

Abstract
Receptor-mediated Gq signaling promotes hypertrophic growth of cultured neonatal rat cardiac myocytes and is postulated to transduce in vivo cardiac pressure overload hypertrophy. Although initially compensatory, hypertrophy can proceed by unknown mechanisms to cardiac failure. We used adenoviral infection and transgenic overexpression of the alpha subunit of Gq to autonomously activate Gq signaling in cardiomyocytes. In cultured cardiac myocytes, overexpression of wild-type Gαq resulted in hypertrophic growth. Strikingly, expression of a constitutively activated mutant of Gαq, which further increased Gq signaling, produced initial hypertrophy, which rapidly progressed to apoptotic cardiomyocyte death. This paradigm was recapitulated during pregnancy in Gαq overexpressing mice and in transgenic mice expressing high levels of wild-type Gαq. The consequence of cardiomyocyte apoptosis was a transition from compensated hypertrophy to a rapidly progressive and lethal cardiomyopathy. Progression from hypertrophy to apoptosis in vitro and in vivo was coincident with activation of p38 and Jun kinases. These data suggest a mechanism in which moderate levels of Gq signaling stimulate cardiac hypertrophy whereas high level Gq activation results in cardiomyocyte apoptosis. The identification of a single biochemical stimulus regulating cardiomyocyte growth and death suggests a plausible mechanism for the progression of compensated hypertrophy to decompensated heart failure.
Heterotrimeric guanine nucleotide-binding proteins (G proteins) of the Gq family transduce signals from a variety of widely expressed membrane receptors to generate diverse, tissue-specific effects (1). In many target tissues, receptor-mediated activation of Gq regulates physiological responses such as contraction and secretion. A role for Gq-coupled receptors in regulation of cell growth has become apparent only more recently (2). Cardiac muscle expresses Gq-coupled receptors that do not appear to play a primary role in modulating cardiac contractile function. Rather, the relevant physiological role for Gq-coupled receptor agonists may be stimulation of cardiac hypertrophy. Indeed, multiple Gq-coupled receptor agonists stimulate hypertrophy of cultured neonatal rat cardiac myocytes (3–6).
In the intact heart, cardiac hypertrophy is typically a compensatory response to increased hemodynamic load. The resulting increase in cardiac mass improves cardiac performance in the short term by reducing wall stress (7). When the hemodynamic load is not relieved however, the hypertrophied heart ultimately dilates and fails in a phenomenon termed “decompensation.” A role for Gq-signaling in the development and decompensation of hypertrophy is supported by the effects of transgenic overexpression of α1 adrenergic and angiotensin II receptors in cardiomyocytes (8, 9). In fact, heterozygous transgenic overexpression of Gαq in cardiomyocytes induces cardiac enlargement with many of the molecular, structural and functional characteristics of pressure overload hypertrophy (10). Of interest, higher level Gαq overexpression in dual heterozygous mice causes a lethal dilated congestive cardiomyopathy with myocyte loss in the absence of inflammation, suggesting a role for apoptosis. Although cardiomyocyte apoptosis has been implicated in human cardiomyopathy (11, 12) and animal models of myocardial injury (13–15), a role for Gq signaling in apoptotic cardiomyocyte death has not been explored, and the relationship between apoptosis and cardiac failure has not been defined adequately.
The current study explores the role of Gq signaling in cardiomyocyte hypertrophy and apoptosis by using cultured adenovirus-infected neonatal cardiac myocytes or transgenic mice with enhanced Gαq signaling. Our results demonstrate that sustained high level activation of Gαq can produce apoptotic cardiomyocyte death and can lead to heart failure.
MATERIALS AND METHODS
Preparation of Adenoviral Constructs and Adenoviral Infection of Cultured Cardiomyocytes.
The wild-type Gαq (Gαq WT) and constitutively active GTPase-deficient mutant Gαq (Q209L) expression plasmids developed and characterized by Johnson et al. (16) were kindly provided by John Exton (Vanderbilt Univ.). Gαq WT and Q209L cDNAs were cloned into PACCMVpLpA(+) shuttle vector. Recombinant adenovirus was generated by homologous recombination of cotransfected pJM17 and PACCMV in human embryonal kidney (HEK293) cells, was confirmed by restriction digests, was amplified in HEK293 cells, and was titrated by agarose overlay (17). Viral supernatants were harvested and used directly for myocyte infection. Similar results were obtained by using cesium chloride purified adenovirus. Neonatal rat ventricular myocytes (6, 17) were plated overnight in serum-containing media, were washed, and were incubated in serum-free media 6 hr before addition of adenovirus at a titer of 10 plaque forming units per cell. Cells were washed, and serum-free media was replaced 16–18 hr later.
