

Abstract
Leukocytes are recruited early and abundantly to experimentally injured vessels, in direct proportion to cell proliferation and intimal growth. Activated circulating leukocytes and Mac-1 (CD11b/CD18, αMβ2) expression are markers of restenosis risk in patients undergoing angioplasty. As angioplastied vessels lack endothelium but have extensive fibrin(ogen) and platelet deposition, we hypothesized that Mac-1-dependent adhesion to fibrin(ogen) is an important determinant of leukocyte recruitment and function, which may in turn promote intimal growth. To study this hypothesis we administered M1/70, an anti-CD11b blocking mAb, to rabbits (1 mg/kg i.v.) immediately before, and every 48 hr for 3, 6, or 14 days after, iliac artery balloon denudation or deeper stent-induced injury. M1/70, which bound to isolated rabbit monocytes and dose-dependently inhibited Mac-1-mediated fibrinogen binding in vitro, reduced leukocyte recruitment more than 2-fold 3, 6, and 14 days after injury. Neointimal growth 14 days after injury was markedly attenuated by treatment with M1/70 (intimal area after balloon injury, 0.12 ± 0.09 mm2, compared with 0.32 ± 0.08 mm2 in vehicle-treated controls, P < 0.01, and 0.38 ± 0.08 mm2 in IgG-treated controls, P < 0.005; intimal area after stent injury, 0.56 ± 0.16 mm2, compared with 0.84 ± 0.13 mm2 in vehicle-treated controls, P < 0.05, and 0.90 ± 0.15 mm2 in IgG-treated controls, P < 0.02). Mac-1 blockade reduces experimental neointimal thickening, suggesting that leukocyte recruitment to and infiltration of injured arteries may be a valid target for preventing intimal hyperplasia.
Keywords: restenosis/vascular injury/monocytes
Monocytes occupy a central role in the pathogenesis and promotion of atherosclerosis. In contrast, the importance of these cells in proliferative vascular disease after arterial injury is less established. Experimental models of accelerated arteriopathies have focused on vascular smooth muscle cells proliferating within the arterial media and migrating from media to neointima, where further proliferation and extracellular matrix production combine to thicken the intimal layer. Treatments have been devised that interrupt one or more of these phases, but have uniformly failed to lessen restenosis after mechanical arterial injury in humans. We recently reported that monocytes are recruited early and abundantly to denuded vessels in an experimental model wherein endovascular metal stents impose deep arterial trauma (1, 2). Furthermore, the presence of these cells correlates with smooth muscle cell proliferation and neointimal growth, and inhibition of intimal thickening with heparin is paralleled by reduced monocyte recruitment (3). Clinical investigation in patients undergoing coronary angioplasty also has identified circulating activated leukocytes as a marker of heightened risk for restenosis (4). Based on such evidence that leukocytes may be central promoters and constituents of intimal growth after vascular injury, we asked whether specific inhibition of leukocyte adhesion and/or function after arterial injury would lessen intimal growth.
Leukocyte adhesion to injured arteries devoid of endothelium may occur through a variety of selectin- and integrin-dependent mechanisms involving platelets and extracellular matrix proteins (5). In particular, leukocyte recruitment to areas of extravascular inflammation is mediated by the β2-integrin family of receptors sharing a common β2 subunit (CD18), but each with a unique α subunit: LFA-1 (CD11a/CD18, αLβ2), Mac-1 (CD11b/CD18, αMβ2), and p150,95 (CD11c/CD18, αXβ2) (6, 7). Recent clinical reports have implicated up-regulation of Mac-1, but not LFA-1 or p150,95, with restenosis after coronary angioplasty (8–10). The multivalent binding properties of Mac-1 make this receptor uniquely poised to regulate adhesive and inflammatory processes after vascular injury. Mac-1 is capable of binding fibrin(ogen) (FGN) (11–13), intercellular adhesion molecule-1 (ICAM-1, CD54) (14), and factor X (15), ligands abundant in the injured wall. Mac-1 is, in fact, the primary FGN receptor on leukocytes, facilitating the adhesion and transmigration of neutrophils and monocytes at sites of fibrin and platelet deposition (16).
Because mAbs to Mac-1 can interrupt the adhesive and migratory capability of leukocytes and reduce tissue injury in models of inflammation (17–20), we used this technique to examine the effects on vascular injury of blocking leukocyte recruitment and function. M1/70 is a rat-derived IgG2b mAb directed to the unique α subunit of mouse Mac-1 (CD11b) (21) with broad species cross-reactivity, which blocks adhesion, coagulation, homotypic aggregation, and complement-dependent binding and phagocytosis (15, 22–24). Specifically inhibiting leukocyte adhesion and function is an approach to modulating arterial responses to injury, and if effective might offer a means for reducing restenosis after vascular intervention.
METHODS
Materials.
Plasminogen-free FGN (Enzyme Research Laboratories, South Bend, IN) was iodinated with Iodobeads (Pierce), as previously described (25). Fluorescein isothiocyanate (FITC)-conjugated M1/70 and isotype control rat IgG2b were obtained from Caltag (San Francisco, CA). M5/114, a rat IgG2b mAb to mouse major histocompatibility complex II, was provided by Evelyn Schneeberger (Massachusetts General Hospital, Boston). RAM 11 and anti-bromodeoxyuridine (BrdUrd) antibodies were purchased from Dako. Immunoperoxidase staining was performed by using a Vector Elite Avidin Biotin Complex kit (Vector Laboratories, Burlingame, CA). All other reagents were purchased from Sigma unless otherwise indicated.
mAb Purification.
