

Abstract
Skewing of X chromosome inactivation (XCI) can occur in normal females and increases in tissues with age. The mechanisms underlying skewing in normal females, however, remain controversial. To better understand the phenomenon of XCI in nondisease states, we evaluated XCI patterns in epithelial and hematopoietic cells of over 500 healthy female mother-neonate pairs. The incidence of skewing observed in mothers was twice that observed in neonates, and in both cohorts, the incidence of XCI was lower in epithelial cells than hematopoietic cells. These results suggest that XCI incidence varies by tissue type and that age-dependent mechanisms can influence skewing in both epithelial and hematopoietic cells. In both cohorts, a correlation was identified in the direction of skewing in epithelial and hematopoietic cells, suggesting common underlying skewing mechanisms across tissues. However, there was no correlation between the XCI patterns of mothers and their respective neonates, and skewed mothers gave birth to skewed neonates at the same frequency as nonskewed mothers. Taken together, our data suggest that in humans, the XCI pattern observed at birth does not reflect a single heritable genetic locus, but rather corresponds to a complex trait determined, at least in part, by selection biases occurring after XCI.
Introduction
In mammals, dosage compensation of X-linked genes in females is achieved by the transcriptional silencing of 1 of the 2 X chromosomes during early development, a process known as X chromosome inactivation (XCI) (1). Once XCI has occurred, the heterochromatin-enriched inactive X chromosome is stably transmitted to each daughter cell through subsequent mitoses. In human and mouse embryos, either of the 2 X chromosomes (paternal or maternal) can be inactivated. As a consequence, females from both species present a mosaic pattern of 2 cell lines, one expressing maternal X-linked genes and the other expressing paternal X-linked genes. The XCI ratio (or XCI pattern) corresponds to the relative proportion of each cell line. This ratio can be determined by the analysis of expression (2), transcription (3), or differential methylation (4) of polymorphic X-linked genes, such as the human androgen receptor (HUMARA) gene (5). Significant deviation from the theoretical 1:1 ratio between the 2 parental alleles is called skewing. As it was first suggested by pioneers of X-inactivation analysis, including Fialkow (6) and Vogelstein (7), in most studies, skewing and extreme skewing are arbitrarily defined as greater than or equal to 75% and greater than or equal to 90% of cells expressing the same X chromosome, respectively.
Skewed patterns of XCI were initially found in female carriers of specific X-linked mutations (8, 9) where skewing resulted from the negative selection of cells harboring the lethal allele in the active state. Skewing of XCI was also reported in rare cases of balanced X/autosome translocations, which perturbed the normal mechanisms of XCI (10). Furthermore, skewing has been associated with some complex traits such as recurrent abortion (11) and mental retardation (12), although the causative role of skewing in these situations has not been established. Skewing is a common feature of tumorigenic processes in which an acquired somatic mutation occurs in a single progenitor cell that subsequently undergoes clonal expansion and gives rise to progeny with the same XCI pattern (13, 14).
Skewed patterns of XCI are not restricted to disease states as they can also be seen in normal females (15). Skewing in normal females can be categorized as primary skewing (occurring at the time of XCI; ref. 16) and secondary skewing (occurring after primary skewing and potentially caused by cell-selection effects; ref. 16).
The incidence of skewing in normal females varies according to age, tissue, and the definition of skewing used by investigators. Using the skewing definition employed by the majority of studies (≥75% of a predominant allele), the incidence of skewing is relatively low at birth (4.9%–8%) (17–19) and in adult nonhematopoietic tissues (~10%–30%) (20, 21), but higher and age dependent in hematopoietic tissues (~40% in 60-year-old women, ref. 22; 67% in centenarians, ref. 23). Although a recent report suggested that buccal epithelial cells were also subjected to increased skewing with time (24), the incidence of skewing in hematopoietic versus nonhematopoietic tissues of aging individuals is generally poorly correlated (correlation ratio 0.19–0.74) (25–27) except in some cases of extreme skewing, suggesting a more stable incidence of skewing in nonhematopoietic tissue. In general, skewing does not have any biologic consequence; however, in rare cases where skewing occurs toward a nonlethal mutated allele, it can expose a female carrier of an X-linked trait to the pathological phenotype. This has been shown in very rare cases of hemophilia A (28) and in some cases of X-linked sideroblastic anemia (29).
The mechanisms underlying XCI skewing in normal females remain contentious. It was originally believed that XCI occurred randomly in the mammalian embryo (30) and that primary skewing was the passive and stochastic consequence of the small number of cells present in the embryo at the time of XCI. Variant alleles of the X chromosome controlling element (Xce) were subsequently found to bias XCI in mice and cause skewing in heterozygous mice, suggesting that primary skewing was genetically controlled, at least in this species (31). In humans, no Xce-like element has been identified to date, though a few isolated reports have favored a genetic influence on primary skewing: (a) a rare mutation in the X (inactive)–specific transcript (XIST) minimal promoter was found to favor primary skewing (32, 33); (b) an alteration in X chromosome inactivation center (XIC) was suggested to be responsible for the skewing of XCI observed in 3 related females (34); (c) twin studies reported a good concordance of XCI patterns between monozygous twins (35–37); and (d) other studies have documented the heritability of the skewing trait (38–40). Most of these studies either reported rare genetic events or were conducted in blood cells of adult females (likely affected by age-associated skewing). A human neonate cohort (theoretically minimally affected by age-associated skewing) would be the preeminent population to study the etiology of primary skewing.
The present study aimed to further characterize XCI occurring in normal females by evaluating whether XCI is a tissue-specific or a body-wide phenomenon, and by determining whether primary skewing is stochastic or determined by a strong in-cis genetic element.
Results
Cohort.
A total of 502 mother-neonate pairs were recruited for this study between July 2003 and October 2004. The majority (72%) of neonates had both parents of French Canadian origin, but 13% and 15% of them had 1 or no parent of French Canadian origin, respectively. Mothers were aged between 18 and 43 years (mean age: 28.9 ± 5.3 years old). One monozygous and six dizygous pairs of twins were recruited. Only 1 randomly chosen twin from each pair was studied.
Validation of XCI ratio determination assays.
To validate the HUMARA assay for the analysis of neonate specimens, we compared XCI ratio results obtained using this assay with results obtained using the transcription assay at the iduronate-2-sulfatase (IDS) gene in 94 neonates heterozygous at both the HUMARA and IDS loci. The mean difference between the 2 assays was 5.82%, and the agreement coefficient between them was ±13.5%. The Pearson correlation (comparing XCI ratios from both assays) was 0.92, which confirms that the HUMARA assay was a valid method to study XCI ratio in neonates (Figure 1).
