

Abstract
Poor ambient air quality is associated with increased morbidity and mortality, including respiratory infections. However, its effects on various host-defense mechanisms are poorly understood. This study utilized an in vitro model to study the effect of particulate matter (PM2.5) on one antimicrobial mechanism of host defense in the airway, β-defensin-2 and its bovine homologue, tracheal antimicrobial peptide (TAP) induction in response to lipopolysaccharide (LPS) and IL-1β. Our model utilized cultured primary bovine tracheal epithelial (BTE) cells and the human alveolar type II epithelial cell line, A549, treated with 0–20 μg/cm2 residual oil fly ash (ROFA) for 6 h. The cells were then washed and stimulated for 18 h with 100 ng/ml LPS or for 6 h with 100 ng/ml IL-1β. ROFA inhibited the LPS-induced increase in TAP mRNA and protein without inducing significant cytotoxicity. As little as 2.5 μg/cm2 of ROFA inhibited LPS-induced TAP gene expression by 30%. The inhibitory activity was associated with the soluble fraction and not the washed particle. The activity in the leachate was attributed to vanadium, but not nickel or iron. SiO2 and TiO2 were utilized as controls and did not inhibit LPS induction of TAP gene expression in BTE. ROFA also inhibited the increase of IL-1β–induced human β-defensin-2, a homologue of TAP, in A549 cells. The results show that ROFA, V2O5, and VOSO4 inhibit the ability of airway epithelial cells to respond to inflammatory stimuli at low, physiologically relevant doses and suggest that exposure to these agents could result in an impairment of defense against airborne pathogens.
Keywords: defensins, epithelial cells, vanadium, metals, residual oil fly ash, particulate matter, human, bovine, lung, innate immunity
INTRODUCTION
Fine particulate air pollution is associated with an increased mortality due to respiratory infection and cardiovascular disease (Pope et al., 2004; Samet et al., 2000). Chronic exposure to particles that are less than 2.5 μm in diameter (PM2.5) also has been associated with an increase in chronic disease, including lung cancer and cardiovascular morbidity (Pope et al., 2002), as well as increasing the incidence of emergency room visits and respiratory infections (Peel et al., 2005).
The airway has many mechanisms to inhibit invasion and colonization of microorganisms from the environment, yet it is poorly understood how particulate air pollution affects innate immune host defenses of airway epithelium. Inducible host responses, such as upregulation of cytokines, lactoferrin, and lysozyme, are stimulated by air pollution particles in vitro (Ghio et al., 1998; Kennedy et al., 1998; Longphre et al., 2000; Quay et al., 1998). Complex PM2.5 air pollutant particles such as residual oil fly ash (ROFA) have been shown to increase production of IL-8 in primary normal human bronchial epithelial cells and cell lines (Carter et al., 1997; Kennedy et al., 1998; Stringer and Kobzik, 1998; Veronesi et al., 1999). Samet et al. 1998 showed that the upregulation of IL-8 in human bronchial airway cells occurred in response to the soluble metal components of ROFA, vanadium and zinc, and resulted in increased mitogen-activated protein kinase pathway activation at a concentration of 500μM. IL-6 and IL-8 upregulation by particulate air pollution in the airway was preceded by activation of NF-κB (Kennedy et al., 1998).
β-Defensins are cationic, broad-spectrum antimicrobial peptides widely expressed in epithelia (Diamond et al., 2000a; Kaiser, 2000). Tracheal antimicrobial peptide (TAP) was the first β-defensin discovered (Diamond et al., 1991) and is predominantly expressed in ciliated respiratory epithelium (Diamond et al., 1993). Expression of TAP and its human homologue, hBD-2 is stimulated by bacterial lipopolysaccharide (LPS) through interaction with toll-like receptors (TLRs) and activation of NF-κB (Becker et al., 2000; Diamond et al., 1996) and participates in host defense against bacterial infections of the airway (Caverly et al., 2003). We hypothesized that exposure of cultured respiratory epithelial cells to ROFA and its components decrease the induction of the innate immune response in the airway, as measured by β-defensin gene expression. Here, we show that ROFA and one of its soluble metal components, vanadium, modulate this mediator of innate immune host defense of airway epithelium, which could diminish the host defense against infection. These effects occur both in differentiated primary bovine airway epithelial cells and in a human type II alveolar epithelial cell line, A549, at low levels likely to be encountered in vivo.
MATERIALS AND METHODS
Reagents
ROFA was generously provided by Dr Allen Ledbetter, Dr Kevin Dreher, and Dr Daniel Costa, Pulmonary Toxicology Branch, U.S. Environmental Protection Agency (EPA), National Health and Environmental Effects Research Laboratory, Research Triangle Park, NC. ROFA particles were collected by the Southern Research Institute (Birmingham, AL) at a temperature of 204°C on a Teflon-coated glass fiber filter, downstream of a cyclone from a power plant burning low sulfur number 6 residual oil. The composition and mass median aerodynamic diameter (MMAD) have been well characterized (MMAD = 1.95 μm) (Dreher et al., 1997; Hatch et al., 1985). Vanadium pentoxide (V2O5), vanadium sulfate (VOSO4), silicone dioxide (SiO2), titanium dioxide (TiO2), ferric sulfate [Fe2(SO4)3], nickel sulfate (NiSO4), and Pseudomonas aeruginosa LPS were purchased from Sigma-Aldrich Corp. (St Louis, MO). Palmitoyl-3-cysteine-serine-lysine-4 (PamCSK4) was acquired from InVivoGen (San Diego, CA). Recombinant human TNF-α and IL-1β were purchased from R&D Systems (Minneapolis, MN). Each component metal and SiO2 were baked at 180°C for 3 h to render it LPS-free. Both unbaked ROFA and baked ROFA, as well as SiO2 and V2O5 were shown by Limulus amoebocyte lysate assay (Associates of Cape Cod, Falmouth, MA) to be endotoxin-free (≤ 25 EU/ml).
Cell culture
Primary cultures of differentiated bovine tracheal epithelial (BTE) cells were harvested from freshly obtained bovine tracheas as previously described (Diamond et al., 1996; Legarda et al., 2005). Cultures were maintained on Vitrogen-coated six-well plates (Cohesion Technologies Inc., Palo Alto, CA) in FDU medium (48% DMEM, 48% F-12, 2% Ultroser G [Biosepra, Cergy-Saint-Christophe, France], 1% antibiotic-antimycotic solution, 0.5% Fungizone, 5 μg/100 ml gentamicin solution [Gibco BRL, Carlsbad, CA]). In some cases, cells were grown in an air-liquid interface in 24-well transwell inserts (Costar) as described (Becker et al., 2000). The type II alveolar epithelial cell line A549 (ATCC) was derived from a human lung carcinoma. Cells were maintained as recommended by ATCC in F-12 with 2mM L-glutamine (Mediatech, Herdon, VA), modified with 0.15% (wt/vol) sodium bicarbonate (Sigma-Aldrich Corp.) and supplemented with 10% (vol/vol) FCS in a humid 37°C, 5% CO2 incubator. Cell viability was determined by trypan blue exclusion.