M1/70 was purified from the M1/70.15.1 HL subcloned line (American Type Culture Collection) by using a spinner flask system and protein-free hybridoma medium (PFHM-II, GIBCO/BRL and Life Technologies, Gaithersburg, MD), as previously described (26). Under these culture conditions, the line yields ≈20 mg of rat IgG/1 liter of protein-free medium.
Cell Lines and Culture Conditions.
Peripheral blood mononuclear cells (PBMC) were isolated from anticoagulated (13 mmol/liter trisodium citrate) rabbit blood by centrifugation with NycoPrep 1.077 Animal (GIBCO/BRL and Life Technologies). To minimize platelet contamination, PBMC were washed with PBS containing 5 mmol/liter of EDTA and then resuspended in RPMI medium 1640 supplemented with 10% (vol/vol) fetal calf serum (FCS). Monocytes were separated from lymphocytes by adherence to plastic dishes placed in a water-jacketed incubator for 2 hr at 37°C and maintained in RPMI medium 1640 supplemented with 10% FCS. Culture media was supplemented with 20 mM Hepes, 2 mM l-glutamine, 100 units/ml of penicillin, and 100 mg/ml of streptomycin.
FITC-Conjugated mAb Binding Assay.
Rabbit monocytes were cultured overnight on microtiter wells (100,000 cells/well) and washed with RPMI medium 1640. Monocytes then were incubated with FITC-conjugated M1/70 or the isotype control, FITC-conjugated rat IgG2b, for 60 min on ice. After washing, FITC-conjugated mAb binding was determined by measuring cell-associated fluorescence on a fluorescent microplate reader (CytoFluor II, Perseptive Biosystems, Framingham, MA). To determine whether rabbit IgG influenced monocyte M1/70 binding, monocytes were preincubated with RPMI medium 1640 containing 10% autologous rabbit serum for 1 hr at 37C before the addition of FITC-conjugated mAbs.
Mac-1-Mediated FGN Binding and Degradation Assays.
Mac-1-dependent binding and degradation of FGN by rabbit monocytes was performed as previously described (25). Briefly, 125I-labeled FGN (1.0 μM) was added to ADP-stimulated adherent monocytes (105/well) for 4 hr at 37°C in the presence or absence of the M1/70 or an isotype control mAb M5/114 (1–5 μg/ml). After removal of an aliquot of the incubation mixture, trichloroacetic acid (10% wt/vol) was added, the incubation mixture was centrifuged, and soluble radioactivity (i.e., 125I-labeled iodotyrosine-containing peptides) was assayed as an index of FGN binding and turnover. Nonspecific degradation was determined in the presence of a 20-fold molar excess of unlabeled FGN, and specific degradation (total-nonspecific) was expressed as μg of FGN protein degraded/105 cells per hr.
Vascular Injury.
We used a model of stent-induced injury in rabbit iliac arteries, where monocyte presence parallels cell proliferation and intimal thickening (2, 3, 28), as well as less deep injury of endothelium-denuding balloon withdrawal, to examine the effects of Mac-1 inhibition on intimal growth. Male New Zealand White rabbits (Millbrook Farm Breeding Labs, Amherst, MA), 3.0–3.3 kg, were fed rabbit chow and water ad libitum and housed individually. To limit stent thrombosis, aspirin (0.07 mg/cc) was added to drinking water beginning 1 day before surgery. Under anesthesia with xylazine (5 mg/kg i.m.; Miles) and ketamine (35 mg/kg i.m.; Fort Dodge Laboratories, Fort Dodge, IA), the femoral arteries were exposed and ligated, and the iliac arterial endothelium was removed bilaterally by using three passes with a 3F balloon embolectomy catheter (Baxter Healthcare, Santa Ana, CA). A 7-mm long stainless steel stent (Multi-Link, Advanced Cardiovascular Systems/Guidant, Santa Clara, CA) mounted on a 3-mm angioplasty balloon (Advanced Cardiovascular Systems/Guidant) then was passed retrograde into one (for 14-day experiments) or both (for 3- and 6-day experiments) iliac arteries and expanded with a 15-sec 8 atm inflation. All animals received heparin (100 units/kg i.v.; Elkin-Sinn, Cherry Hill, NJ) at the time of surgery. Animal care and procedures were in accordance with guidelines of the American Association for the Accreditation of Laboratory Animal Care and National Institutes of Health.
Antibody Administration.
M1/70 (1 mg/kg i.v. via marginal ear vein) was administered 2 hr before surgery, and at 48-hr intervals thereafter throughout the 3-, 6-, or 14-day experiments. The dose and frequency of administration were chosen based on previous reports demonstrating plasma IgG concentrations in rabbits 24 hr or longer after IgG administration (29). We also performed a pharmacokinetic study in two animals: serum concentrations of M1/70 were measured by standard ELISA techniques at 10 min, and 2, 4, 8, and 24 hr after administration of M1/70, 1 mg/kg i.v. Briefly, high-protein binding microtiter plates (Nunc-Immuno Plate, Nalge Nunc International) were coated with polyclonal rabbit anti-rat IgG (1:500) in 100 mM sodium bicarbonate, pH 8.2. Diluted serum samples were added to wells; M1/70 was detected with rabbit anti-rat peroxidase conjugate (1:1,000), and then quantified by measuring A 492 nm using peroxidase substrate (o-phenylenediamine dihydrochloride). In four animals, M1/70 or rat IgG was administered (1 mg/kg i.v.) and blood collected 2 hr later for assay of serum inhibitory activity.