Figure 1. Correlation of Psup XCI ratios for 94 neonates as determined by 2 different methods: a methylation-based assay (HUMARA) versus a transcription-based assay.

Pearson correlation coefficient was 0.92.
Validation of the purity of buccal swabs and of cord blood samples.
To evaluate the possibility of blood cell contamination of buccal swabs, cells obtained by gentle scrapping of the inner cheek of 10 candidates were spread out on hematology slides and stained with Wright-Giemsa. Microscope analysis revealed the exclusive presence of epithelial cells (without any blood cell contamination).
As most neonates were breastfed, we evaluated the possibility that maternal epithelial cells were present in the mouth of neonates by analyzing the HUMARA genotype of mother-neonate pairs and searching for an extra allele (nontransmitted) in the genotype of infants. Neonatal genotypes did not show any maternal nontransmitted allele.
It has been shown that cord blood samples may be contaminated by the presence of maternal cells. Using FISH, Tsang et al. (41) documented the presence of maternal cells in 9 of 58 cord blood samples. However, 7 of 9 had contamination levels of less than 1%, and a single cord blood sample showed greater than 5% (5.25%) contamination by maternal cells. Although such levels of maternal cell contamination would not have altered the conclusions of our study, we nonetheless carefully analyzed the genotype (at HUMARA) of each mother-neonate pair for the presence of the nontransmitted maternal allele. No nontransmitted maternal allele was visualized on the chromatograph obtained from each neonatal cord blood sample.
Incidences of skewing.
Of the 502 mother-neonate pairs tested, 454 neonates (91.2%) and 448 mothers (90.0%) were heterozygous at the HUMARA locus and, hence, available for evaluation by the HUMARA assay. XCI ratio was assessed in both hematopoietic and epithelial cells. The percentage of predominant allele was used to detect skewing. In neonates, the median value of this score was 59.5% in epithelial cells and 62.2% in blood cells, while in mothers, it was 60.9% and 65.7%, respectively. In accordance with the literature (42–45), skewing was defined as equal to or greater than 75% of the predominant allele. Using this criterion, 7.6% (33 of 436) and 14.2% (64 of 450) of neonates showed a skewed XCI pattern in their epithelial and blood cells, respectively (Table 1). A higher proportion of mothers met this criterion, as 13.5% (60 of 445) and 27.9% (124 of 444) of them exhibited skewed XCI in epithelial and blood cells, respectively (Table 1). The χ2 test confirmed a significantly higher skewing incidence in the epithelial (P = 0.004) and blood (P < 0.001) cells of mothers, relative to neonates. Moreover, blood cells were significantly more skewed than epithelial cells in both neonates (P = 0.002) and mothers (P < 0.001). An extremely skewed XCI ratio (defined as ≥90% of the predominant allele) was uncommon in both groups, being found in 0.5% (2 of 436) and 2.7% (12 of 450) of neonates and 1.3% (6 of 445) and 4.5% (20 of 444) of mothers, in epithelial and blood cells, respectively.
Table 1 .
Incidences of skewing (≥75% and ≥90% of predominant alleles) in epithelial and hematopoietic cells of neonates and mothers

Concordance of XCI ratios between epithelial and hematopoietic cells.
For both neonates and mothers, the tissue specificity of XCI was assessed by comparing the proportion of the superior HUMARA allele (Psup score; see Methods for details) in epithelial and blood cells, as this score indicates whether or not skewing is in favor of the same allele. We performed a linear regression analysis and tested for Pearson correlation. The normality of the distributions was tested with the Kolmogorov-Smirnov normality test. The XCI ratios of 433 neonates and 441 mothers are shown in Figure 2. A significant positive correlation was found between the Psup score of blood and epithelial cells for both neonates (Pearson, r = 0.715; P < 0.001) and their mothers (Pearson, r = 0.779; P < 0.001). The mean absolute difference between neonatal blood and epithelial Psup scores was 9.8%, and this difference ranged from 0.1% to 42.0%, while for mothers, the mean difference was 10.2%, ranging from 0.0% to 44%. As blood and epithelial cells have a different embryologic origin, these findings suggest similar etiology of skewing occurring prior to the differentiation of each tissue from the primordial cell pool.
Figure 2. Scatter plots and correlations of blood versus buccal epithelial tissue XCI ratios.

Psup scores were used to correlate the XCI ratios of the 2 tissues for neonates (A) and mothers (B).
Transmission of skewing in mother-neonate pairs.
To test for a genetic contribution to skewing, in particular for the possibility of a cis-acting gene such as the mouse Xce, we evaluated the transmission of XCI ratio between mother and neonate. A total of 407 pairs of mother-neonate were assessed, in which both members of the pair were HUMARA heterozygotes. We utilized the percentage of predominant allele scores to qualitatively evaluate the transmission of skewing. Skewing was defined as above (≥75% of the predominant allele), whereas nonskewing was defined as a percentage of predominant allele between 50%–65%. We assessed whether skewed mothers were more likely to give birth to skewed neonates compared with nonskewed mothers (Table 2). Of the 53 mothers with skewing in epithelial cells, only 3 gave birth to a skewed neonate (5.7%), whereas of the 242 nonskewed mothers, 20 gave birth to a skewed neonate (8.3%). The Fisher’s exact test showed no significant difference between the groups (P = 0.777). Similar conclusions were reached upon analysis of blood cells (14.7% versus 13.6%; P = 0.800). These results thus show that neonate skewing is not directly determined by the skewing status of her mother. Moreover, the incidence of skewing in neonates of skewed or nonskewed mothers was similar to that found in the whole neonate population (Table 1). In accordance with these results, skewed neonates were not more likely to have a skewed mother (Table 3). The same type of qualitative analysis was also performed using a more stringent criterion for skewing (≥90% of the predominant allele). Again, there was no difference in the rate of skewing between skewed/nonskewed neonates and mothers (data not shown).
Table 2 .
Incidences of skewing in neonates of skewed versus nonskewed mothers

Table 3 .