In Situ Terminal Deoxytransferase Assays.
Cardiomyocytes on laminin coated coverslips were fixed in 4% paraformaldehyde, were permeabilized and labeled with digoxigenin-11-deoxyuridine triphosphate by using terminal deoxynucleotide transferase (18), and were stained with alkaline phosphatase-conjugated anti-digoxigenin. Myocardial sections were deparaffinized, were labeled with fluorescein-12-digoxigenin-11-deoxyuridine triphosphate (terminal deoxynucleotidyltransferase-mediated UTP end labeling, (TUNEL), assay), and were counterstained with propidium iodide (0.5 μg/ml). Labeled nuclei were counted to determine the apoptotic index (no. labeled nuclei/no. total myocyte nuclei × 100). For internucleosomal DNA cleavage assays, DNA preparation and agarose electrophoresis were performed essentially as described (12).
Transgenic Mice.
FVB/N mice expressing murine Gαq under control of the α myosin heavy chain promoter have been described (10). For peripartum studies, 8-week-old female Gαq-25 mice were bred and were observed daily for signs of congestive heart failure. Mice were considered to have died of heart failure if post mortem examination revealed cardiomegaly, pleural effusions, and pulmonary congestion or if dyspnea and cyanosis was of such severity that the animal was clearly terminal. Methods for RNA dot blot analysis, cardiac morphometric, and cardiac myocyte cross sectional area measurements and cardiac histology are as reported (10, 19). Organ weights were indexed to tibial length rather than body weight because body weight varied with gestational duration.
Cell Signaling Assays.
Infected cells were labeled for 8 hr with 2 μCi/ml [3H]myoinositol, and inositol phosphate formation was determined as described (5). Jun N-terminal kinase (JNK) or p38 mitogen activated protein (MAP) kinase activities were measured as described (20) except that myelin basic protein was used as substrate for p38.
Statistical Analysis.
Results are presented as mean ± SEM. Multiple groups were compared by one way analysis of variance followed by the Bonferroni test for comparison of individual means. Statistical significance was accepted at P < 0.05.
RESULTS
Gαq Stimulates Hypertrophy and Apoptosis in Cultured Neonatal Cardiac Myocytes.
Hypertrophy induced by Gq-coupled agonists in cultured neonatal cardiomyocytes is characterized by cell enlargement, organization of myofilaments, and expression of atrial natriuretic factor (ANF) (3–6). To define more clearly the role of Gαq in cardiomyocytes, recombinant adenoviral expression vectors encoding wild-type or activated Gαq Q209L were generated. Cultured myocytes were infected with adenovirus constructs at a multiplicity of infection of 10 plaque-forming units per cell at nearly 100% infection efficiency (β-galactosidase staining of LacZ-infected myocytes) without cytotoxicity (not shown).
Immunoblot analysis demonstrated overexpression of Gαq in myocytes infected with adenoviral constructs encoding Gαq WT or Q209L (Fig. 1a). Endogenous Gαq expression was observed in longer immunoblot exposures (not shown). Expression of Gαq WT or Q209L autonomously activated phospholipase C, resulting in 12- and 65-fold increases in [3H]-inositol phosphate accumulation respectively (Fig. 1b). As reported with microinjection of activated Gαq (21), adenoviral expression of Gαq WT or Q209L increased ANF immunoreactivity (Fig. 2 d and f). We further observed myocyte enlargement and increased sarcomeric organization in Gαq WT-expressing cells (Fig. 2c). Unexpectedly, myocytes expressing mutant Q209L manifested a typical hypertrophic response at 8 hr (Fig. 2 e and f) but underwent cellular shrinkage, loss of sarcomeric organization, and cell death at later times (Fig. 2 g and h).
Figure 1.

Adenovirus-mediated Gαq expression and signaling in cultured cardiac myocytes. (a) Expression of Gαq in neonatal rat cardiac myocytes (representative Western blot of three separate experiments). (b) Overexpression of Gαq stimulates phosphoinositide hydrolysis. P < 0.01 (∗) compared with LacZ; P < 0.001 (#) compared with Gαq WT (n = 9).