Treatment of BTE and human cells with particulate air pollutants and metal components
ROFA and individual component metals were resuspended in RPMI at a stock concentration of 18 mg/ml, sonicated for 10 min and stored at 4°C for up to 18 h. The solutions were sonicated again for 10 min and vortexed thoroughly before each sample was removed for use. The appropriate concentration of metal or pollutant was then added to fresh FDU and vortexed to ensure homogeneity before adding to the cells. Confluent cultures of BTE cells were washed once in 1×phosphate buffered saline (PBS) and then treated with the metal or pollutant solutions and incubated in a humid 37°C incubator with 5% CO2. After the specified incubation period, the cells were washed three times in 1×PBS and received fresh FDU. The cells were then stimulated with 100 ng/ml LPS for 18 h (the time required to achieve maximal stimulation of mRNA levels; Diamond et al., 1993) or 100 ng/ml IL-1β for 6 h. The medium was removed, and then either the mRNA or protein was harvested from lysed cells as described below.
For one set of experiments, an 18 mg/ml ROFA suspension was centrifuged for 15 min at maximum speed (15,000 RPM) in a microcentrifuge. The leachate was removed, and the remaining particles were washed twice and then resuspended in a volume of RPMI equivalent to that removed for the leachate before performing the procedure described above on the BTE cells.
Analysis of gene expression
Cells were lysed and homogenized as per manufacturer’s instructions with QIAshredder columns (Qiagen, Valencia, CA). Total RNA was isolated using the RNeasy Mini Kit with on-column DNase (Qiagen) and stored at −80°C until analysis. For the initial northern blot analysis, total mRNA was electrophoresed and hybridized with 32P-labeled oligonucleotide probes to TAP and β-actin as described in (Diamond et al., 2000b).
RT-PCR for both bovine samples was performed in a Bio-Rad iCycler (Hercules, CA). To make cDNA, approximately 1 μg of total RNA was reverse-transcribed using Invitrogen reagents (Invitrogen Life Technologies, Carlsbad, CA); the final reaction conditions were 1×Invitrogen PCR buffer, 5mM MgCl2, 1mM each dNTP, 0.5 μg Oligo(dT)12–18, 40 units (U) RNaseOUT ribonuclease inhibitor, and 200 U Superscript II RT reverse transcriptase. The reaction was incubated at 42°C for 15 min, 99°C for 5 min, and 5°C for 5 min.
PCR primers for TAP (sense: 5′ GCGAGCATGAGGCCTCCAT 3′; antisense: 5′ AACAGGTGCCAATCTGT 3′) and GAPDH (sense: 5′ TGGCAAAGTGGACATCGTCG 3′; antisense: 5′ TGGCGTGGACAGTGGTCATAAGTC 3′) were titrated for primer concentration and cycle number to determine the number of cycles necessary to be within log phase of amplification for both unstimulated and LPS-stimulated samples. Primers were synthesized by the Molecular Resource Facility, UMDNJ-NJMS (Dr Robert Donnelly, director). For each PCR reaction, 50 ng of cDNA was amplified with 1×PCR buffer, 2.5mM MgCl2, 20nM each dNTP, primers (300nM for TAP primers, and 50nM for GAPDH), and 2 U of Taq polymerase. The samples were amplified at 94°C for 2 min, then 94°C for 30 s, 60°C for 1 min, and 72°C for 2 min for 25 cycles, and a final extension at 72°C for 15 min. Both sets of TAP and GAPDH primers were run in the same reaction tube. The PCR products were visualized in a 2% agarose gel with 300 ng/ml ethidium bromide using the Typhoon 8600 imaging system (Amersham Biotech, Piscataway, NJ). All PCR products were ligated into a pTOPO-TA plasmid (Invitrogen), and the inserts were sequenced by dye-termination sequencing at the UMDNJ-Molecular Resource Facility to verify that the desired product was amplified. In either case, the results are expressed as fold increase over control, where the control is the unstimulated sample with no particle or metal. Due to the nature of the primary cultures, the level of stimulation by LPS is variable between experiments, from 2- to 15-fold.
For the quantitative polymerase chain reaction (QPCR) reaction, 500 ng of cDNA was transcribed with 1×SYBR Green PCR Master Mix and either hBD-2 primers (300nM forward: 5′ TGCCTCTTCCAGGTGTTTTTG3′/300nM reverse: 5′ GGCTCCACTCTTAAGGCAGGTA 3′) or hPRT primers (300nM forward: 5′ CAGCCCTGGCGTCGTGATTAGT 3′/50nM reverse: 5′ GCAAGACGTTCAGTCCTGTCCATAA 3′) in a final volume of 20 μl. At these primer concentrations, both sets of primers were determined by to have the same efficiency; therefore they were all run using the same cycling conditions. The samples were first incubated at 50°C for 2 min to activate the UTPase which degrades dUTP from the cDNA reaction, followed by one cycle of 95°C for 1 min to inactivate the UTPase. The cDNA was then amplified at 50 cycles of 15 s at 95°C and 1 min at 60°C, measuring fluorescence continuously, and ending with an additional cycle of 95°C for 1 min. Lastly, the samples were incubated for 41 cycles of 30 s at 55°C, increasing one degree with each cycle to 95°C. During these cycles, three end point measurements were taken to determine the dissociation curves of the products.
Mass spectrometry of TAP
Cytoplasmic extracts were isolated using a modified protocol from Clontech (Palo Alto, CA). Specifically, treated cells were harvested in 1×trypsin-EDTA as described above and spun at 450 ×g at 4°C for 10 min. Cells were washed twice in 1×PBS. Next, cells were resuspended in 5×cell pellet volume of lysis buffer (10mM HEPES pH 7.9, 1.5mM MgCl2, 10mM KCl, 1μM DTT, 1mM phenylmethylsulfonyl fluoride [PMSF]) and incubated for 15 min on ice. Samples were then centrifuged for 5 min at 420 ×g at 4°C. The supernatant was discarded and the cells were resuspended in 2×cell pellet volume of lysis buffer and passed 10 times through a 27-gauge needle. Lysates were centrifuged again at 11,000 ×g at 4°C for 20 min. The supernatant is the cytoplasmic extract, which was transferred to a clean tube and stored at −80°C for analysis. Protein concentrations of cytoplasmic extracts were analyzed by Bradford assay, using the Bio-Rad Protein Assay as described by the manufacturer. Cytoplasmic extracts were analyzed by mass spectrometry. For the unstimulated and LPS 18-h samples, 200 μg of each sample was analyzed, whereas for the vanadium pentoxide pretreated samples, 500 μg of each sample was analyzed. These samples were concentrated by Millepore ZipTipC18 (Billerica, MA). The accurate masses of the component peptides were determined by matrix assisted laser desorption/ ionization-time of flight-mass spectroscopy (MALDI-TOF-MS) using a-Cyano-4-hydroxycinnamic acid as the energy-absorbing matrix. The mass of TAP was calculated to be 4083.0780 Da; the masses of prepro-TAP and pro-TAP were previously described (Diamond et al., 1991).