Incidences of skewing in mothers of skewed versus nonskewed neonates

To quantitatively study the transmission of XCI ratios, we tested the correlation between mothers and neonates using scores based on the proportion of cells having the maternal HUMARA allele on the active X chromosome (Pmat) and the proportion of cells having the transmitted HUMARA allele on the active X chromosome (Ptrans). These scores were suitable for transmission studies, as XCI ratio calculations are based on the X chromosome shared by mother and daughter. A linear regression analysis (Figure 3) did not show any correlation between the XCI ratios of mother and daughter pairs, either in epithelial cells (r = 0.074; n = 347; P = 0.172) or in blood cells (r = 0.058; n = 357; P = 0.272), thereby corroborating the qualitative data. The empiric heritability could be estimated from the regression slopes (46). Although the values of the regression slopes were 0.064 for epithelial cells and 0.047 for blood cells and gave estimated heritability values of 12.8% and 9.3%, this was not considered significant as the slopes were not significantly different from 0. Moreover, the correlation of XCI ratios between mothers and neonates using normalized percentage of predominant allele (which does not take into account the direction of skewing but only the degree of deviation from a 50% XCI pattern) was null and nonsignificant (data not shown).
Figure 3. Linear regressions and correlations of mother versus neonate XCI ratios.

Ptrans and Pmat scores were used to show the proportion of cells having the transmitted (mothers) or the maternal (neonates) allele active in mother-neonate duos. Studies were performed in buccal epithelium (A) and blood (B). H2, heritability value.
Statistical power of the study.
To assess the power of our cohort, and thus, validate the transmission tests outlined above, a power study was conducted using PASS 2002. This power study was based on a linear regression analysis of Pmat and Ptrans scores and showed that our cohort was capable of detecting heritabilities in greater than 25% in epithelial cells and greater than 23% in hematopoietic cells.
Association between XIST SNPs and skewing.
The search for Xce-like cis-acting genes in humans can be approached by using DNA markers, such as SNPs, to test for an association with the trait. Five SNPs in the region surrounding XIST were tested in this study. Prior to performing the analysis, the Hardy-Weinberg equilibrium of each genotyped SNP was validated with PEDSTATS. The association was tested using UNPHASED QTPHASE, by comparing unrelated individuals. We took advantage of the percentage of predominant allele, as this expresses the magnitude of skewing without taking into account its direction. As UNPHASED uses a parametric test that requires normalized data, we used the box-cox normalized scores of neonates. Analyzed separately, none of the SNPs showed an association with skewing, neither in buccal epithelium nor in blood (data not shown). The SNPs were also analyzed by haplotypes of 2, 3, 4, and 5 markers. None of these analyses revealed an association with the degree of skewing expressed as a percentage of the predominant allele (see data for the 5-marker haplotypes analysis in Table 4).
Table 4 .
Unphased analysis of 5-marker haplotypes versus the percentage of predominant allele score for 154 neonates

Discussion
Forty-five years ago, Mary Lyon proposed the insightful hypothesis that 1 of the 2 X chromosomes in each female cell is randomly inactivated during early embryogenesis (1). Since that time, tremendous research effort has been deployed to decipher the basic mechanisms governing XCI. A number of cis-acting genetic elements (such as Xist, ref. 47; Xite, ref. 48; and Xce, ref. 49) and trans-acting factors (such as CTCF; ref. 50) have been identified, and their specific role has been defined. However, there are still several questions related to the biology of XCI that remain unanswered. One of them is why don’t all normal females (without any known X-linked mutation or disease) have balanced XCI — as it was postulated by Lyon? Investigating the cause of XCI skewing in the normal human females has been a major challenge for most investigators, as the expression of XCI has proven to be more complex than initially postulated. There are several limitations to the study of XCI skewing in normal human females: (a) early post-conception events cannot be studied; (b) it is difficult to sample several tissues from the same individual; (c) post-XCI changes occur with aging; (d) different tissues may be differently subjected to post-XCI selection biases; (e) heritability investigation is limited to the matrilineage as men are uninformative; and (f) the incidence of skewing in the neonate population is relatively low, and large cohorts need to be studied to get meaningful results. Furthermore, data on the etiology of skewing obtained from animal models and human cohorts have been contradictory: studies in mice suggest that an Xic-linked gene (Xce) biases XCI (51, 52), studies in cats suggest an age-dependent hemizygous cell selection (53), and studies in humans suggest various etiologies for skewing (cis-control of XCI [refs. 54, 55], stochastic [refs. 56, 57] or X-linked [ref. 58] cell selections, or a combination of these processes [ref. 59]).
With these limitations in mind, we designed a study that could significantly further characterize the etiology of skewing in normal human females. We used an epidemiogenetic approach based on the analysis of XCI ratios in a large cohort of neonates and their mothers. Analysis of the neonate cohort was mandatory to eliminate, or at least reduce, the confounding effect of age-associated skewing known to affect adult hematopoietic cells. Data obtained in neonates allowed us to have a less biased glance at events occurring during embryogenesis. The concordance of XCI ratios between hematopoietic and nonhematopoietic cells was ascertained by evaluating both blood and buccal cells from each subject (neonates and mothers). To evaluate the potential genetic basis of XCI skewing, we studied the transmission of the trait from mother to daughter and tested for association between genetic polymorphisms (SNPs) in the vicinity of the XIST gene and the skewing trait. As an excellent concordance was found between the methylation-based HUMARA assay and a transcription-based assay, this ruled out the possibility that the XCI ratios determined by the HUMARA assay were biased by aberrant methylation. We ascertained that there was no significant blood cell contamination in buccal swabs, as this could have induced a pseudoconcordance between the 2 tissues. Similarly, we verified that there was no maternal cell contamination of cord blood samples that could have led to pseudoconcordance between mother and neonate.
Age-associated skewing occurs in hematopoietic as well as in nonhematopoietic cells.
The incidence of skewing in blood cells of neonates (14.2%) and young women (27.9%) was similar to that reported in previous studies (59, 60) when taking into account the criteria used to define skewing (≥75% of the predominant allele) and the methods employed to calculate XCI ratios. Extreme skewing (≥90%) was rare in both neonates (2.7%) and mothers (4.9%). Blood cells of young women presented a substantially higher incidence of skewing than those of neonates, suggesting that hematopoietic cells are already affected by age-associated skewing in early adulthood (i.e., that age-associated skewing is not limited to the elderly). Analysis of buccal epithelial cells from both age groups allowed us to document that the incidence of skewing in epithelial tissue increases significantly from birth to early adulthood (7.6% versus 13.5%; P = 0.004). Age-associated skewing in epithelial cells has recently been reported in a small female cohort aged between 19 and 90 years old (24). Our results confirm that age-associated skewing occurs in nonhematopoietic cells and suggest that it is a lifelong process.
The direction of skewing (toward maternal versus paternal allele) is concordant in hematopoietic and nonhematopoietic cells.