The control group in the 3-day experiment received rat IgG (1 mg/kg i.v. every 48 hr) to exclude nonspecific antibody effects, and in the 6-day experiment NaCl (0.5 ml i.v, every 48 hr). In the 14-day experiment, control groups received either rat IgG or NaCl. Each 3-day experimental group consisted of two animals, each 6-day group of three animals, and each 14-day group of four animals. All animals survived until the time of planned sacrifice.
Tissue Harvest and Analysis.
Three, 6, or 14 days after vascular injury, anesthesia was administered as above, animals were sacrificed, and pressure was perfused with lactated Ringer’s solution via left ventricular puncture followed by Carnoy’s solution (60% methanol, 30% chloroform, 10% glacial acetic acid) at 100 mmHg pressure for 5 min. Iliac arteries were excised and immersed in Carnoy’s solution for 4 hr, then in ethanol. All animals received BrdUrd, (50 mg/kg i.v.) 1 hr before sacrifice to allow immunocytochemical identification of proliferating cells.
Arterial segments from each iliac artery were embedded and sectioned as previously described (2, 3). For animals sacrificed after 6 days, a balloon-injured segment of each iliac artery far distant from the stent was embedded separately. Five-micrometer cross sections were cut with a tungsten carbide knife from three equally spaced locations at the middle and ends of each stented or balloon-injured segment, all three sites were analyzed, and the results were averaged. Adherent mural thrombus (after 3 days in stented arteries) or neointimal area (after 6 or 14 days in balloon-injured or stented arteries) was measured histomorphometrically in verHoeff tissue elastin-stained sections by using a computer-assisted digital system. Adherent leukocytes were examined in sections stained with hematoxylin/eosin and counted under ×600 magnification as previously reported (2, 3, 28). Deep stent-related arterial injury was graded as previously described (30).
Immunohistochemical identification of rabbit tissue macrophages with a cell-specific antibody (RAM 11) and proliferating cells incorporating BrdUrd (anti-BrdUrd antibody) was accomplished by using standard immunocytochemical protocols (2, 3, 31). Briefly, sections were incubated with a primary antibody and then a species-specific biotinylated secondary antibody. Cells were labeled with avidin-biotin peroxidase complex and 3,3-diaminobenzidine or alkaline phosphatase. Total intimal or medial cell number was determined, and the fraction identified as macrophages or proliferating cells was calculated.
Statistics.
All data are presented as the mean ± SD. Comparisons between treatment groups used a nonpaired t test. P values less than 0.05 were considered significant.
RESULTS
Binding and Inhibition of Rabbit Mac-1 by M1/70.
Although targeted to mouse Mac-1, M1/70 has broad species specificity (21–24, 32). To determine whether it would serve as a useful blocker of rabbit Mac-1, we first performed FITC-conjugated mAb binding studies and confirmed that M1/70 bound to rabbit peripheral blood monocytes (mean fluorescence number: 298 ± 69 for M1/70, n = 6, versus 47 ± 47 for isotype control mAb, n = 6, P = 0.0001). Preincubation of monocytes with rabbit serum did not diminish M1/70 binding (mean fluorescence number: 263 ± 41, n = 4, P = NS), suggesting Fc-independent binding.
To confirm that M1/70 binding was associated with inhibition of Mac-1 function, the effect of M1/70 on Mac-1 ligand binding in rabbit monocytes was examined. Activated human (myelo)monocytic cells possess a FGN clearance pathway that uses Mac-1 (25–27). We found that rabbit ADP-stimulated peripheral blood monocytes also bound and degraded soluble FGN (2.5 ± 0.5 μg/105 cells per hr, n = 3). M1/70 significantly inhibited this Mac-1-dependent FGN binding and degradation by rabbit monocytes in a concentration-dependent manner (Fig. 1) with 70% inhibition at 5 μg/ml of M1/70.
Figure 1.

Bar graph shows Mac-1-dependent FGN binding, uptake, and degradation by rabbit monocytes in the presence of M1/70 (1–5 μg/ml) or isotype-matched control mAb M5/114 (5 μg/ml). M1/70 inhibited FGN turnover in a concentration-dependent manner. The effect of mAb treatment is depicted as the percentage of control nonantibody-treated monocyte FGN turnover present in antibody-treated monocytes. ∗, P < 0.01 compared with control or M5/114.
Previous pharmacokinetic experiments have demonstrated that the bolus infusion of an intact IgG antibody to the β2 (CD18)-subunit of Mac-1 results in an antibody concentration greater than 5 μg/ml for more than 24 hr (29). We found that i.v. administration of M1/70 (1 mg/kg) produced peak levels of 18.2 ± 1.3 μg/ml, which fell to 7.0 ± 0.3 μg/ml after 8 hr, with an estimated T1/2 of 3.9 hr. Finally, rabbit serum obtained 2 hr after bolus administration of M1/70 inhibited Mac-1-dependent FGN binding and turnover by 54%.
Effect of Mac-1 Inhibition on Vascular Repair Intimal Thickening.
To examine the role of Mac-1-mediated leukocyte adhesion on vascular repair, two forms of vascular injury were used: standard balloon-withdrawal denudation injury or stent-induced deep injury known to provoke prolonged recruitment of cells of monocyte/macrophage lineage. Fourteen days after balloon injury, circumferential intimal thickening was seen in the iliac arteries of control animals treated with NaCl (intimal area 0.32 ± 0.08 mm2) or IgG (intimal area 0.38 ± 0.08 mm2, P = NS). In animals treated with M1/70 intimal growth was reduced 60–70% (intimal area 0.12 ± 0.09 mm2, P < 0.01 compared with NaCl-treated group, P < 0.005 compared with IgG-treated group, Figs. 2 and 3).