Statistics
ANCOVA was performed to evaluate the effects of ROFA at varying doses, with and without a stimulus factor (LPS) as a function of dose. Statistical analysis was performed using StatView Se+ Graphics (Abacus Concepts, Berkley, CA) and SuperANOVA (Abacus Concepts). For the washed particle and A549 QPCR data, the Student’s t-test was used to compare the effect of each dose of ROFA on hBD-2 induction by LPS and IL-1β, respectively. TAP gene expression induced by IL-1β in BTE cells pre-exposed to V2O5 was also evaluated using the Student’s t-test. Significance was considered to be p ≤ 0.05. All graphs represent triplicate experiments unless otherwise stated.
RESULTS
ROFA Inhibition of TAP is Time-Dependent and Dose-Dependent
In order to characterize the effects of ROFA on the immune response of the airway epithelium, BTE cells were first preincubated with 5 μg/cm2 of ROFA for increasing periods of time (2–6 h), washed, and then stimulated with 100 ng/ml LPS for 18 h. The mRNA was then harvested and analyzed by northern blot for TAP gene expression. TAP mRNA levels were normalized to those of α-tubulin, and expressed as fold increase relative to levels from unstimulated cultures with no ROFA. At this relatively low concentration, ROFA inhibited the induction of TAP gene expression by LPS even after only 2 h exposure; furthermore, this inhibition is amplified with increasing duration of ROFA exposure (Fig. 1).
FIG. 1.
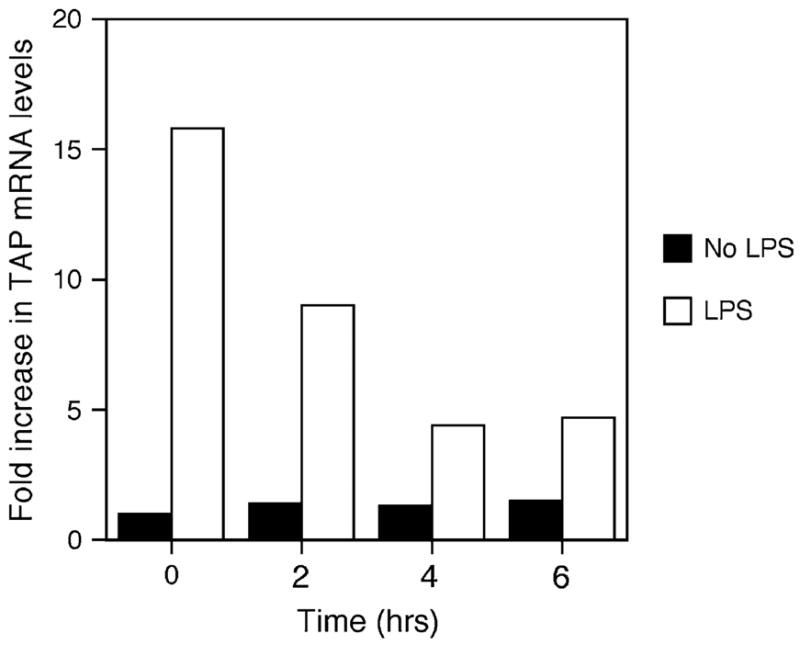
Time course of TAP gene expression in response to ROFA suspension. BTE cells were treated with 5.0 μg/cm2 of ROFA suspension for increasing periods of time, then stimulated with or without LPS (100 ng/ml) for 18 h. Total mRNA was isolated and analyzed by northern blot. Blots were probed for TAP and normalized to α-tubulin, and expressed as fold increase over unstimulated TAP mRNA levels in the absence of ROFA. Maximum suppression of LPS-induced TAP mRNA occurred from 4 to 6 h. The results are representative of three separate experiments.
Next, BTE cells were pretreated for 6 h with increasing doses of ROFA, between 2.5 and 20 μg/cm2, washed, and then stimulated with 100 ng/ml LPS as above. Total mRNA was analyzed by semi-quantitative RT-PCR, and TAP expression was normalized to GAPDH. Figure 2 illustrates that BTE cells exposed to increasing concentrations of baked ROFA exhibit a decrease in basal TAP gene expression, with a significant dose-dependent decrease in LPS-induced TAP gene expression. Similar results were observed when unbaked ROFA was added to the apical surface of differentiated cells in transwell plates, and the LPS was added to the medium on the basolateral side (Diamond et al., 2000a). Both types of culturing systems yielded similar dose-response curves. Together these results demonstrate that LPS contamination was not a factor and that baking the ROFA did not destroy the activity of the particle in this biological assay.
FIG. 2.
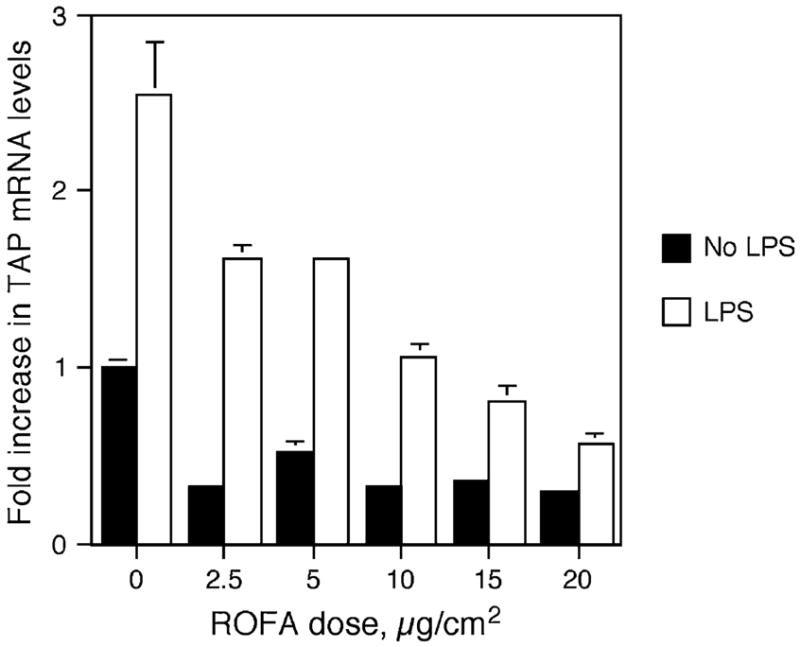
ROFA-suspension dose response. BTE cells were treated with increasing doses of ROFA-suspension for 6 h and then stimulated with or without LPS (100 ng/ml) for 18 h. mRNA was isolated and analyzed by semi-quantitative RT-PCR for TAP and normalized to GAPDH. Results are expressed as fold increase over unstimulated TAP mRNA levels in the absence of ROFA. Experiments were carried out in triplicate in six-well plates. Error bars represent 1 SEM. The interaction between ROFA dose and LPS stimulus is significant as measured by ANCOVA (p ≤ 0.001), illustrating an increased inhibition of TAP gene expression with ROFA dose only in the presence of LPS.