The blood and epithelial cells of neonates showed a strong positive correlation (r = 0.715; P < 0.001) in the direction of skewing, in accordance with the concept that primary XCI occurs during early development, prior to the splitting of the inner cell mass and to tissue specification, as previously described in mice (61). Blood cells of neonates exhibited a higher incidence of skewing relative to their epithelial counterparts (7.6% versus 14.2%, respectively; P = 0.002). This might be a sign that age-associated skewing in hematopoietic cells begins as early as conception (and increases from conception to birth), or that there is an unequal splitting of the inner cell mass with significantly more cells committed to the epithelial tissue (therefore demonstrating less skewing). Unfortunately, investigating this possibility remains elusive in the human population.
In mothers, the incidence of skewing in epithelial versus hematopoietic tissue was statistically different (13.5% versus 27.9%, respectively; P < 0.001), but skewing direction was concordant among the 2 tissues (r = 0.779; P < 0.001). Given that mothers presented an incidence of skewing that was twice that of neonates, but that the direction of skewing in maternal buccal and blood cells was the same, we suggest that age-associated skewing occurs chiefly in the same direction in both cell types. If the principal cause of age-associated skewing is hemizygous cell selection caused by polymorphic X-linked gene(s), the candidate gene(s) should similarly affect both tissues (where it/they could potentially influence cell division, growth, apoptosis, etc.). Our data is in step with such XCI selection biases. Intriguingly, when older (>60 years old) females are studied (62), this correlation is lost, probably because the incidence of skewing in hematopoietic cells continues to increase, whereas that in epithelial cells has plateaued. This may be due to hematopoietic-specific continued genetic selection or, more likely, to the plasticity of the hematopoietic cells, which are more vulnerable to selective biases than an anatomically restricted tissue such as the buccal mucosa.
The skewing trait is not heritable.
If primary skewing of XCI (occurring at the time of XCI) in humans is influenced by an Xce-like element (as it is in mice), and if the skewing observed at birth predominantly reflects primary skewing, then transmission of the skewing trait would be expected to be observed in a significant proportion of mother-neonate pairs. To test this hypothesis, we studied whether skewed patterns of XCI were transmitted from mother to daughter. Using a qualitative analysis (categorizing females as skewed if they expressed ≥75% of the predominant allele or nonskewed if the percentage of the predominate allele was comprised between 50%–65%), we looked for an increased incidence of skewing in neonates of skewed mothers (compared with neonates of nonskewed mothers). We did not observe any enrichment of the proportion of skewed neonates born from skewed versus nonskewed mothers (P = 0.777 and 0.800 for epithelial and hematopoietic cells, respectively). Using a quantitative analysis (comparing mother and neonate degrees of skewing toward the shared X chromosome), we confirmed the lack of correlation between both groups (r = 0.074 for epithelial tissue, P = 0.172; r = 0.058 for blood tissue, P = 0.272). These results are clearly not in favor of transmission of the skewing trait.
We acknowledge that this approach is limited by several factors; for example, it is not possible to analyze paternal contribution or the presence of age-associated skewing in mothers. Furthermore, the concordance in direction of skewing in the mother-neonate pairs is informative only for putative genes that are in linkage disequilibrium with the HUMARA locus at Xq12, such as XIST, Xce, and others residing at the X-inactivation center at Xq13.2. The quantitative analysis took into account the direction of skewing for the transmitted mother allele (Figure 3) and is sensitive for genes in linkage disequilibrium with Xq12. On the other hand, the qualitative analysis (Tables 2 and 3) would have revealed any relationship between a skewed mother and neonate regardless of the direction of skewing. Therefore, we are confident that the analyses performed in this study are robust, complementary, and inclusive of any mechanisms of transmission of skewing. In fact, this study has the power to detect a genetic contribution of at least 23%, which is sufficient to detect a significant Xce-like effect. We calculated that for these results to be reversed it would require the analysis of over 6,000 mother-neonate pairs.
Our results were further supported by the evaluation of the association between the degree of skewing and the genotypes at 5 polymorphic loci covering 1 candidate region, the XIST gene. The drawback of testing a small number of polymorphisms was overcome by the fact that this study was performed on a founder population where the haplotype blocks are generally large (63) and that the size of the candidate region is believed to be relatively small (64). Our data showed no correlation between the degree of skewing and the genotypes at 5 polymorphic loci within the XIST gene (see Table 4), excluding the implication of the region surrounding the XIST gene in XCI skewing, and corroborated the results of our transmission study. Therefore, the results of the transmission and the association analyses are not compatible with a strong single-locus (Xce-like; ref. 64) effect that would be responsible for the majority of XCI skewing present at birth. Our data, hence, suggest that the mechanisms governing XCI in humans are strikingly different from those controlling XCI in mice.
The lack of evidence for a strong Xce-like effect brings forward a number of alternative hypothetical mechanisms as the cause of skewing at birth. One of them is that inactivation of the X chromosome is simply stochastic in humans, as was first proposed by Lyon (30). Another hypothetical mechanism is that the skewing seen at birth is already predominantly caused by age-associated skewing that has occurred during foetal development. If certain X-linked genes affect the rate of cellular growth and division, the rapid cellular division rate occurring during embryogenesis would be particularly sensitive to polymorphic allele variants that confer growth advantage to cells having these on the active X chromosome. In support of this hypothesis, we demonstrated in this study that age-associated skewing was already present in young adults, in both their hematopoietic and nonhematopoietic cells. Age-associated skewing is thus not an elderly- and hematopoietic-specific trait, as previously thought. Moreover, the incidence of skewing in blood cells was twice that in buccal epithelial cells in neonates, suggesting that age-associated skewing is likely to be responsible for at least 50% of the skewing phenotype observed at birth in these cells. The concordance of skewing between blood and buccal cells at birth and the maintenance of this concordance in young adults suggest a rather homogeneous trait dominated by the age-effect phenomenon. If this is the case, the incidence of true primary skewing may not be measurable after birth, not even in neonates. Furthermore, if both forms of skewing are genetically determined, it is unlikely that the causal genes originate from the same locus and that the strongest allele is from the same parental X chromosome. The complex interaction of 2 different loci would preclude a positive result for transmission of skewing in such a study as the present one. The absence of positive transmission results suggests that skewing is determined, at least in part, by post-XCI selection biases, some of which may be transmissible. Accordingly, a number of studies performed in non-neonatal populations have demonstrated a certain degree of heritability of skewing (65–67). In light of these considerations, we propose that neonatal skewing is caused in part, but not exclusively, by selection biases operating after primary XCI.