Figure 2.

Bar graph shows neointimal cross-sectional area in rabbit iliac arteries 14 days after balloon injury (hatched bars) or stent injury (solid bars). Administration every 48 hr of M1/70, an antibody directed against the α subunit of Mac-1, reduced intimal size. ∗, P < 0.05 compared with NaCl-treated group, P < 0.02 compared with IgG-treated group. †, P < 0.01 compared with NaCl-treated control group, P < 0.005 compared with IgG-treated control group.
Figure 3.
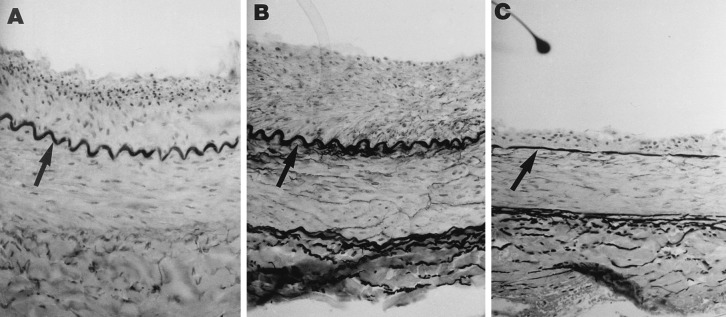
Photomicrographs of rabbit iliac arteries stained with verHoeff tissue elastin stain 14 days after denuding balloon withdrawal injury. Neointima separates the internal elastic lamina (arrows) from the lumen. Rabbits were treated every 48 hr with (A) NaCl, (B) IgG, or (C) M1/70, directed against the α subunit of Mac-1. Original magnification: ×200.
Stent-induced deep injury resulted in 3-fold more neointimal hyperplasia than balloon injury (0.84 ± 0.13 mm2 for NaCl-treated controls, and 0.90 ± 0.15 mm2 for IgG-treated controls, P = NS). Treatment with M1/70 reduced neointimal growth after stenting to 0.56 ± 0.16 mm2 (P < 0.05 compared with NaCl-treated group, P < 0.02 compared with IgG-treated group, Figs. 2 and 4). The absolute reduction in neointimal area achieved by M1/70 in balloon-injured arteries was nearly identical to that achieved in stented arteries. There were no differences in the degree of deep vascular laceration caused by stent placement (30) in the different experimental groups.
Figure 4.
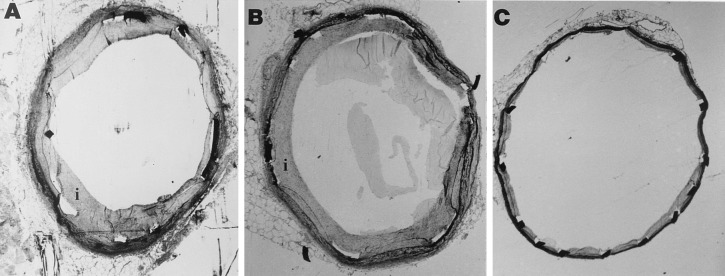
Photomicrographs of rabbit iliac arteries stained with verHoeff tissue elastin stain 14 days after endovascular stenting. Stent struts appear as filled or open rectangles. Neointima separates the internal elastic laminae from the lumen. Rabbits were treated every 48 hr with (A) NaCl, (B) IgG, or (C) M1/70, directed against the α subunit of Mac-1. Original magnification: ×30.
Inflammation.
Both adherent mononuclear cells and immunocytochemically identified tissue-infiltrative macrophages have been used as measures of inflammation after stent implantation. Three days after stent-induced injury, a layer of leukocytes carpeted the arterial lumen. The number of neutrophils per arterial cross section was 104 ± 54 in animals treated with IgG and 20 ± 7 in animals treated with M1/70, P < 0.02, and the number of mononuclear cells per section 169.3 ± 5.2 in IgG-treated animals, compared with 113.6 ± 33.2 in M1/70-treated animals, P < 0.04. The accumulation of mural thrombus adjacent to stent struts is the hallmark of response to this form of injury over the first few days (3). The area of mural thrombus, measured histomorphometrically in cross sections, was not significantly different between M1/70-treated and IgG-treated animals (0.16 ± 0.08 mm2 and 0.21 ± 0.03 mm2, respectively).
Mononuclear cell invasion of the neointima peaks between 3 and 7 days after balloon- or stent-induced injury of rabbit arteries. We examined this aspect of vascular response to injury after 6 days. Surface mononuclear cell adhesion and neointimal monocyte/macrophage infiltration 6 days after stent-induced injury were markedly curtailed by M1/70 treatment. Adherent mononuclear cells/section were reduced from 66 ± 63 in NaCl-treated control animals to 17 ± 6 in M1/70-treated animals (P = 0.09), and tissue-infiltrative (RAM-11+) cells from 1.34 ± 1.31 to 0.03 ± 0.02% of cells, P < 0.03, Fig. 5). Tissue infiltrative cells were far less frequent after balloon injury than after stent-induced injury, (0.12 ± 0.19% of cells in NaCl-treated group), and there was a nonsignificant reduction in their number by M1/70 (0.05 ± 0.08% of cells).
Figure 5.