Figure 2.

Stimulation of myocyte hypertrophy and death by Gαq overexpression. Myocytes infected with indicated adenovirus constructs were fixed, permeabilized, and stained with rhodamine-conjugated phalloidin or polyclonal ANF antiserum. Myocyte hypertrophy is evidenced by increased ANF expression, increased sarcomeric structure, and increased cell size in Gαq-expressing cells at 8 hr. At 24 hr, Q209L expressing cells show shrinkage and death.
Growth promoting and transforming effects of Gq signaling are well established, but the lethal effect of Q209L in cardiac myocytes was not anticipated. In situ DNA end-labeling studies showed a 3- to 4-fold increase in labeled nuclei in Q209L myocytes but a decrease in Gαq WT myocytes compared with LacZ infected cells (Fig. 3 a and b), suggesting enhancement of apoptosis with Q209L but protection with Gαq WT. This pattern of apoptosis was confirmed by nucleosomal laddering of genomic DNA, which was increased in Q209L-infected myocytes but decreased in Gαq WT compared with LacZ-expressing cells (Fig. 3c).
Figure 3.
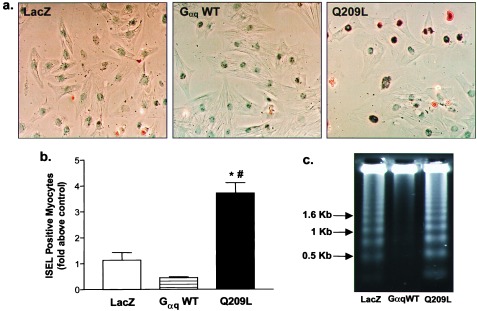
Constitutive activation of Gαq signaling causes apoptosis in myocytes. (a) In situ DNA end labeling (ISEL) of myocytes infected with indicated adenovirus constructs. ISEL positive myocytes have reddish brown nuclei. (b) Quantitative analysis of ISEL studies (n = 200 cells per experimental group from three separate experiments). P < 0.001 (∗) compared with LacZ; P < 0.001 (#) compared with Gαq WT. (c) DNA was isolated from myocytes 48 hr after infection with the indicated adenovirus constructs. DNA fragmentation was detected by ethidium bromide fluorescence.
Gαq Stimulates Hypertrophy, Apoptosis, and Heart Failure in Transgenic Mice.
An in vivo counterpart of Gq-mediated apoptosis was identified in Gαq-overexpressing mice. Heterozygous Gαq WT expression in cardiac myocytes resulted in stable cardiac hypertrophy (10). By crossing two independent Gαq WT-expressing lines, dual heterozygotes, mice expressing Gαq WT at 8× control levels (compared with 4- and 5-fold for the parent lines) were obtained; these mice died of heart failure at 11 ± 2 weeks. TUNEL assays labeled ≈8% of ventricular cardiomyocytes in failing dual heterozygotes (vs. <0.5% in nontransgenic and heterozygous mice), indicating that apoptosis was occurring (not shown) and suggesting a relationship between the extent of Gαq signaling, cardiomyocyte apoptosis, and heart failure.
A more striking example of apoptotic decompensation in heterozygous Gαq-overexpressing mice was observed in a lethal peripartum cardiomyopathy. The survival of Gαq transgenic mice as a function of number of pregnancies is depicted in Fig. 4a. The peak incidence of heart failure occurred within 1 week after delivery (Fig. 4a Inset). Necropsy examination demonstrated massive cardiac enlargement involving all four cardiac chambers (Fig. 4b) and pulmonary congestion (Table 1; lung weight indexed to tibial length). Indices of cardiac hypertrophy, which are increased in nonfailing Gαq overexpressors (cardiac myocyte cross-sectional area and heart weight indexed to tibial length), were increased further in the cardiomyopathic peripartum hearts, as was hypertrophy-associated fetal cardiac gene expression (Table 1). Protein kinase C activity also was augmented (Table 1). These data support the notion that peripartum cardiomyopathy in these mice is associated with exaggerated Gαq signaling.
Figure 4.
