respectively). No significant lysis was observed by mock-electroporated PBLs or of control 888 and 938 melanoma lines that were HLA-A2−.
The additional engineered disulfide bond cannot substitute for the native interchain one. In three independent experiments with a mutant version of F4-Cys, in which we eliminated the original bond, we observed much lower TCR and Vβ12 surface expression as well as impaired cytokine secretion in coculture with specific tumors (data not shown). These results are consistent with previous reports that showed that the original interchain disulfide bond is essential for TCR activity (7, 8).
Enhanced expression and activity is a general property of cysteine-modified TCRs
To investigate the generality of these results, we similarly mutated the following TCRs by adding cysteine residues: F5, a second MART-1–specific TCR (9) and both humanized (p53-H) and wild-type (p53-M) forms of the murine anti-p53 TCR (previously described in refs. 3, 10).
All cysteine-engineered TCRs showed enhanced surface expression and improved biological activity when compared with their wild-type versions (Fig. 3). This effect was not restricted only to TCR expressed in CD8+ cells because OKT3-stimulated CD4+ lymphocytes (>98% purity) that were electroporated with the cysteine-modified version of the CD8-independent receptor F5 (i.e., F5-Cys) stained a greater proportion of cells than the wild-type F5 receptor (85% versus 67%, respectively; Fig. 3A) and triggered higher cytokine secretion (IFN-γ: Fig. 3B and GM-CSF: data not shown). The enhancement of CD4+ cell function and their recruitment at tumor sites may be especially important because CD4 helper responses can be essential for mediating efficient antitumor activity as well as the maintenance of functional CD8+ T-cell memory (11).
Figure 3.

Enhanced properties of different cysteine-modified TCRs. A, purified CD8+ or CD4+ human PBLs (>98% purity) were electroporated with F5 or F5-Cys. Twenty-four hours after electroporation, we assessed MART-1 tetramer binding. Percentage of positive cells and the relative MFI (in brackets). B, electroporated cells were cocultured with melanoma cell lines (HLA-A2+: 526 and 624 and control HLA-A2−: 888 and 938). Twenty-four hours after the beginning of the coculture, the concentration of IFN-γ secreted in the medium was measured using an ELISA procedure. The difference between F5 and F5-Cys, based on 10 independent experiments, was found to be statistically significant (P < 0.001). C, human PBLs were electroporated with different p53-specific TCR: the humanized p53-H, cysteine-modified humanized p53-Hcys, wild-type (fully murine) p53-M, and cysteine-modified murine p53-Mcys. Twenty-four hours after electroporation, we assessed p53 pentamer binding. Percentage of positive cells and the relative MFI (in brackets). D, electroporated cells were cocultured with the tumors p53+/HLA-A2+ (H2087, MDA-MB-231, and Saos-2/*143) and the p53−/HLA-A2+ (Saos-2) cell lines (3). Twenty-four hours after the beginning of the coculture, the concentration of IFN-γ secreted in the medium was determined by ELISA. The difference between p53-H and p53-HCys, based on five independent experiments, was found to be statistically significant (P < 0.001).
Murine TCRs directed to human epitopes isolated from HLA-A2 transgenic mice have become valuable tools for antitumor adoptive immunotherapy (10, 12) because they provide a means to circumvent natural tolerance to self-antigens. Although the putative antigenicity of murine domains when expressed in patient lymphocytes remains to be examined, we previously showed that the partial humanization of such TCRs, by replacing their constant domains with human ones, reduced their biological activity (3). We sought to determine if we could compensate for the loss of activity of the humanized p53-TCR (p53-H) compared with the wild-type receptor (p53-M) by modifying its human constant regions using the same approach described above. Hence, we generated a cysteine-modified humanized p53-TCR (i.e., p53-Hcys). We electroporated PBLs with p53-H, p53-Hcys, and p53-M and stained them with a p53/HLA-A2 pentamer. p53-Hcys exhibited intermediate TCR expression (p53-H, 29%; p53-Hcys, 50%; and p53-M, 78%; Fig. 3C). Correspondingly, the levels of cytokines triggered by p53-HCys in cocultures with tumor lines were greater than those by p53-H but less than by p53-M (e.g., for the H2087 cell-line, IFN-γ: p53-H, 12,315 pg/mL; p53-Hcys, 21,347 pg/mL; and p53-M, 33,770 pg/mL in Fig. 3D; GM-CSF: data not shown). These results indicated that it is possible to partly overcome the loss of biological activity due to the partial humanization of murine TCRs using cysteine modifications.