Quantitative immunohistochemical examination of stented rabbit iliac arteries 6 and 14 days after implantation of endovascular stents. After 6 or 14 days, treatment with M1/70 (hatched bars), directed against the α subunit of Mac-1, reduced the number of tissue macrophages (RAM-11+ cells) compared with arteries treated with NaCl (solid bars). ∗, P < 0.03, †, P < 0.004.
After 14 days, the pronounced reduction in tissue-infiltrative macrophages by M1/70 was still evident in stented arteries, from 1.4 ± 0.2 or 1.5 ± 0.2% of cells in NaCl- or IgG-treated controls, respectively, to 0.6 ± 0.3% of cells in M1/70-treated animals (P < 0.004 compared with either control group, Fig. 5). The number of adherent mononuclear cells seen 14 days after stent implantation, a marker of persistent cell recruitment, was also lower in M1/70-treated (8 ± 4 cells per section) than in NaCl-treated or IgG-treated arteries (14 ± 2 or 23 ± 13 cells per section, P = 0.08 or P = 0.07, respectively).
Cell Proliferation.
Cell proliferation in the neointima and media also peaks between 3 and 7 days after balloon- or stent-induced injury of rabbit arteries (3, 33), and many pharmacologic inhibitors of experimental arterial hyperplastic disease inhibit cell proliferation. We chose to examine the effect of M1/70 on these aspects of vascular repair 6 days after balloon- or stent-induced injury by using incorporation of BrdUrd at the time of sacrifice. The only location in which M1/70-reduced proliferation compared with NaCl-treated controls was in the media of stent-injured vessels (0.5 ± 0.2 vs. 0.8 ± 0.4% of cells, respectively, P < 0.05). M1/70 had no effect on intimal cell proliferation, where rates are 10-fold higher than in the media: balloon injury, 5.8 ± 1.2 vs. 4.7 ± 0.9% of cells for M1/70 and NaCl-treated vessels, respectively; stent injury, 2.9 ± 0.9% vs. 3.6 ± 1.3%, respectively, P = NS for both).
Fourteen days after injury, intimal cell proliferation had fallen 10-fold and was not significantly altered by treatment with M1/70: balloon injury, 1.3 ± 1.2, 0.4 ± 0.2, and 0.4 ± 0.2% of cells for M1/70-, NaCl-, and IgG-treated vessels, respectively; stent injury, 0.3 ± 0.1 vs. 0.4 ± 0.1, and 0.5 ± 0.1% of cells, respectively).
DISCUSSION
Our study demonstrates that specific inhibition of leukocyte adhesion reduces neointimal growth after balloon- and stent-induced vascular injury. This effect after either superficial denuding injury or deep arterial laceration attests to the importance of leukocytes as participants in vascular repair (34). Our hypothesis that inhibition of integrin-mediated leukocyte adhesion would favorably modulate vascular repair was based on experimental and clinical evidence. We had demonstrated previously that after deep experimental arterial injury imposed by endovascular stents monocyte recruitment is brisk and prolonged, that the abundance of monocytes within the vessel wall is correlated with intimal growth and vascular cell proliferation, and finally that reduction in intimal thickening with an archetypal inhibitor of neointimal hyperplasia, heparin, is accompanied by equal reduction in mononuclear cell recruitment (3). Clinical evidence supporting our hypothesis includes the observations that leukocyte activation and Mac-1 expression at the time of coronary angioplasty correlate with the likelihood of restenosis in the ensuing months (4, 8, 10), and that the number of macrophages in arteries healing after angioplasty again correlates with the amount of tissue growth (35).
Others have demonstrated modulation of responses to vascular injury through targeting cell adhesion molecules. Molossi and coworkers (36) showed that blockade of VLA-4 integrin (CD49d/CD29, α4β1) binding to fibronectin reduces accelerated arteriopathy in rabbit cardiac allografts. Kling et al. (37) showed that mononuclear leukocytes invade the tunica intima of injured rabbit arteries predominantly via VLA-4 and to a lesser extent via CD-18-dependent mechanisms and may stimulate smooth muscle cell migration, although effects on neointimal thickening were not examined, as the experiments were terminated after 36 hr. Finally, Yasukawa and colleagues (38) administered antibodies to intercellular adhesion molecule-1 (ICAM-1) and LFA-1 after injury of rat carotid arteries, demonstrating inhibition of intimal thickening by anti-ICAM-1, but not anti-LFA-1, antibodies (37). Although a possible effect of anti-ICAM-1 antibodies on mononuclear cell invasion was considered, a greater proportion of the effect was attributed to altered smooth muscle cell kinetics.
Mac-1, although a marker of restenosis after angioplasty, has not been causally linked to the biology of neointimal hyperplasia. Yet, Mac-1 is a strong candidate to mediate leukocyte recruitment to arteries denuded of endothelium. Mac-1 is the primary FGN receptor on monocytes and neutrophils (11–13) and is responsible for firm neutrophil accumulation on and migration across platelets deposited at sites of vascular injury (16). Furthermore, Mac-1 is physically linked with the urokinase receptor (uPAR, CD87) on myelomonocytic cells (39, 40), and directly regulates uPAR-dependent adhesion to vitronectin (41), which may be present at sites of arterial disease (41). Mac-1-dependent modulation of vascular injury also may be bolstered by clinical trials with 7E3 mAb Fab fragment (abciximab) directed against platelet integrin IIb/IIIa (CD41/CD61, αIIbβ3) (43). 7E3 cross-reacts with Mac-1 (32, 44), suggesting that inhibition of both IIb/IIIa- and Mac-1-dependent functions by 7E3 may jointly contribute to the regulation of vascular repair and to sustained clinical benefits observed in some studies with 7E3 after angioplasty.