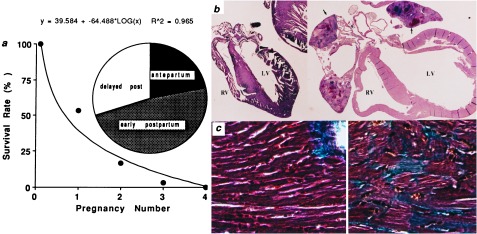
Characteristics of peripartum cardiomyopathy in Gαq-overexpressing mice. (a) Survival curve relating mortality to number of pregnancies. (Inset) Chronology of heart failure development relative to parturition defined as within 7 days of expected delivery (antepartum), within 7 days after delivery (early postpartum), or 7–14 days after delivery (delayed post). (b) Gross morphology (4×) of peripartum nontransgenic (Left) and Gαq-overexpressing (Right) mouse hearts showing cardiomyopathic dilatation of cardiac chambers (LV, left ventricle; RV, right ventricle) with mural thrombi in atria (arrows). (c) Trichrome stain (400×) of hearts depicted in b shows interstitial fibrosis and myocyte replacement (blue-stained cells), without inflammation in transgenic heart (Right).
Table 1.
Morphometric and molecular characteristics of nonpregnant or pregnant mice
| NTG | NTG-P | Gq-25 | Gq-25/P | |
|---|---|---|---|---|
| Morphometry | ||||
| Heart/tibial length | 0.91 ± 0.4 | 1.05 ± 0.02 | 1.16 ± 0.04* | 1.92 ± 0.08† |
| Lung/tibial length | 1.06 ± 0.07 | 0.97 ± 0.10 | 1.11 ± 0.06 | 1.54 ± 0.07† |
| Liver/tibial length | 7.8 ± 0.27 | 9.45 ± 0.39† | 7.16 ± 0.27 | 10.6 ± 0.35† |
| Myocyte CSA, μm2 | 165 ± 3 | 171 ± 19 | 316 ± 24* | 433 ± 88† |
| Gene expression | ||||
| ANF | 1 | 0.7 | 7.8* | 13.1*† |
| βMHC | 1 | 0.8 | 3.1* | 7.2*† |
| αSk actin | 1 | 1.5 | 2.9* | 3.6* |
| Protein kinase C activity | 1 | 0.9 | 1.4* | 1.8*† |
Morphometric values are expressed as mean ± SEM of 8 to 17 animals. Organ weights indexed to tibial length are expressed in mg/mm. Gene expression and protein kinase C activation are expressed as fold increased over nontransgenic (means of four or five animals per group). NTG, nontransgenic (control); Gq-25, Gαq overexpressor; Gq-25/P, peripartum; CSA, cross sectional area; MHC, myosin heavy chain; sk, skeletal.
P < 0.05 compared with NTG.
P < 0.05 compared with nonpregnant mouse of same genotype.
Histologic examination of peripartal Gαq hearts showed interstitial and replacement fibrosis (Fig. 4c) consistent with myocyte loss (22) but no myocardial inflammation. Apoptosis was evident by widespread TUNEL labeling of ventricular cardiomyocyte nuclei (Fig. 5 a and b), with apoptotic indices of 26 ± 12% in peripartum Gαq vs. 0.2 ± 0.1% in peripartum controls (n = 5 each, P < 0.05). Cells with apoptotic nuclei were identified as cardiac myocytes by propidium iodide staining (Fig. 5b) or labeling with α sarcomeric actin (not shown) and TUNEL-stained nuclei demonstrated nuclear chromatin clumping and fragmentation (Fig. 5c). The presence of apoptosis in peripartum cardiomyopathic hearts was confirmed by DNA laddering in ventricular extracts (Fig. 5d).
Figure 5.
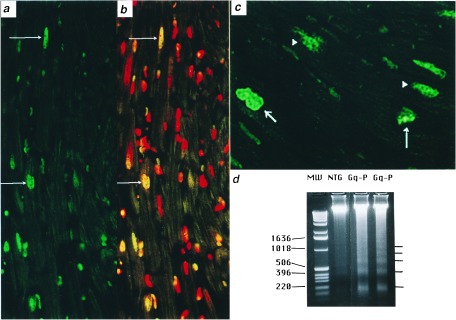
DNA strand breaks in cardiac myocyte nuclei of Gαq peripartal hearts. (a) Low power (200×) and (c) high power (1,000×) images showing nuclear DNA labeling visualized by fluorescein fluorescence. (b) a counterstained with propidium iodide (red). Apoptotic nuclei appear green or yellow (arrows), occur only in cardiac myocytes, and demonstrate varying degrees of condensed nuclear chromatin (arrows in c). Normal myocyte nuclei are red with a diffuse chromatin staining pattern. (d) DNA laddering of Gαq cardiac DNA. MW, DNA size markers.
p38 MAP Kinase and Jun Kinase Activation in Gαq-Mediated Cardiomyocyte Apoptosis.