To control for the possibility that the buffering capacity of culture media may mitigate the ability of free-oxygen radicals to acidify the local cellular environment, the effect of 18 mg/ml stock solutions of ROFA suspended in either isotonic saline or RPMI on TAP gene expression was compared. The pH of the stock saline solution was between 2 and 3, while the pH of the RPMI solution was between 6 and 7. However, the pH of the culture media containing up to 196 μg/ml ROFA (representing a cellular dose of 20 μg/cm2) was between 6 and 7 for both RPMI- and saline-resuspended ROFA solutions. Moreover, no difference was observed in the ability of the saline-resuspended versus the RPMI-resuspended ROFA samples to inhibit inducible TAP gene expression (data not shown). Therefore, ROFA did not cause significant acidification of the cellular milieu in vitro. There was no significant effect of ROFA on cellular viability at lower doses as measured by trypan blue exclusion. At doses higher than 20 μg/cm2, cultures exhibited 80% viability.
Inhibition of TAP Gene Upregulation Occurs in Response to the Soluble Components of ROFA
Previous research has suggested that acute lung injury and upregulation of inflammatory cytokines in the airway epithelium occur in response to the soluble components of ROFA, but not in response to the insoluble components (Dreher et al., 1997; Dye et al., 1999). In order to determine whether the inhibitory effect was due to ROFA-derived leachate or the residual washed particles, BTE cells were pretreated for 6 h with a volume of either ROFA-leachate or washed particles equivalent to that used for the ROFA suspension dose response, followed by 100 ng/ml LPS for 18 h. ROFA-leachate significantly inhibited the induction of TAP expression in response to LPS, as is observed with the complete ROFA suspension (Fig. 3A). In contrast, the washed particles of ROFA significantly increased the inducible expression of TAP at doses of 2.5 to 10.0 μg/cm2 (Fig. 3B), suggesting that the insoluble components are not inert.
FIG. 3.
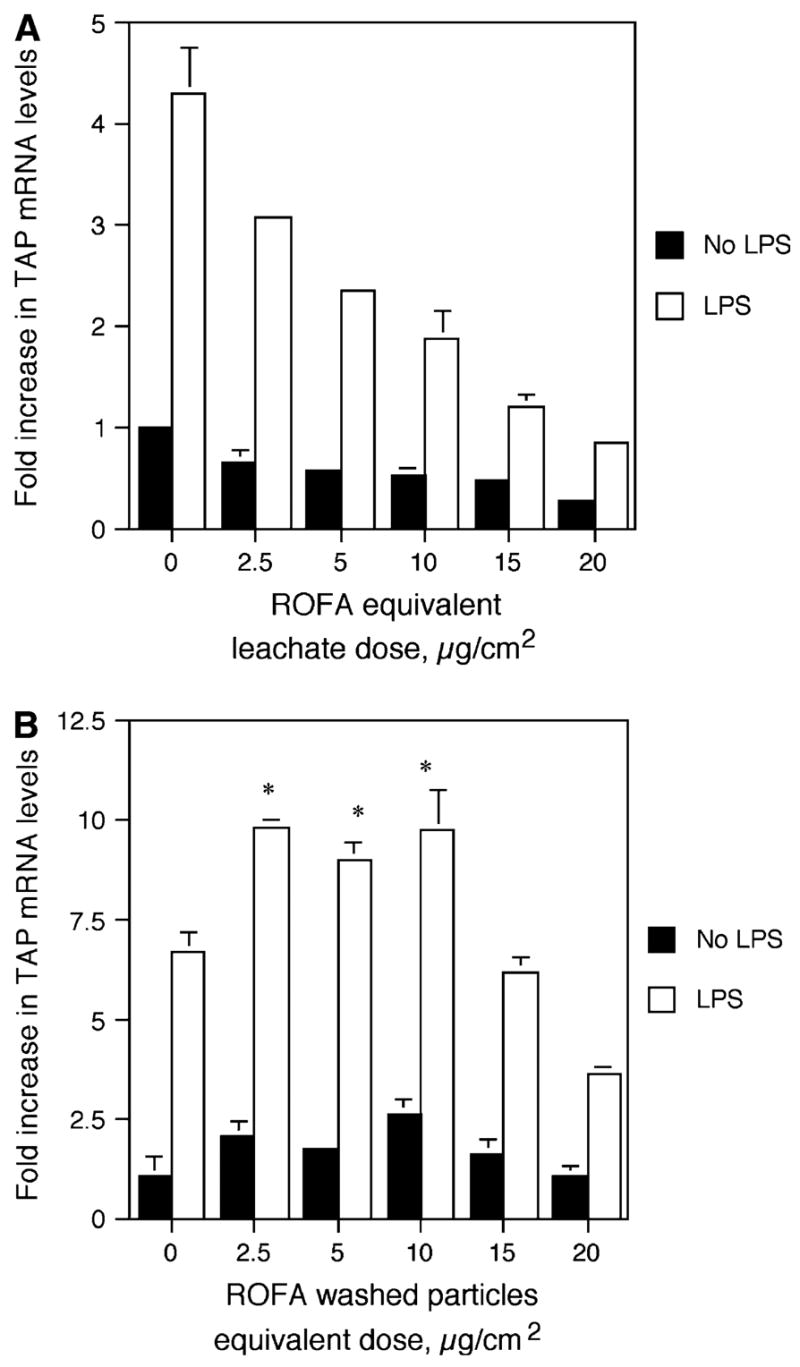
TAP gene expression in response to ROFA components. BTE cells were treated with increasing doses of ROFA-leachate (A) or washed particles (B) for 6 h and then treated with or without LPS for 18 h. mRNA was isolated and analyzed by semi-quantitative RT-PCR for TAP and normalized to GAPDH. Results are expressed as fold increase over unstimulated TAP mRNA levels in the absence of ROFA components. Experiments were carried out in triplicate. In (A), the interaction between ROFA-leachate equivalent dose and LPS stimulus is significant as measured by ANCOVA (p ≤ 0.001), illustrating an increased inhibition of TAP gene expression with ROFA-leachate equivalent dose only in the presence of LPS. In (B), asterisks represent significant increase in TAP gene expression in response to washed particles (Student’s t-test, p ≤ 0.05). Error bars represent ± SEM.