More specific blockade of Mac-1 is possible through several inhibitory mAbs. We chose a M1/70, a rat-derived mAb to mouse Mac-1, because of its known broad species cross-reactivity and ability to block multiple Mac-1 ligands (15, 21–24). After first confirming that M1/70 inhibited rabbit mononuclear cell Mac-1 function, we administered M1/70 to rabbits in a dose of 1 mg/kg i.v. and confirmed that this dose achieved and maintained inhibitory levels. We then found that M1/70 reduced leukocyte recruitment 3 days after stent-induced injury, lessened mononuclear cell infiltration after 6 and 14 days, and reduced overall intimal thickening. There was no effect of M1/70 after 6 or 14 days in neointimal cell proliferation, and a small effect on medial smooth muscle cell proliferation after 6 days. These findings were not nonspecific Ig effects, as rat IgG-treated controls were no different from NaCl-treated controls.
Several mechanisms may account for the beneficial effect of Mac-1 inhibition on neointimal thickening. Abundant experimental evidence from in vivo studies using Mac-1-blocking mAbs (17–20) or Mac-1-deficient mice (45, 46) suggests that Mac-1 plays a central role in the inflammatory response. Diverse ligand engagement by Mac-1 regulates many leukocyte functions, including adhesion (15), migration (16), proteolysis (25, 27), phagocytosis (47), and degranulation/respiratory burst (45). However, it has been proposed that excessive activation of Mac-1 can have deleterious consequences, promoting tissue damage and ischemia-reperfusion injury (48). mAbs to Mac-1 have been shown to interrupt the adhesive and migratory capability of leukocytes and reduce tissue injury in a variety of animal models of inflammation (17–20). Furthermore, Mac-1 both directly and indirectly regulates the expression of procoagulant activity. Mac-1 binds factor X and coordinates the activation of factor X independent of tissue factor (15). At the same time, Mac-1 has been shown to enhance cytokine-stimulated tissue factor expression by macrophages (49). Therefore, mAb blockade of Mac-1 may reduce local thrombus formation that may directly affect neointimal area.
Of particular interest, M1/70 also exerted a profound inhibitory effect on neointimal growth after simple balloon injury as well. (Myelo)monocytes are present but not common within the neointima after experimental balloon injury (33, 34). Nevertheless, their sparse number must belie a potent promoting effect, as inhibition with M1/70 resulted in marked reduction of intimal growth. Possible mechanisms whereby (myelo)monocytes may contribute to neointimal thickening include direct bulk within the intima, the elaboration of factors chemotactic or mitogenic for other (myelo)monocytes or vascular smooth muscle cells (50–54), production of enzymes capable of degrading extracellular constituents favoring cell migration (55, 56), or generation of reactive oxygen species injurious to other vascular cells (57, 58). The molecular underpinnings of (myelo)monocyte recruitment after mechanical vascular injury are not known. Greater numbers of these cells in stented arteries may reflect foreign material introduced into the vessel wall as well as the chronicity and depth of injury. Understanding these determinants will help direct more precise use of agents to modulate (myelo)monocyte function.
Study Limitations.
Because intimal thickening in the experimental models used in this study peaks 14 days after injury (1), blockade of Mac-1 was achieved by infusing antibody throughout the 14-day period. Whether a shorter duration of antibody administration to, for example, only block leukocyte adhesion during the peak period of leukocyte recruitment (3–7 days) (3) also would limit intimal thickening is unknown. Also, inhibition of neointimal growth was achieved by using an intact Fc-containing antibody, although our ex vivo assays did not demonstrate an effect of either isotype-matched IgG2b or rat IgG on Mac-1 function and our in vivo antibody control groups used nonspecific rat IgG, suggesting an Fc-independent effect of M1/70. Although cell proliferation was examined 6 days after injury (during the peak proliferative phase) and only minor effects of M1/70 were identified, it is possible that integrated over the entire experimental period antiproliferative effects might achieve a more substantial level. Finally, although inhibition of Mac-1 carries the theoretical concern of impairing host defenses and predisposing to infection, no animal in our study developed overt infection, consistent with the findings in Mac-1-deficient mice (45, 46).
Conclusions.
Myriad agents limit neointimal growth in experimental models of vascular injury but fail clinical scrutiny, a paradox often attributed to redundant signals directing vascular cell proliferation and migration. Although other investigators have administered nonspecific anti-inflammatory agents to modulate vascular healing, no previous approach has specifically targeted leukocytes as central elements of vascular repair. Inhibiting the mechanisms whereby these cells adhere immediately after injury has the potential to remove the source of many secondary redundant mechanisms. Inhibition of leukocyte functions mediated by Mac-1 therefore may offer a novel approach for reducing restenosis after vascular injury, free from the complexity of directly regulating vascular cell proliferation.
Acknowledgments
We are grateful to Philip Seifert, Danielle Bornstein, and Hui Xu for technical assistance. This work was supported in part by grants from the American Heart Association National Center (95004400 to C.R.), the American Heart Association Massachusetts Affiliate (13-526-945 to D.I.S.), the National Institutes of Health (HL03104 to C.R., GM/HL49039 to E.R.E., and HL02768 and HL57506 to D.I.S.), the Burroughs Wellcome Fund for Experimental Therapeutics, Durham, NC (E.R.E.), and the Whitaker Foundation, Rosslyn, VA (E.R.E.).