The above in vitro and in vivo studies suggested that activation of Gαq-coupled signaling pathways in excess of levels that cause cardiomyocyte hypertrophy can lead to cardiomyocyte apoptosis. We therefore postulated that a common downstream signaling pathway might be activated in the cultured cell and transgenic models of Gαq-stimulated apoptosis. Because the JNK and p38 MAP kinases have been implicated as apoptotic mediators in other systems, we assayed JNK and p38 activities in infected cardiomyocytes and transgenic hearts representing a spectrum of Gαq signaling. In cultured cardiomyocytes expressing Gαq WT, as in heterozygous transgenic mice expressing Gαq WT, the increase in JNK and p38 activity was minimal (Fig. 6). In contrast, p38 and JNK activities were increased significantly both in cells expressing Q209L and in failing peripartal Gαq-overexpressing hearts (Fig. 6). Thus, both in vitro and in vivo paradigms of cardiomyocyte apoptosis exhibited coordinate activation of JNK and p38 kinases.
Figure 6.

JNK and p38 MAP kinase activities in Gαq-overexpressing cardiomyocytes (a) and transgenic mice (b). There is coactivation of both kinases in apoptotic Q209L expressing cardiomyocytes and peripartum (Gαq-25/P) cardiomyopathic mice. P < 0.05 (∗) compared with control (LacZ infected) myocytes or control nontransgenic (NTG) mice, and P < 0.01 (#) compared with control (LacZ, NTG) or Gαq WT (Gαq WT, Gαq-25) groups (n = 6–9 determinations).
DISCUSSION
These results demonstrate a continuum of response to increasing Gαq-mediated signaling, which suggests that Gαq is an early amplitude-modulated sensor for signals that initiate cardiomyocyte growth and death. This paradigm provides a possible cellular mechanism for the progression of “compensated” hypertrophy to heart failure. By using adenoviral infection to achieve nearly complete expression of wild-type or activated Gαq in cultured myocytes, we show that Gαq WT expression increases phospholipase C activity (12-fold) and leads to increased cell size and myofilament organization. These data extend the prior observation that microinjection of an activated Gαq expression plasmid into cardiomyocytes increased a genetic marker of hypertrophy (ANF) (21). The effects of Gαq WT expression are thus identical to those observed in agonist-stimulated cardiac myocyte hypertrophy (3–6) and are similar to those seen in the Gαq WT transgenic mice (10).
Surprisingly, when we expressed a constitutively activated mutant form of Gαq, which markedly increased phospholipase C activity, hypertrophy was followed reproducibly by apoptotic cell death over 24–48 hr. These observations are similar to those of Althoefer et al. (24) showing that activated Gαq can cause apoptosis in Chinese hamster ovary and Cos-7 cells and they provide a plausible mechanism for decompensation of hypertrophied hearts in vivo. Accordingly, we characterized cardiomyopathies in Gαq WT-overexpressing mice. Whereas transgenic mice expressing Gαq at 4-fold control levels develop cardiac hypertrophy, mice expressing 8-fold normal levels developed a dilated cardiomyopathy with myocyte apoptosis. Using the more available peripartum cardiomyopathy Gαq model, we compiled several lines of evidence that heart failure is associated with myocyte apoptosis, including increased in situ end labeling of cardiac myocyte nuclei, microscopic identification of nuclear chromatin condensation, endonucleolytic DNA fragmentation, and evidence of cardiomyocyte loss without necrosis or inflammation. Notably, apoptosis and heart failure were not observed in nonpregnant female Gαq-overexpressing mice followed for 72 weeks (ref. 10; unpublished data). It is also noteworthy that signs of peripartal heart failure in Gαq-overexpressing mice occurred most commonly after delivery, when hemodynamic changes associated with pregnancy are resolving (25), and that chronic volume overload per se does not induce heart failure (unpublished results). Of importance, other transgenic murine models of hypertrophy have not been associated with peripartum cardiomyopathies (8, 26). Thus, neither preexisting hypertrophy nor the hemodynamic effects of pregnancy appear to be sufficient to cause an apoptotic cardiomyopathy. Rather, the exaggerated cardiomyocyte hypertrophy and apoptosis of peripartal Gαq overexpressors implicate a pregnancy-associated stimulus that augments Gαq signaling. This notion is supported by increased protein kinase C activation and a pattern of MAP kinase activation (coincident activation of p38 kinase and JNK) that is the same as that observed in apoptotic Q209L myocytes. Although the postulated in vivo stimulus for apoptosis has not been identified, chronic infusion of angiotensin II caused a rapidly progressive apoptotic cardiomyopathy in preliminary studies with these mice (unpublished data).