In conclusion, skewing of XCI ratios in normal human female neonates is not caused by a strong in cis, Xce-like element, as is the case in mouse. The results presented in this paper suggest that the skewing pattern observed at birth is a complex trait determined by the combinatorial effect of stochastic events and selection biases occurring after primary XCI. The later affect similarly hematopoietic and epithelial cells and may be caused by X-linked allelic variants of genes affecting cellular growth. Future studies are needed to attest the genetic basis of these hemizygous selections and eventually identify the genes implicated.
Methods
Subjects.
Research protocol was approved by the Ethical Research Committee of the Maisonneuve-Rosemont Hospital. Female neonates and their mothers were prospectively recruited at the hospital’s Obstetric and Gynecology department, following written informed consent from mothers (for themselves and their neonate) and fathers (for neonates).
Specimens.
Buccal epithelial samples were obtained using an Omniswab for buccal scraps (Whatman). DNA was extracted from swabs using a lithium chloride method according to the manufacturer’s instructions. Cord blood (neonates) or peripheral blood (mothers) was collected in EDTA blood tubes. A standard Triton X-100, proteinase K, and phenol-chloroform extraction protocol was used to isolate DNA from total blood. RNA isolation was performed using TriPure isolation reagent (Roche Applied Science) and the manufacturer’s protocol. Buccal cells were stained with Wright-Giemsa to verify that there was no blood cell contamination.
Phenotypic determination of XCI ratios — methylation assay.
HUMARA assays were essentially performed as previously described (68). Briefly, each specimen was digested with either RsaI alone (henceforth termed undigested fraction) or with RsaI and HpaII (digested fraction) and then submitted to PCR amplification of the first exon of the HUMARA locus (containing a CAG repeat) using fluorochrome-coupled primers. Amplification products were migrated on an ABI PRISM 3100 Automatic Genetic Analyzer (Applied Biosystems), and allele calling was performed using Genescan software (v.3.7 NT) (Applied Biosystems). Area under the curve (AUC) was measured with Genotyper software (v.3.7 NT) (Applied Biosystems) and used to calculate the XCI ratio. The XCI ratio of the digested fraction was corrected with that of the undigested fraction to allow for preferential amplification of the smallest allele (i.e., the allele containing less CAG repeats) (68). When the 2 alleles of an individual were separated by only 1–2 CAG repeats, XCI ratio was corrected for shadow banding. Results presented herein correspond to the mean of triplicates.
XCI ratios are reported using percentage of predominate allele.
The percentage (%) of the parental allele that is predominantly expressed ranges from 50% to 100%. Skewing is said to be present when the percentage of the predominant allele exceeds 74% (i.e., is ≥75%). This arbitrary definition has been used by a number of other groups (69–72). A percentage of predominant allele between 90% and 100% is considered extreme skewing. Individuals with allele ratio between 50% and 65% were considered nonskewed for the qualitative analysis. In this study, individuals between 66% and 74% were considered undetermined.
XCI ratios are reported using Psup.
The proportion of cells presenting the superior HUMARA allele on the active X chromosome was determined. The superior allele is defined as that which has the greater number of CAG repeats. The Psup score directly specifies the direction of skewing: Psup > 0.5 indicates that most cells express the X chromosome bearing the superior allele, while Psup < 0.5 indicates the opposite. Scores of 0 and 1 indicate complete skewing, and a score of 0.5 indicates nonskewing. As any given female has the same superior allele in each of her cells, Psup can be used to establish intra-individual correlations.
Equation 1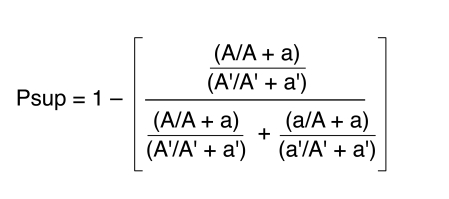
(A and A′ represent the AUC of the superior HUMARA allele from the digested and undigested sample, respectively. The AUC of the inferior HUMARA allele for the digested and undigested samples are represented by a and a′, respectively.)
XCI ratios are reported using Pmat and Ptrans.
The proportion of cells having the maternal or transmitted allele on the active X chromosome gives scores for neonates and mothers, respectively. This score is similar to Psup, but rather than considering the superior HUMARA allele, it considers the HUMARA allele that the mother transmitted to her daughter. Pmat and Ptrans are calculated the same way as Psup, except that A corresponds to the HUMARA allele shared between mother and daughter. A score of 1 indicates that all cells express the X chromosome bearing the shared allele, while a score of 0 indicates that all cells express the other X chromosome. Pmat and Ptrans were used for the quantitative assessment of XCI ratio transmission from mother to neonate. For example, a neonate sample carrying HUMARA alleles 20 and 22 (the number represents the extent of CAG repeat) and showing a Psup score of 0.8 will have a Pmat score of 0.2 if the maternal allele is allele 20.
Phenotypic determination of XCI ratios — transcriptional assay.
Transcriptional-based assays using the IDS locus were performed (73) upon RNA isolated from the blood of 94 neonates selected for their heterozygosity at both the IDS and the HUMARA loci. In brief, cDNA was synthesized from RNA using random hexamers and Taqman Reverse Transcription Reagents (Applied Biosystems), following which a SNP within an exon of the IDS gene (reference ID: rs1141608) was detected using the Taqman SNP assay (Applied Biosystems). This assay included probes capable of distinguishing each allele of the SNP on cDNA and was carried out on a real-time ABI Prism 7000 machine (Applied Biosystems).
SNP allele frequencies were measured using a standard curve. Cells from 2 individuals homozygous for a different allele were mixed in various proportions, and RNA extracted from these samples was used as standard. A standard curve was established by plotting allele frequency against ΔCts (Ct allele 1–Ct allele 2). All samples were measured in triplicate, and mean ΔCt value was used to calculate allele frequency.
XIST polymorphisms.
A total of 5 SNPs were chosen, covering approximately 40 kb of the region surrounding XIST. Three of them (reference IDs: rs1620574, rs1794213, and rs1082089) were genotyped using the TaqMan Assay-by-Design technology provided by Applied Biosystems. The other 2 (reference IDs: rs1009948 and rs1894271) were genotyped using a PCR-RFLP assay developed in our laboratory. Briefly, genomic DNA was amplified using PCR primers flanking the polymorphic restriction site. The oligonucleotides used to detect the first SNP were 5′-TTGACATAAAGGTTTCCTCATG-3′ (forward primer) and 5′-GCTAATGGTCATCCTGTTGCT-3′ (reverse primer), while those used to detect the second SNP were 5′-TGAAGGACAGCATGGTTGGT-3′ (forward primer) and 5′-ACATGGAATGAGCAGTGTGC-3′ (reverse primer). The amplification product was digested overnight with the appropriate restriction enzyme (RsaI for the first SNP, MfeI for the second), run on an agarose gel, and genotyped according to the band patterns.