Vanadium Pentoxide Inhibition of Inducible TAP Gene Expression is Dose-Dependent and Time-Dependent
In order to elucidate the mechanism behind the inhibition of inducible TAP expression by the air pollutant ROFA, we investigated the effects of soluble metal components on BTE cells. Vanadium pentoxide (V2O5) previously has been demonstrated in ROFA-leachate (Dreher et al., 1997) and has been shown to alter the expression of genes involved in the inflammatory response and innate immunity of airway epithelial cells (Carter et al., 1997; Ghio et al., 1998). BTE cells were pretreated with 5 μg/cm2 V2O5 over increasing periods of time and then washed thoroughly to remove any remaining metal. The cells were then incubated with or without 100 ng/ml LPS for 18 h. Total RNA was analyzed by semi-quantitative RT-PCR for TAP gene expression and normalized to GAPDH expression. A reproducible trend, although not statistically significant, of decreased inducible TAP gene expression was observed with increasing durations of pre-exposure to V2O5 (Fig. 4A). BTE cells were then incubated with increasing concentrations of V2O5 over a period of 6 h, and then the cells were stimulated with or without 100 ng/ml of LPS as above. Using molar equivalent doses of vanadium, based on the measured concentration of vanadium in ROFA (42 μg vanadium/mg ROFA, at 84% solubility; Costa and Dreher, 1997), we observe the same dose-dependent inhibition of LPS- stimulated TAP expression in BTE cells (Fig. 4B) as seen with ROFA (Fig. 2). There was no significant decrease in cell viability after 6-h treatment with vanadium at concentrations lower than 2.5 μg/cm2. At this concentration, viability was reduced to 73.5% of control with V2O5.
FIG. 4.
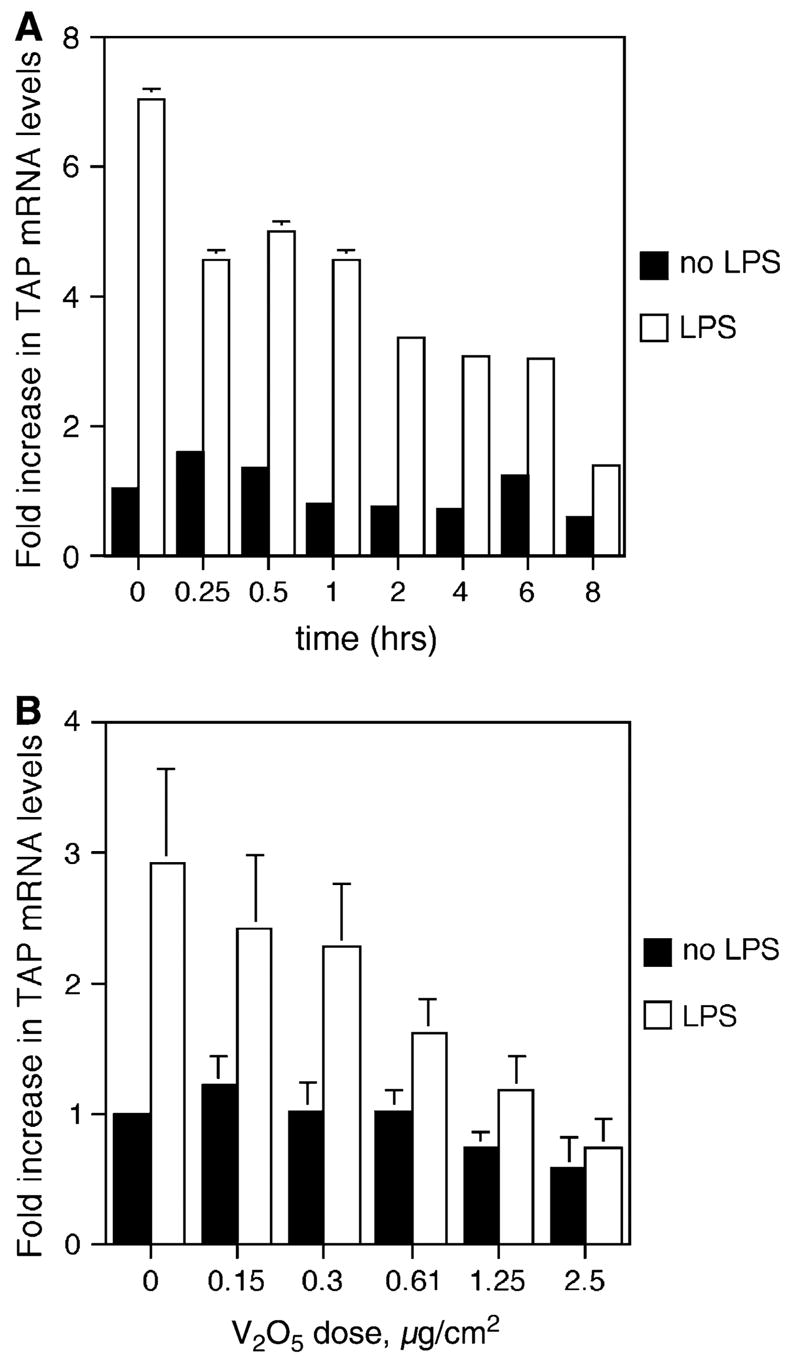
TAP gene expression in response to vanadium. (A) Time course: BTE cells were treated with 5.0 mg/cm2 of V2O5 increasing periods of time and then stimulated with or without LPS for 18 h. Total RNA was extracted from harvested BTE cells and analyzed by semi-quantitative RT-PCR for TAP and normalized to GAPDH. (B) Dose response: BTE cells were treated with increasing doses of V2O5 at molar equivalents of those found in ROFA for 6 h and then stimulated with or without LPS (100 ng/ml) for 18 h. Error bars represent ± SEM. Samples are significant for the interaction between V2O5 dose and LPS stimulus (ANCOVA, p ≤ 0.01), indicating an increased inhibition of TAP gene expression with increasing doses of V2O5 only in the presence of LPS.