There is broad support for the notion that activation of JNK and p38 kinase stress-activated MAP kinases is associated with programmed cell death. Early studies implicated JNK and p38 as critical mediators of apoptosis in PC12 cells and lymphocytes (27–29). Subsequently, p38 was shown to increase during ischemia/reperfusion and JNK during reperfusion of isolated rat hearts (30, 31), suggesting a relationship of stress-activated MAP kinase activation and ischemic injury. Recently, Wang, et al. (17) demonstrated distinct roles for each of two isoforms of p38 (α and β) in controlling hypertrophy and apoptosis in cardiac myocytes. Additionally, a specific upstream activator of JNK (MKK7) induced marked hypertrophy and, when expressed together with activators of p38 (MKK3 or 6), induced apoptosis (23). The mediation of Gαq-stimulated cardiomyocyte apoptosis by combined activation of JNK and p38 is also consistent with other studies demonstrating that sustained or concurrent activation of stress-activated MAP kinase family members is required for development of apoptosis (27, 32, 33).
A substantial body of information exists concerning biochemical determinants of cardiac hypertrophy whereas a generally accepted mechanism for hypertrophy decompensation does not exist. Conventional wisdom has held that decompensated hypertrophy reflects the transition from a physiologic to a pathologic state. The current studies suggest that, although hypertrophy is the initial result of Gq activation, this same stimulus, with time and sufficient signal strength, mediates progression to a pathological condition in which apoptosis occurs. Thus, compensated hypertrophy and heart failure may represent two different physiologic states, but we suggest that they are phases of the same process and are initiated by common biochemical stimuli. The possibility that Gq signals the transition from cardiac hypertrophy to cardiac failure under conditions of increased hemodynamic load is being studied.
Acknowledgments
The authors would like to thank Reene Cantwell for secretarial assistance. This work was supported in part by National Institutes of Health Grants HL49267, HL58010, and HL52318 (G.W.D.) and HL28143 and HL46345 (J.H.B.) and a Veterans Administration Merit Review Grant (G.W.D.). J.W.A. is supported by an American Heart Association Western States Affiliate Research Fellowship.
ABBREVIATIONS
- Gαq WT
wild-type Gαq
- JNK
Jun N-terminal kinase
- MAP
mitogen activated protein
- ANF
atrial natriuretic factor, TUNEL, terminal deoxynucleotidyltransferase-mediated UTP end labeling
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Simon M I, Strathmann M P, Gautam N. Science. 1991;252:802–808. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- 2.Post G R, Brown J H. FASEB J. 1996;10:741–749. doi: 10.1096/fasebj.10.7.8635691. [DOI] [PubMed] [Google Scholar]
- 3.Sadoshima J-I, Izumo S. Circ Res. 1993;73:413–423. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- 4.Shubeita H E, McDonough P M, Harris A N, Knowlton K U, Glembotski C C, Brown J H, Chien K R. J Biol Chem. 1990;265:20555–20562. [PubMed] [Google Scholar]
- 5.Knowlton K U, Michel M C, Itani M, Shubeita H E, Ishihara K, Brown J H, Chien K R. J Biol Chem. 1993;268:15374–15380. [PubMed] [Google Scholar]
- 6.Adams J W, Migita D S, Yu M K, Young R, Hellickson M S, Castro-Vargas F E, Domingo J D, Lee P H, Bui J S, Henderson S A. J Biol Chem. 1996;271:1179–1186. doi: 10.1074/jbc.271.2.1179. [DOI] [PubMed] [Google Scholar]