Statistics.
Pearson correlations, Kolmogorov-Smirnov normality tests, and graphs and linear regression analyses were conducted using SPSS software version 10.1 for Microsoft Windows and NCSS 2004. Frequencies were compared by Fisher’s exact test or χ2 test. Agreement between methylation and transcription assays was assessed using the Bland-Altman method (74). A power study was conducted by a linear regression analysis using PASS 2002. Hardy-Weinberg equilibrium was assessed for each SNP using PEDSTATS (75). Haplotype association tests were conducted by using UNPHASED QTPHASE version 2.402 (76). As the percentage of predominant allele scores are not normally distributed, a box-cox transformation was performed to normalize the data using SAS 9.1.3.
Genetic profiling for twin zygosity determination.
Seven different single tandem repeat (STR) markers were used on DNA obtained from cord blood samples to assess the zygosity of twin pairs. If a pair of twins had identical alleles at these 7 markers, 3 additional markers were tested. All DNA profiles showed clear single individual patterns, and no sign suggesting mixed blood.
Acknowledgments
We gratefully thank Rolande Ste-Marie for coordinating recruitment, Eve Pomerleau and Myriam Beaupré for technical assistance, the employees of the Obstetric and Gynecology department of Maisonneuve-Rosemont Hospital for their collaboration, and all the study participants. This study was funded by Fonds de la recherche en santé du Québec (FRSQ) (L. Busque) and an unrestricted grant from Hemax Genome Inc. L. Mollica is a scholar from FRSQ.
Footnotes
Nonstandard abbreviations used: HUMARA, human androgen receptor; IDS, iduronate-2-sulfatase gene; Pmat, proportion of cells having the maternal HUMARA allele on the active X chromosome; Psup, proportion of cells having the superior HUMARA allele on the active X chromosome; Ptrans, proportion of cells having the transmitted HUMARA allele on the active X chromosome; Xce, X chromosome controlling element; XCI, X chromosome inactivation; XIST, X (inactive)–specific transcript.
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 118:333–341 (2008). doi:10.1172/JCI33166
Pierre Chagnon’s present address is: Institute for Research in Immunology and Cancer, University of Montreal, Montreal, Quebec, Canada.
See the related Commentary beginning on page 20.
References
- 1.Lyon M.F. Gene action in the X-chromosome of the mouse [Mus musculus L.]. . Nature. 1961;190:372. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- 2.Beutler E., Yeh M., Fairbanks V.F. The normal human female as a mosaic of X–chromosome activity: Studies using the gene gor G–6–PD deficieny as a marker. Proc. Natl. Acad. Sci. U. S. A. 1962;48:9. doi: 10.1073/pnas.48.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prchal J.T., Guan Y.L., Prchal J.F., Barany F. Transcriptional analysis of the active X-chromosome in normal and clonal hematopoiesis [letter]. Blood. 1993;81:269–271. [PubMed] [Google Scholar]
- 4.Vogelstein B., Fearon E.R., Hamilton S.R., Feinberg A.P. Use of restriction fragment length polymorphisms to determine the clonal origin of human tumors. Science. 1985;227:642–645. doi: 10.1126/science.2982210. [DOI] [PubMed] [Google Scholar]
- 5.Allen R.C., et al. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 6.Fialkow P.J. Primordial cell pool size and lineage relationships of five human cell types. Ann. Hum. Genet. 1973;37:39–48. doi: 10.1111/j.1469-1809.1973.tb01813.x. [DOI] [PubMed] [Google Scholar]
- 7.Vogelstein B., et al. Clonal analysis using recombinant DNA probes from the X-chromosome. Cancer Res. 1987;47:4806–4813. [PubMed] [Google Scholar]
- 8.Felix J.S., DeMars R. Purine requirement of cells cultured from humans affected with Lesch-Nyhan syndrome (hypoxanthine-guanine phosphoribosyltransferase deficiency). Proc. Natl. Acad. Sci.U. S. A. 1969;62:536–543. doi: 10.1073/pnas.62.2.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gartler S.M. X-chromosome inactivation and selection in somatic cells. Fed. Proc. 1976;35:2191–2194. [PubMed] [Google Scholar]
- 10.Zabel B.U., Baumann W.A., Pirntke W., Gerhard-Ratschow K. X-inactivation pattern in three cases of X/autosome translocation. Am. J. Med. Genet. 1978;1:309–317. doi: 10.1002/ajmg.1320010307. [DOI] [PubMed] [Google Scholar]
- 11.Pegoraro E., et al. Familial skewed X inactivation: a molecular trait associated with high spontaneous-abortion rate maps to Xq28. Am. J. Hum. Genet. 1997;61:160–170. doi: 10.1086/513901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plenge R.M., Stevenson R.A., Lubs H.A., Schwartz C.E., Willard H.F. Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders. Am. J. Hum. Genet. 2002;71:168–173. doi: 10.1086/341123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linder D., Gartler S.M. Glucose-6-phosphate dehydrogenase mosaicism: utilization as a cell marker in the study of leiomyomas. Science. 1965;150:67–69. doi: 10.1126/science.150.3692.67. [DOI] [PubMed] [Google Scholar]
- 14.Beutler E., Collins Z., Irwin L.E. Value of genetic variants of glucose-6-phosphate dehydrogenase in tracing the origin of malignant tumors. N. Engl. J. Med. 1967;276:389–391. doi: 10.1056/NEJM196702162760706. [DOI] [PubMed] [Google Scholar]
- 15.Gale R.E., Wheadon H., Linch D.C. Assessment of X-chromosome inactivation patterns using the hypervariable probe M27 beta in normal hemopoietic cells and acute myeloid leukemic blasts. Leukemia. 1992;6:649–655. [PubMed] [Google Scholar]
- 16.Clerc P., Avner P. Random X-chromosome inactivation: skewing lessons for mice and men. Curr. Opin. Genet. Dev. 2006;16:246–253. doi: 10.1016/j.gde.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Busque L., et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- 18.Tonon L., et al. Unbalanced X-chromosome inactivation in haemopoietic cells from normal women. Br. J. Haematol. 1998;102:996–1003. doi: 10.1046/j.1365-2141.1998.00867.x. [DOI] [PubMed] [Google Scholar]