We additionally mutated the native fully murine anti-p53 TCR by adding cysteines at the corresponding positions in mouse constant regions (p53-MCys). The latter TCR showed higher surface expression than p53-M (84% versus 78% pentamer-positive cells) and greater cytokine-mediated secretion (e.g., for the H2087 cell line, IFN-γ: 41,252 versus 33,770 pg/mL in Fig. 3C and D). These results suggested that this region of the TCR constant subunits exhibits similar structural features both in human and murine receptors, and that this approach might be further applied to enhance the efficacy of TCR gene transfer in mouse models.
Preferential pairing of cysteine-modified TCR chains
The mispairing of the introduced TCR subunits with endogenous TCR chains may result in the reduction of the cell surface density of the exogenous TCR and therefore greatly impairs its function (3, 13). In contrast, we showed an increased proportion of MHC/multimer–positive cells that expressed the cysteine-modified form of any TCR tested, rather than its wild-type form (Figs. 2 and 3), which may suggest a more effective pairing of the cysteine-modified constant regions with themselves, leading to improved biological function.
To test this hypothesis, we did two types of experiments. First, we electroporated OKT3-stimulated PBLs with titrated amounts of mRNA encoding either the F5 or F5-Cys receptor (from 0.1 to 3 μg for each chain). We then cocultured those lymphocytes with melanoma tumors and compared cytokine secretion 24 h after electroporation. Figure 4A shows that lower amounts of F5-Cys are required to achieve similar levels of IFN-γ secretion compared with F5. This was paralleled by improved MART-1 tetramer staining of lymphocytes expressing the F5-Cys receptor compared with F5 (data not shown). Comparable results were obtained in titration experiments with F4 and F4-Cys (data not shown). These observations suggested that specific pairing of cysteine-modified α and β chains was more efficient and accounted for the enhanced biological activity.
In addition, we did a TCR competition experiment, as previously described (3), by electroporating the TCR-deficient cell line Jurkat/RT3-T3.5 with either the wild-type F4 or F4-Cys and added a second mRNA encoding a different TCR as a competitor (p53-H and two different NY-ESO-1–specific TCRs 1G4 and TE8). Twenty-four hours after electroporation, we stained the cells with MART-1 tetramer to compare the different levels of MART-1 TCR expression relative to controls without competitor TCR.
As shown in Fig. 4B, F4-Cys was relatively insensitive to the addition of competitive TCR. In contrast, the expression of the native receptor F4 was significantly reduced by the presence of competitive TCR. We stained the electroporated cells for Vβ12 surface expression and observed similar levels for both MART-1 TCRs (data not shown), suggesting that the decrease in tetramer staining of F4 was not due to a lower TCR chain expression but more likely to nonspecific pairing with the competitor chains. Competition seemed to be dose dependent because we observed an increase in fluorescence intensity for both F4-H and F4-Hcys when we lowered the amount of the p53-H competitor to half (1:0.5). Additionally, we noted diverse levels of competition by different competitor TCRs, which may reflect preferential interactions of certain TCRs with the F4 variable regions, leading to different expression efficiencies (14, 15) or a “functional allelic exclusion” at the protein level (16). Comparable results were obtained when we compared the expression of p53-H and p53-HCys in similar TCR competition experiments (data not shown).