- 7.Grossman W, Jones D, McLaurin L P. J Clin Invest. 1975;55:56–64. doi: 10.1172/JCI108079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milano C A, Dolbert P C, Rockman H A, Bond R A, Venable M E, Allen L F, Lefkowitz R J. Proc Natl Acad Sci USA. 1994;91:10109–10113. doi: 10.1073/pnas.91.21.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hein L, Stevens M E, Barsh G S, Pratt R E, Kobilka B K, Dzau V J. Proc Natl Acad Sci USA. 1997;94:6391–6396. doi: 10.1073/pnas.94.12.6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Angelo D D, Sakata Y, Lorenz J N, Boivin G P, Walsh R A, Liggett S B, Dorn G W, II. Proc Natl Acad Sci USA. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Narula J, Haider N, Virmani R, DiSalvo T G, Kolodgie F D, Hajjar R J, Schmidt U, Semigran M J, Dec G W, Khaw B-A. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- 12.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara J A, Quaini E, Loreto C D, Beltrami C A, Krajewski S, et al. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 13.Fliss H, Gattinger D. Circ Res. 1996;79:949–956. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Cigola E, Cheng W, Kajstura J, Olivetti G, Hintze T H, Anversa P. Lab Invest. 1995;73:771–787. [PubMed] [Google Scholar]
- 15.Kajstura J, Cheng W, Sarangarajan R, Li P, Li B, Nitahara J A, Chapnick S, Reiss K, Olivetti G, Anversa P. Am J Physiol. 1996;271:H1215–H1228. doi: 10.1152/ajpheart.1996.271.3.H1215. [DOI] [PubMed] [Google Scholar]
- 16.Qian N X, Winitz S, Johnson G L. Proc Natl Acad Sci USA. 1993;90:4077–4081. doi: 10.1073/pnas.90.9.4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Huang S, Sah V P, Ross J R, Jr, Brown J H, Han J, Chien K R. J Biol Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- 18.Blaschke A J, Staley K, Chun J. Development (Cambridge, UK) 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- 19.Sakata Y, Lorenz J, Hoit B D, Liggett S B, Walsh R A, Dorn G W, II. Circulation. 1998;97:1488–1495. doi: 10.1161/01.cir.97.15.1488. [DOI] [PubMed] [Google Scholar]
- 20.Ramirez M T, Sah V P, Zhao X-L, Hunter J J, Chien K R, Brown J H. J Biol Chem. 1997;272:14057–14061. doi: 10.1074/jbc.272.22.14057. [DOI] [PubMed] [Google Scholar]
- 21.LaMorte V J, Thorburn J, Absher D, Spiegel A, Brown J H, Chien K R, Feramisco J R, Knowlton K U. J Biol Chem. 1994;269:13490–13496. [PubMed] [Google Scholar]
- 22.Weber K T, Brilla C G. Circulation. 1991;83:1849–1865. doi: 10.1161/01.cir.83.6.1849. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Su B, Sah V P, Brown J H, Hah J, Chien K R. J Biol Chem. 1998;273:5423–5426. doi: 10.1074/jbc.273.10.5423. [DOI] [PubMed] [Google Scholar]
- 24.Althoefer H, Eversol-Cire P, Simon M I. J Biol Chem. 1997;272:24380–24386. doi: 10.1074/jbc.272.39.24380. [DOI] [PubMed] [Google Scholar]
- 25.Clark S L, Cotton D B, Lee W, Bishop C, Hill T, Southwick J, Pivarnik J, Spillman T, DeVore G R, Phelan J, et al. Am J Obstet Gynecol. 1989;161:1439–1442. doi: 10.1016/0002-9378(89)90900-9. [DOI] [PubMed] [Google Scholar]
- 26.Hunter J J, Tanaka N, Rockman H A, Ross J, Jr, Chien K R. J Biol Chem. 1995;270:23173–23178. doi: 10.1074/jbc.270.39.23173. [DOI] [PubMed] [Google Scholar]
- 27.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 28.Karin M. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 29.Johnson N L, Gardner A M, Diener K M, Lange-Carter C A, Gleavy J, Jarpe M B, Minden A, Karin M, Zon L I, Johnson G L. J Biol Chem. 1996;271:3229–3237. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- 30.Bogoyevitch M A, Gillespie-Brown J, Ketterman A J, Fuller S J, Ben-Levy R, Ashworth A, Marshall C J, Sugden P H. Circ Res. 1996;79:162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 31.Knight R J, Buxton D B. Biochem Biophys Res Commun. 1996;218:83–88. doi: 10.1006/bbrc.1996.0016. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y-R, Meyer C F, Tan T-H. J Biol Chem. 1996;271:631–634. doi: 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y-R, Wang X, Templeton D, Davis R J, Tan T-H. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]