TAP Gene Expression in Response to IL-1β is Also Inhibited by V2O5
Pretreatment of BTE cells with V2O5 also significantly inhibited TAP induction in response to the inflammatory cytokine IL-1β (Fig. 5). Subsequent to V2O5 treatment for 6 h, cells were washed, and then stimulated with 100 ng/ml IL- 1β for 6 h. Inhibition was similar to the V2O5 inhibition of LPS-induced TAP. A similar inhibition of induction by TNF-α was also observed (data not shown). Previous work in our laboratory has demonstrated that TAP gene expression is increased in response to challenge with live cultures of the respiratory pathogen, Bordetella bronchiseptica (Legarda et al., 2005). A preliminary experiment where BTE cells were pretreated with V2O5 as above, and subsequently cocultured with the WD3 strain of B. bronchiseptica at a multiplicity of infection of 100:1 for 6 h as described (Legarda et al., 2005) indicated a similar inhibition (data not shown).
FIG. 5.
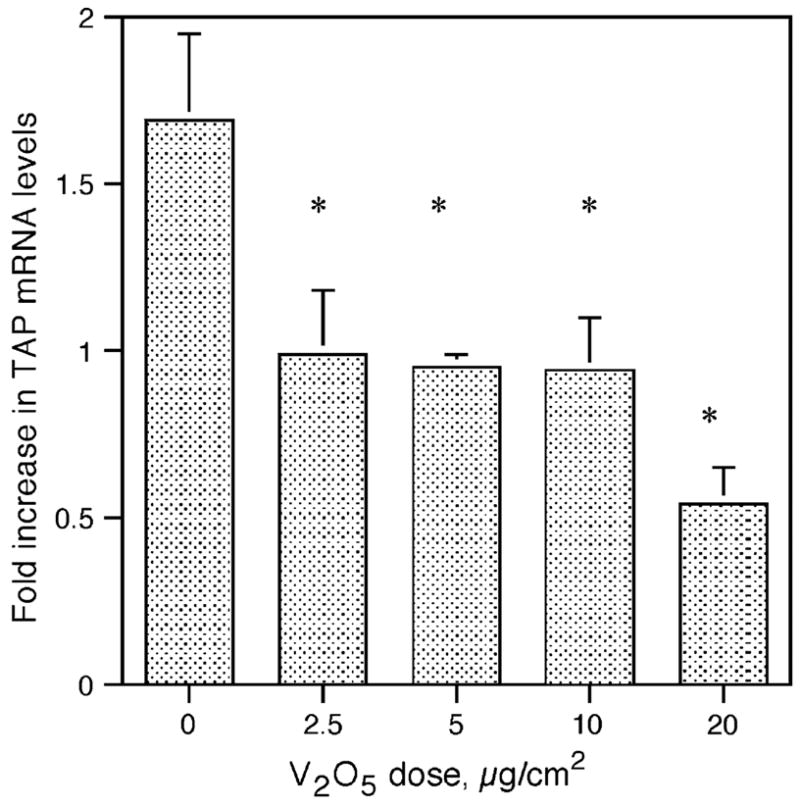
Effect of V2O5 on TAP gene induction by the proinflammatory cytokine, IL-1β. BTE cells were stimulated with increasing doses of V2O5 for 6 h and then treated with or without IL-1β (100 ng/ml) for 6 h. Total RNA was extracted from harvested BTE cells and analyzed by semi-quantitative RT-PCR for TAP and normalized to GAPDH. Experiments were carried out in triplicate. Asterisks represent a significant decrease in TAP levels in response to washed particles (Student’s t-test, p ≤ 0.05). Error bars represent ± SEM.
Effects on TAP Gene Expression by Other Soluble Metal Components of ROFA and Control Particles: Nickel Sulfate, Ferric Sulfate, Titanium Dioxide, and Silicon Dioxide
In order to assess the contribution of other ROFA components to the inhibition of TAP upregulation, BTE cells were stimulated with increasing doses of nickel sulfate (NiSO4), ferric sulfate [Fe2(SO4)3], titanium dioxide (TiO2), and silicon dioxide (SiO2) as above. While the interaction between stimulus and dose for NiSO4 is not significantly different, there is a trend toward decreased TAP gene expression with increased dose of NiSO4, similar to that observed with V2O5 (Fig. 6A). In comparison, Fe2(SO4)3 had no effect on inducible TAP gene expression (Fig. 6B), nor did control particles TiO2 and SiO2 (Figs. 6C and 6D).
FIG. 6.

Effect of metals and silica on TAP gene induction. BTE cells were treated with increasing doses of NiSO4 (A), Fe2(SO4)3 (B), TiO2 (C), or SiO2 (D) for 6 h, and then stimulated with or without LPS (100 ng/ml) for 18 h. mRNA was harvested and analyzed by semi-quantitative RT-PCR for TAP and normalized to GAPDH. No significance for the interaction between any of these components and LPS stimulus (ANCOVA). Experiments were carried out in triplicate. Error bars represent 1 SD.
ROFA and Vanadium Inhibit the Inducible Expression hBD-2 in A549
To investigate if ROFA had similar immunomodulatory effects in the human airway, hBD-2 gene expression was determined using QPCR in a human epithelial type II alveolar cell line. This cell line was chosen as it has been shown that human alveolar epithelial cells can be stimulated to express hBD-2 in response to monocyte-derived cytokines (Tsutsumi-Ishii and Nagaoka, 2003), and thus represent a good model for the induction of an innate immune response in the human airway. To identify a specific mediator for hBD-2 induction, A549 cells were first stimulated with 100 ng/ml LPS, 50 ng/ml TNF-α, 100 ng/ml IL-1β and 300 ng/ml of the TLR2 ligand, PAM3CSK4 (Fig. 7A). IL-1β induced the highest amount of hBD-2 in A549 cells. When A549 cells were preincubated with 2.5, 5.0, 10.0, or 20.0 μg/cm2 of ROFA for 6 h, washed, and subsequently stimulated with 100 ng/ml IL-1β for 6 h (Fig. 7B), a significant inhibition of hBD-2 induction was observed. Using molar equivalent doses of V2O5, we observe the same dose-dependent inhibition of IL-1β–induced hBD-2 mRNA levels (Fig. 7C). Since V2O5 only exhibits partial solubility, we reproduced the experiment with the soluble salt VOSO4. The results demonstrated that treatment with the same molar equivalents of vanadium produced a similar dose-dependent inhibition of hBD-2 expression (Fig. 7D). There was no effect on cell viability of either salt at concentrations of up to 2.5 μg/cm2.
FIG. 7.