ABBREVIATIONS
- FGN
fibrinogen
- FITC
fluorescein isothiocyanate
- BrdUrd
bromodeoxyuridine
References
- 1.Rogers C, Parikh S, Edelman E R. Circulation. 1995;92:I–300. [Google Scholar]
- 2.Rogers C, Parikh S, Seifert P, Edelman E R. Circulation. 1996;94:2909–2914. doi: 10.1161/01.cir.94.11.2909. [DOI] [PubMed] [Google Scholar]
- 3.Rogers C, Welt F G P, Karnovsky M J, Edelman E R. Arterioscler Throm Vasc Biol. 1996;16:1312–1318. doi: 10.1161/01.atv.16.10.1312. [DOI] [PubMed] [Google Scholar]
- 4.Pietersma A, Kofflard M, de Wit L E A, Stinjen T, Koster J F, Serruys P, Sluiter W. Circulation. 1995;91:1320–1325. doi: 10.1161/01.cir.91.5.1320. [DOI] [PubMed] [Google Scholar]
- 5.Springer T A. Nature (London) 1990;346:425–434. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 6.Hynes R O. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- 7.Hynes R O. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 8.Mickelson J K, Lakkis N M, Villarreal-Levy G, Hughes B, Smith C W. J Am Coll Cardiol. 1996;28:345–353. doi: 10.1016/0735-1097(96)00164-7. [DOI] [PubMed] [Google Scholar]
- 9.Neumann F-J, Ott I, Gawaz M, Puchner G, Schomig A. J Am Coll Cardiol. 1996;27:819–824. doi: 10.1016/0735-1097(95)00563-3. [DOI] [PubMed] [Google Scholar]
- 10.Inoue T, Sakai Y, Morooka S, Hayashi T, Takayanagi K, Takabatake Y. J Am Coll Cardiol. 1996;28:1127–1133. doi: 10.1016/S0735-1097(96)00308-7. [DOI] [PubMed] [Google Scholar]
- 11.Altieri D C, Morrissey J H, Edgington T S. Proc Natl Acad Sci USA. 1988;85:7426–7466. doi: 10.1073/pnas.85.20.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altieri D C, Mannucci P M, Capitano A M. J Clin Invest. 1986;78:968–976. doi: 10.1172/JCI112687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright S D, Weitz J S, Huang A J, Levin S M, Silverstein S C, Leike J D. Proc Natl Acad Sci USA. 1988;85:7734–7738. doi: 10.1073/pnas.85.20.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diamond M S, Staunton D E, Morlin S D, Springer T A. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- 15.Altieri D C, Bader R, Mannucci P M, Edgington T S. J Cell Biol. 1988;107:1893–1900. doi: 10.1083/jcb.107.5.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diacovo T G, Roth S J, Buccola J M, Bainton D F, Springer T A. Blood. 1996;88:146–157. [PubMed] [Google Scholar]
- 17.Vedder N B, Winn R K, Rice C L, Chi E Y, Arfors K E, Harlan J M. J Clin Invest. 1988;81:939–944. doi: 10.1172/JCI113407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson P J, Todd R F, Fantone J C, Mickelson J K, Griffin J D, Lucchesi B R. J Clin Invest. 1988;81:624–629. doi: 10.1172/JCI113364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosen H, Gordon S. J Exp Med. 1987;166:1685–1701. doi: 10.1084/jem.166.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosen H, Gordon S. Am J Respir Cell Mol Biol. 1990;3:3–10. doi: 10.1165/ajrcmb/3.1.3. [DOI] [PubMed] [Google Scholar]
- 21.Springer T A, Galfre G, Secher D S, Milstein C. Eur J Immunol. 1979;9:301–306. doi: 10.1002/eji.1830090410. [DOI] [PubMed] [Google Scholar]
- 22.Ault K A, Springer T A. J Immunol. 1981;120:359–364. [PubMed] [Google Scholar]
- 23.Anderson D C, Miller L J, Schmalsteig F C, Rothlein R, Springer T A. J Immunol. 1986;137:15–27. [PubMed] [Google Scholar]
- 24.Sanchez-Madrid F, Simon P, Thompson S, Springer T A. J Exp Med. 1983;158:586–602. doi: 10.1084/jem.158.2.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simon D I, Ezratty A M, Francis S A, Rennke H, Loscalzo J. Blood. 1993;82:2414–2422. [PubMed] [Google Scholar]
- 26.Tarleton R L, Beyer A M. BioTechniques. 1991;11:590–593. [PubMed] [Google Scholar]
- 27.Simon D I, Ezratty A M, Loscalzo J. Biochemistry. 1994;33:6555–6563. doi: 10.1021/bi00187a024. [DOI] [PubMed] [Google Scholar]
- 28.Rogers C, Edelman E R. Circulation. 1995;91:2995–3001. doi: 10.1161/01.cir.91.12.2995. [DOI] [PubMed] [Google Scholar]
- 29.Price T H, Beatty P G, Corpuz S R. J Immunol. 1987;139:4174–4177. [PubMed] [Google Scholar]
- 30.Schwartz R S, Huber K C, Murphy J G, Edwards W D, Camrud A R, Vlietstra R E, Holmes D R. J Am Coll Cardiol. 1992;19:267–274. doi: 10.1016/0735-1097(92)90476-4. [DOI] [PubMed] [Google Scholar]