- 19.Amos-Landgraf J.M., et al. X chromosome-inactivation patterns of 1, 005 phenotypically unaffected females. Am. J. Hum. Genet. 2006;79:493–499. doi: 10.1086/507565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gale R.E., Wheadon H., Boulos P., Linch D.C. Tissue specificity of X-chromosome inactivation patterns. Blood. 1994;83:2899–2905. [PubMed] [Google Scholar]
- 21.Sharp A., Robinson D., Jacobs P. Age- and tissue-specific variation of X chromosome inactivation ratios in normal women. Hum. Genet. 2000;107:343–349. doi: 10.1007/s004390000382. [DOI] [PubMed] [Google Scholar]
- 22.Busque L., et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- 23.Kristiansen M., et al. Twin study of genetic and aging effects on X chromosome inactivation. Eur. J. Hum. Genet. 2005;13:599–606. doi: 10.1038/sj.ejhg.5201398. [DOI] [PubMed] [Google Scholar]
- 24.Knudsen G.P., Pedersen J., Klingenberg O., Lygren I., Orstavik K.H. Increased skewing of X chromosome inactivation with age in both blood and buccal cells. Cytogenet. Genome Res. 2007;116:24–28. doi: 10.1159/000097414. [DOI] [PubMed] [Google Scholar]
- 25.Gale R.E., Wheadon H., Boulos P., Linch D.C. Tissue specificity of X-chromosome inactivation patterns. Blood. 1994;83:2899–2905. [PubMed] [Google Scholar]
- 26.Azofeifa J., Waldherr R., Cremer M. X-chromosome methylation ratios as indicators of chromosomal activity: evidence of intraindividual divergencies among tissues of different embryonal origin. Hum. Genet. 1996;97:330–333. doi: 10.1007/BF02185765. [DOI] [PubMed] [Google Scholar]
- 27.Sharp A., Robinson D., Jacobs P. Age- and tissue-specific variation of X chromosome inactivation ratios in normal women. Hum. Genet. 2000;107:343–349. doi: 10.1007/s004390000382. [DOI] [PubMed] [Google Scholar]
- 28.Bicocchi M.P., et al. Familial nonrandom inactivation linked to the X inactivation centre in heterozygotes manifesting haemophilia A. Eur. J. Hum. Genet. 2005;13:635–640. doi: 10.1038/sj.ejhg.5201386. [DOI] [PubMed] [Google Scholar]
- 29.Cazzola M., et al. Familial-skewed X-chromosome inactivation as a predisposing factor for late-onset X-linked sideroblastic anemia in carrier females. Blood. 2000;96:4363–4365. [PubMed] [Google Scholar]
- 30.Lyon M.F. Sex chromatin and gene action in the mammalian X-chromosome. Am. J. Hum. Genet. 1962;14:135–148. [PMC free article] [PubMed] [Google Scholar]
- 31.Cattanach B.M., Isaacson J.H. Controlling elements in the mouse X chromosome. Genetics. 1967;57:331–346. doi: 10.1093/genetics/57.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plenge R.M., et al. A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. Nat. Genet. 1997;17:353–356. doi: 10.1038/ng1197-353. [DOI] [PubMed] [Google Scholar]
- 33.Pereira L.V., Zatz M. Screening of the C43G mutation in the promoter region of the XIST gene in females with highly skewed X-chromosome inactivation. Am. J. Med. Genet. 1999;87:86–87. [PubMed] [Google Scholar]
- 34.Bicocchi M.P., et al. Familial nonrandom inactivation linked to the X inactivation centre in heterozygotes manifesting haemophilia A. Eur. J. Hum. Genet. 2005;13:635–640. doi: 10.1038/sj.ejhg.5201386. [DOI] [PubMed] [Google Scholar]
- 35.Christensen K., et al. X-linked genetic factors regulate hematopoietic stem-cell kinetics in females. Blood. 2000;95:2449–2451. [PubMed] [Google Scholar]
- 36.Vickers M.A., McLeod E., Spector T.D., Wilson I.J. Assessment of mechanism of acquired skewed X inactivation by analysis of twins. Blood. 2001;97:1274–1281. doi: 10.1182/blood.v97.5.1274. [DOI] [PubMed] [Google Scholar]
- 37.Kristiansen M., et al. Twin study of genetic and aging effects on X chromosome inactivation. Eur. J. Hum. Genet. 2005;13:599–606. doi: 10.1038/sj.ejhg.5201398. [DOI] [PubMed] [Google Scholar]
- 38.Naumova A.K., et al. Heritability of X chromosome--inactivation phenotype in a large family. Am. J. Hum. Genet. 1996;58:1111–1119. [PMC free article] [PubMed] [Google Scholar]
- 39.Pegoraro E., et al. Familial skewed X inactivation: a molecular trait associated with high spontaneous-abortion rate maps to Xq28. Am. J. Hum. Genet. 1997;61:160–170. doi: 10.1086/513901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naumova A.K., et al. Genetic mapping of X-linked loci involved in skewing of X chromosome inactivation in the human. Eur. J. Hum. Genet. 1998;6:552–562. doi: 10.1038/sj.ejhg.5200255. [DOI] [PubMed] [Google Scholar]
- 41.Tsang K.S., et al. Implication of maternal-cell contamination in the clinical banking of umbilical cord blood. Cytotherapy. 2002;4:375–383. doi: 10.1080/146532402760271163. [DOI] [PubMed] [Google Scholar]
- 42.Vogelstein B., et al. Clonal analysis using recombinant DNA probes from the X-chromosome. Cancer Res. 1987;47:4806–4813. [PubMed] [Google Scholar]
- 43.Gale R.E., Wheadon H., Linch D.C. X-chromosome inactivation patterns using HPRT and PGK polymorphisms in haematologically normal and post-chemotherapy females. . Br. J. Haematol. 1991;79:193–197. doi: 10.1111/j.1365-2141.1991.tb04521.x. [DOI] [PubMed] [Google Scholar]
- 44.Allen R.C., Nachtman R.G., Rosenblatt H.M., Belmont J.W. Application of carrier testing to genetic counseling for X-linked agammaglobulinemia. Am. J. Hum. Genet. 1994;54:25–35. [PMC free article] [PubMed] [Google Scholar]
- 45.Busque L., et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- 46.Hedrick, P.W. 2000. Quantitative traits and evolution. In Genetics of populations. Jones and Barlett Publishers. Sudbury, Massachusetts, USA/Mississauga, Ontario, Canada/London, United Kingdom. 463–480. [Google Scholar]
- 47.Borsani G., et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature. 1991;351:325–329. doi: 10.1038/351325a0. [DOI] [PubMed] [Google Scholar]