Taken together, these findings indicated that a cysteine-modified TCR is less likely to participate in unproductive mispairing with the endogenous TCR in lymphocytes, which leads to its overrepresentation on the cell surface and improved biological activity. Our findings that the introduction of an additional disulfide bond in five distinct TCRs enhanced expression and activity suggest that this modification is likely to improve the function of any TCR. These observations of preferential paring have potential clinical relevance for ongoing cancer gene therapy trials (2). Moreover, a higher density of TCR may help to reach the critical threshold for T-cell activation as previously shown (17).
In conclusion, we showed that it is possible to improve expression and biological activity of TCRs by promoting the creation of an additional interchain disulfide bond. Such biological improvement has important implications for the treatment of viral disease and cancer using TCR gene transfer to reprogram the specificity of patient lymphocytes.
References
- 1.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–86. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boulter JM, Glick M, Todorov PT, et al. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;16:707–11. doi: 10.1093/protein/gzg087. [DOI] [PubMed] [Google Scholar]
- 5.Topalian SL, Solomon D, Rosenberg SA. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J Immunol. 1989;142:3714–25. [PubMed] [Google Scholar]
- 6.Zhao Y, Zheng Z, Cohen CJ, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13:151–9. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnaud J, Huchenq A, Vernhes MC, et al. The interchain disulfide bond between TCR alpha beta heterodimers on human T cells is not required for TCR-CD3 membrane expression and signal transduction. Int Immunol. 1997;9:615–26. doi: 10.1093/intimm/9.4.615. [DOI] [PubMed] [Google Scholar]
- 8.Li Z, Wu W, Kemp O, Stephen M, Manolios N. The interchain disulfide linkage of T-cell antigen receptor-alpha and -beta chains is a prerequisite for T-cell activation. Cell Immunol. 1998;190:101–11. doi: 10.1006/cimm.1998.1383. [DOI] [PubMed] [Google Scholar]
- 9.Johnson LA, Heemskerk B, Powell D, et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cell and tumor-infiltrating lymphocytes. J Immunol. 2006;177:6548–59. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen CJ, Zheng Z, Bray R, et al. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol. 2005;175:5799–808. doi: 10.4049/jimmunol.175.9.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho WY, Yee C, Greenberg PD. Adoptive therapy with CD8(+) T cells: it may get by with a little help from its friends. J Clin Invest. 2002;110:1415–7. doi: 10.1172/JCI17214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanislawski T, Voss RH, Lotz C, et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat Immunol. 2001;2:962–70. doi: 10.1038/ni1001-962. [DOI] [PubMed] [Google Scholar]
- 13.Roszkowski JJ, Lyons GE, Kast WM, Yee C, Van Besien K, Nishimura MI. Simultaneous generation of CD8+ and CD4+ melanoma-reactive T cells by retroviral-mediated transfer of a single T-cell receptor. Cancer Res. 2005;65:1570–6. doi: 10.1158/0008-5472.CAN-04-2076. [DOI] [PubMed] [Google Scholar]
- 14.Li ZG, Wu WP, Manolios N. Structural mutations in the constant region of the T-cell antigen receptor (TCR)beta chain and their effect on TCR alpha and beta chain interaction. Immunology. 1996;88:524–30. [PMC free article] [PubMed] [Google Scholar]
- 15.Heemskerk MH, Hagedoorn RS, van der Hoorn MA, et al. Efficiency of T-cell receptor expression in dual-specific T cells is controlled by the intrinsic qualities of the TCR chains within the TCR-CD3 complex. Blood. 2007;109:235–43. doi: 10.1182/blood-2006-03-013318. [DOI] [PubMed] [Google Scholar]
- 16.Sant’Angelo DB, Cresswell P, Janeway CA, Jr, Denzin LK. Maintenance of TCR clonality in T cells expressing genes for two TCR heterodimers. Proc Natl Acad Sci U S A. 2001;98:6824–9. doi: 10.1073/pnas.121179998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–6. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 18.Lefranc M-P, Lefranc G. The T-cell receptor facts book. London: Academic Press; 2001. [Google Scholar]