- 31.Wolf E, Roser K, Hahn M, Welkerling H, Delling G. Virchows Archiv A Pathol Anat. 1992;420:17–24. doi: 10.1007/BF01605979. [DOI] [PubMed] [Google Scholar]
- 32.Altieri D C, Edgington T S. J Immunol. 1988;141:2656–2660. [PubMed] [Google Scholar]
- 33.Tanaka H, Sukhova G K, Swanson S J, Clinton S K, Ganz P, Cybulsky M I, Libby P. Circulation. 1993;88:1788–1803. doi: 10.1161/01.cir.88.4.1788. [DOI] [PubMed] [Google Scholar]
- 34.Libby P, Schwartz D, Brogi E, Tanaka H, Clinton S. Circulation. 1992;86:III 47–III 52. [PubMed] [Google Scholar]
- 35.Moreno P R, Bernardi V H, Lopez-Cuellar J, Newell J B, McMellon C, Gold H K, Palacios I F, Fuster V, Fallon J T. Circulation. 1996;94:3098–3102. doi: 10.1161/01.cir.94.12.3098. [DOI] [PubMed] [Google Scholar]
- 36.Molossi S, Elices M, Arrhenius T, Diaz R, Coulber C, Rabinovitch M. J Clin Invest. 1995;95:2601–2610. doi: 10.1172/JCI117962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kling D, Fingerle J, Harlan J M, Lobb R R, Lang F. Circ Res. 1995;77:1121–1128. doi: 10.1161/01.res.77.6.1121. [DOI] [PubMed] [Google Scholar]
- 38.Yasukawa H, Imaizumi T, Matsuoka H, Nakashima A, Morimatsu M. Circulation. 1997;95:1515–1522. doi: 10.1161/01.cir.95.6.1515. [DOI] [PubMed] [Google Scholar]
- 39.Xue W, Kindzelski A L, Todd R F, Petty H R. J Immunol. 1994;152:4630–4640. [PubMed] [Google Scholar]
- 40.Bohuslav J, Horejsi V, Hansmann C, Stockl J, Weilde U H, Majdic O, Bartke I, Knapp W, Stockinger H. J Exp Med. 1995;181:1381–1390. doi: 10.1084/jem.181.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon D I, Rao N K, Xu H, Wei Y, Majdic O, Ronne E, Kobzik L, Chapman H A. Blood. 1996;88:3185–3194. [PubMed] [Google Scholar]
- 42.Niculescu F, Rus H G, Porutiu D, Ghiurca V, Vlaicu R. Atherosclerosis. 1989;78:197–203. doi: 10.1016/0021-9150(89)90223-2. [DOI] [PubMed] [Google Scholar]
- 43.Topol E J, Califf R M, Weisman H F, Ellis S G, Tcheng J E, Worley S, Ivanhoe R, George B S, Fintel D, Weston M, et al. Lancet. 1994;343:881–886. doi: 10.1016/s0140-6736(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 44.Simon D I, Xu H, Ortlepp S, Rogers C, Rao N K. Arterioscler Thromb Vasc Biol. 1997;17:528–535. doi: 10.1161/01.atv.17.3.528. [DOI] [PubMed] [Google Scholar]
- 45.Lu H, Smith C W, Perrard J, Bullard D, Tang L, Shappell S B, Entman M L, Beaudet A L, Ballantyne C M. J Clin Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coxon A, Rieu P, Barkalow F J, Askari S, Sharpe A H, von Adrian U H, Arnaout M A, Mayadas T N. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 47.Talle M A, Westberg E F, Goldstein G, Silverstein S C. Proc Natl Acad Sci USA. 1983;80:5699–5703. doi: 10.1073/pnas.80.18.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Plow E F. J Clin Invest. 1997;99:1145–1146. doi: 10.1172/JCI119267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan S-T, Edgington T S. J Clin Invest. 1991;87:50–57. doi: 10.1172/JCI115000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leibovich S J, Ross R. Am J Pathol. 1976;84:501–514. [PMC free article] [PubMed] [Google Scholar]
- 51.Martin B M, Gimbrone M A, Jr, Unanue E R, Cotran R S. J Immunol. 1981;126:1510–1515. [PubMed] [Google Scholar]
- 52.Glenn K C, Ross R. Cell. 1981;25:603–615. doi: 10.1016/0092-8674(81)90168-9. [DOI] [PubMed] [Google Scholar]
- 53.Falcone D J, McCaffrey T A, Haimovitz-Friedman A, Vergilio J-A, Nicholson A C. J Biol Chem. 1993;268:11951–11958. [PubMed] [Google Scholar]
- 54.Assoian R K, Fleurdelys B E, Stevenson H C, Miller P J, Madtes D K, Raines E W, Ross R, Sporn M B. Proc Natl Acad Sci USA. 1987;84:6020–6024. doi: 10.1073/pnas.84.17.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campbell E J, Silverman E K, Campbell M A. J Immunol. 1989;143:2961–2968. [PubMed] [Google Scholar]
- 56.Garbisa S, Ballin M, Daga-gordini D, Fastelli G, Naturale M, Negro A, Semenzato G, Liotta L A. J Biol Chem. 1986;261:2369–2375. [PubMed] [Google Scholar]
- 57.Carpenter K L H, Brabbs C E, Mitchincon M J. Klin Wochenschr. 1991;69:1039–1045. doi: 10.1007/BF01645155. [DOI] [PubMed] [Google Scholar]
- 58.Peri G, Chiaffarino F, Bernasconi S, Padura I, Mantovani A. J Immunol. 1990;144:1444–1448. [PubMed] [Google Scholar]