- 48.Ogawa Y., Lee J.T. Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol. Cell. 2003;11:731–743. doi: 10.1016/s1097-2765(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 49.Cattanach B.M., Isaacson J.H. Controlling elements in the mouse X chromosome. Genetics. 1967;57:331–346. doi: 10.1093/genetics/57.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chao W., Huynh K.D., Spencer R.J., Davidow L.S., Lee J.T. CTCF, a candidate trans-acting factor for X-inactivation choice. Science. 2002;295:345–347. doi: 10.1126/science.1065982. [DOI] [PubMed] [Google Scholar]
- 51.Cattanach B.M., Isaacson J.H. Controlling elements in the mouse X chromosome. Genetics. 1967;57:331–346. doi: 10.1093/genetics/57.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chadwick L.H., Pertz L., Broman K.W., Bartolomei M.S., Willard H.F. Genetic control of X chromosome inactivation in mice: definition of the Xce candidate interval. Genetics. 2006;173:2103–2110. doi: 10.1534/genetics.105.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abkowitz J.L., et al. An X chromosome gene regulates hematopoietic stem cell kinetics. Proc. Natl. Acad. Sci. U. S. A. 1998;95:3862–3866. doi: 10.1073/pnas.95.7.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Plenge R.M., et al. A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. . Nat. Genet. 1997;17:353–356. doi: 10.1038/ng1197-353. [DOI] [PubMed] [Google Scholar]
- 55.Pugacheva E.M., et al. Familial cases of point mutations in the XIST promoter reveal a correlation between CTCF binding and pre-emptive choices of X chromosome inactivation. Hum. Mol. Genet. 2005;14:953–965. doi: 10.1093/hmg/ddi089. [DOI] [PubMed] [Google Scholar]
- 56.Gale R.E., Fielding A.K., Harrison C.N., Linch D.C. Acquired skewing of X-chromosome inactivation patterns in myeloid cells of the elderly suggests stochastic clonal loss with age. Br. J. Haematol. 1997;98:512–519. doi: 10.1046/j.1365-2141.1997.2573078.x. [DOI] [PubMed] [Google Scholar]
- 57.Sandovici I., Naumova A.K., Leppert M., Linares Y., Sapienza C. A longitudinal study of X-inactivation ratio in human females. Hum. Genet. 2004;115:387–392. doi: 10.1007/s00439-004-1177-8. [DOI] [PubMed] [Google Scholar]
- 58.Christensen K., et al. X-linked genetic factors regulate hematopoietic stem-cell kinetics in females. Blood. 2000;95:2449–2451. [PubMed] [Google Scholar]
- 59.Hatakeyama C., et al. The dynamics of X-inactivation skewing as women age. Clin. Genet. 2004;66:327–332. doi: 10.1111/j.1399-0004.2004.00310.x. [DOI] [PubMed] [Google Scholar]
- 60.Amos-Landgraf J.M., et al. X chromosome-inactivation patterns of 1, 005 phenotypically unaffected females. Am. J. Hum. Genet. 2006;79:493–499. doi: 10.1086/507565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okamoto I., Otte A.P., Allis C.D., Reinberg D., Heard E. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science. 2004;303:644–649. doi: 10.1126/science.1092727. [DOI] [PubMed] [Google Scholar]
- 62.Sharp A., Robinson D., Jacobs P. Age- and tissue-specific variation of X chromosome inactivation ratios in normal women. Hum. Genet. 2000;107:343–349. doi: 10.1007/s004390000382. [DOI] [PubMed] [Google Scholar]
- 63.Arcos-Burgos M., Muenke M. Genetics of population isolates. Clin. Genet. 2002;61:233–247. doi: 10.1034/j.1399-0004.2002.610401.x. [DOI] [PubMed] [Google Scholar]
- 64.Chadwick L.H., Pertz L., Broman K.W., Bartolomei M.S., Willard H.F. Genetic control of X chromosome inactivation in mice: definition of the Xce candidate interval. Genetics. 2006;173:2103–2110. doi: 10.1534/genetics.105.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Naumova A.K., et al. Heritability of X chromosome--inactivation phenotype in a large family. Am. J. Hum. Genet. 1996;58:1111–1119. [PMC free article] [PubMed] [Google Scholar]
- 66.Naumova A.K., et al. Genetic mapping of X-linked loci involved in skewing of X chromosome inactivation in the human. Eur. J. Hum. Genet. 1998;6:552–562. doi: 10.1038/sj.ejhg.5200255. [DOI] [PubMed] [Google Scholar]
- 67.Pegoraro E., et al. Familial skewed X inactivation: a molecular trait associated with high spontaneous-abortion rate maps to Xq28. Am. J. Hum. Genet. 1997;61:160–170. doi: 10.1086/513901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Allen R.C., Zoghbi H.Y., Moseley A.B., Rosenblatt H.M., Belmont J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 69.Vogelstein B., et al. Clonal analysis using recombinant DNA probes from the X-chromosome. Cancer Res. 1987;47:4806–4813. [PubMed] [Google Scholar]
- 70.Busque L., et al. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- 71.Gale R.E., Wheadon H., Linch D.C. X-chromosome inactivation patterns using HPRT and PGK polymorphisms in haematologically normal and post-chemotherapy females. . Br. J. Haematol. 1991;79:193–197. doi: 10.1111/j.1365-2141.1991.tb04521.x. [DOI] [PubMed] [Google Scholar]
- 72.Allen R.C., Nachtman R.G., Rosenblatt H.M., Belmont J.W. Application of carrier testing to genetic counseling for X-linked agammaglobulinemia. Am. J. Hum. Genet. 1994;54:25–35. [PMC free article] [PubMed] [Google Scholar]
- 73.el-Kassar N., Hetet G., Briere J., Grandchamp B. Clonality analysis of hematopoiesis in essential thrombocythemia: advantages of studying T lymphocytes and platelets. Blood. 1997;89:128–134. [PubMed] [Google Scholar]
- 74.Bland J.M., Altman D.G. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1:307–310. [PubMed] [Google Scholar]
- 75.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin — rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 76.Dudbridge F. Pedigree disequilibrium tests for multilocus haplotypes. Genet. Epidemiol. 2003;25:115–121. doi: 10.1002/gepi.10252. [DOI] [PubMed] [Google Scholar]