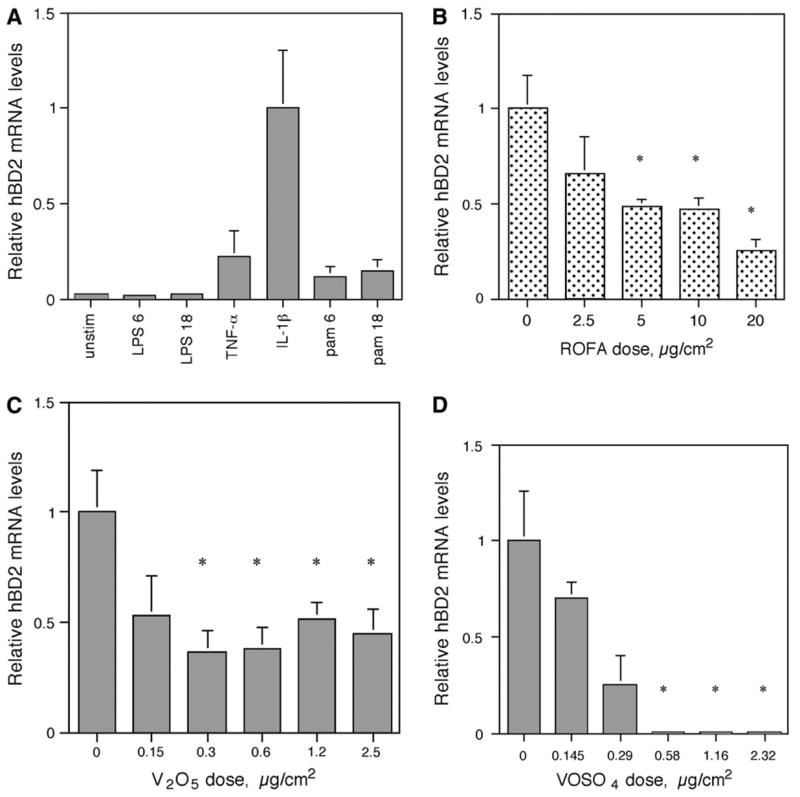
Gene expression of human β-defensin-2 (hBD-2) in A549 cells using quantitative PCR. (A) Induction of hBD-2. A549 cells were either unstimulated or treated with LPS (100 ng/ml) for 6 h (LPS 6) or 18 h (LPS 18), TNF-α (50 ng/ml) for 6 h, IL-1β (100 ng/ml) for 6 h, Pam3CSK4 (300 ng/ml) for 6 h (pam6) or 18 h (pam18). Total RNA was isolated from A549, reverse transcribed, and then amplified by QPCR for hBD-2 with SYBR Green (representative experiment one of three). hBD-2 expression was normalized to HPRT. Error bars = 95% confidence limit of three QPCR reactions per RNA sample. (B) Inhibition of hBD-2 expression by ROFA. A549 cells were treated with or without ROFA for 6 h and then stimulated with IL-1β (100 ng/ml) for 6 h. hBD-2 mRNA levels were quantified as above. For each sample within an experiment, triplicate values were averaged and then the means of the QPCR results from three experiments were compared and analyzed statistically. Each bar containing ROFA represents the mean ± SEM. Asterisks represent significantly lowered induction of hBD-2 by ROFA compared with IL-1β alone (relative level = 1); p ≤ 0.03 by the Student’s t-test. (C) Inhibition of hBD-2 expression by V2O5. A549 cells were treated with or without V2O5 at molar equivalent doses of ROFA as performed in (B), above, for 6 h and then stimulated with IL-1β (100 ng/ml) for 6 h. hBD-2 mRNA levels were quantified as above. For each sample within an experiment, triplicate values were averaged and then the means of the QPCR results from three experiments were compared and analyzed statistically. Error bars represent the mean ± SEM. Asterisks represent significantly lowered induction of hBD-2 by V2O5 compared with IL-1β alone (relative level = 1); p ≤ 0.05 by the Student’s t-test. (D) Inhibition of hBD-2 expression by VOSO4. A549 cells were treated with or without VOSO4 at molar equivalent doses of ROFA as performed in (B), above, for 6 h and then stimulated with IL-1β (100 ng/ml) for 6 h. hBD-2 mRNA levels were quantified as above (n = 3). Error bars represent the mean ± SEM. Asterisks represent significantly lowered induction of hBD-2 by VOSO4 compared with IL-1β alone (relative level =1); p ≤ 0.05 by the Student’s t-test.
V2O5 Inhibits Induction of TAP in BTE Cells
To determine whether V2O5 inhibition of TAP gene induction by LPS resulted in diminished peptide levels, mass spectrophotometric analysis of TAP was performed on cytoplasmic extracts. BTE cells were treated with 10 μg/cm2 V2O5 for 6 h, followed by the induction of TAP by LPS for 18 h, and cytoplasmic extracts were analyzed for the TAP peptide. A peak corresponding to the calculated mass of TAP (m/z = 4083.0780 Da) was detected in the LPS-stimulated cells (Fig. 8B), but not in the unstimulated cells (Fig. 8A). As predicted by the gene expression data, TAP was not observed in cultures treated with V2O5 (Fig. 8C).
FIG. 8.
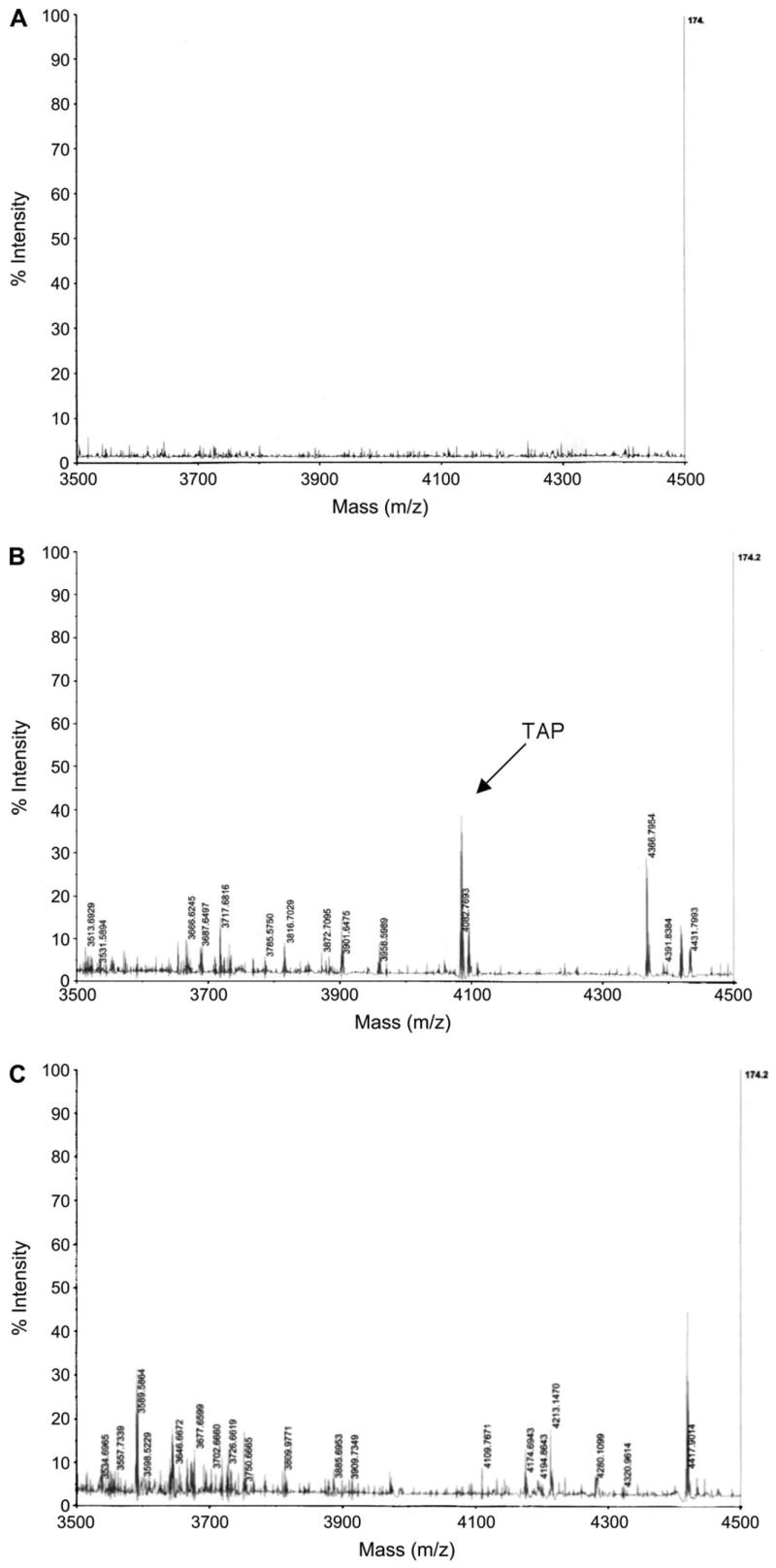
Mass spectrometry of BTE cell cytoplasmic extracts. BTE cells were unstimulated (A) or induced with LPS (100 ng/ml) for 18 h (B), or treated with 10.0 μg/cm2 V2O5 for 6 h prior to incubation with 100 ng/ml LPS for 18 h (C). Cytoplasmic extracts were isolated from 2 × 106 cells as described in ‘‘Materials and Methods’’ section, and subjected to MALDI-TOF-MS. 200 μg of cytoplasmic extract was used for unstimulated and LPS-stimulated samples, while 500 μg was used for the vanadium-treated samples. TAP m/z = 4083.0780 Da.
DISCUSSION
Air pollutant particulate matter has long been associated with increased respiratory infection in children (Ware et al., 1986) and is linked to increased mortality due to pneumonia in elderly individuals (Schwartz, 1994a,b). Experimentally, Hatch et al. (1985) compared ROFA and other urban air particulates with silica, TiO2, and other metals such as ferric oxide (Fe2O3) in their ability to enhance infectivity. They found that ROFA and two samples of ambient air particulate matter intratra-cheally instilled in mice (100 μg per mouse) prior to infection with Group C Streptococcus species caused the highest (> 50% excess mortality) increase in infectivity. Fe2O3 pre-exposure caused 21.2% excess mortality whereas silica, National Institute of Standards and Technology urban air particulate matter from St Louis (No. 1648) urban air particulate matter [UPM] and TiO2 did not. Vanadium and nickel were not tested.
Our initial results on silica and ROFA (Diamond et al., 2000a) and unpublished UPM data by our laboratories correlated with the degree of increased infectivity by these toxic agents in the Hatch et al. study with the degree of inhibition of β-defensin. Of the particles and metals tested in both the Hatch et al. study and in our study, only ROFA inhibited the induction of β-defensin genes. The other particles and metals tested in both studies had no significant effect on β-defensin gene induction by LPS. In addition, although UPM and silica did not have a significant excess infectivity in the Hatch et al. 1985 study, alveolar macrophage cytotoxicity induced by these particles was high, indicating that increased cytotoxicity did not correlate with increased infectivity. The results suggest that ROFA may considerably affect the ability of the host airway to respond to bacterial stimulation and that this impairment is not related to cellular host defense mechanisms, but could be due to other antimicrobial mechanisms such as β-defensin gene induction by bacteria.
ROFA is known to induce lung injury and DNA damage, as well as increasing inflammatory gene expression through the production of free-oxygen radicals (Prahalad et al., 2001; Quay et al., 1998; Roberts et al., 2004). In rats, ROFA enhanced injury and slowed the pulmonary clearance of Listeria monocytogenes (Antonini et al., 2002). The present study demonstrates that in addition to the effects on lung injury, low exposures to ROFA in vitro inhibit the ability of the airway epithelium to increase the expression of β-defensin in response to inflammatory and bacterial stimuli in the absence of cytotoxicity.
The role of β-defensins in host defense against microbial infections of the respiratory tract is increasingly being recognized as an important component of the innate immune response to airway infections (Schutte and McCray, 2002a). These antimicrobial peptides are evolutionarily conserved in their structure, and not only aid in the killing of bacteria but also can act as chemokines, attracting memory T cells and dendritic cells into the site of infection (Yang et al., 1999). TAP was the first β-defensin discovered in bovine tracheal and bronchial epithelial cells (Diamond et al., 1991) and it was found to be inducible with LPS and cytokines in vitro (Diamond et al., 1993, 1996) and with bacterial infection in calves (Caverly et al., 2003) and cows (Schonwetter et al., 1995). Subsequently, other LPS-inducible TAP-like β-defensin homologues were discovered in tracheal epithelial cells of other species such as rat, mouse, pigs, and humans, suggesting that this molecule is necessary for maintaining sterility of the airway (for review see Schutte and McCray, 2002a). The results suggest that even brief exposure to particulate matter is able to interfere with this particular inducible innate immune response.
Initial experiments with LPS-stimulated BTE cells pre-treated with ROFA showed a time-dependent decrease in TAP expression. A 6-h exposure to ROFA and its components were chosen to eliminate the possibility that this decrease was due to cytotoxicity, which was negligible at this time. Low levels of cytotoxicity in epithelial cells treated for 6 h was also reported by another study (Dye et al., 1999).
While it is difficult to extrapolate exposures of ROFA in vitro to actual human exposures encountered in the environment in vivo, the majority of in vitro research has focused on the effects of air pollutants which occur at doses of 20 μg/cm2 or greater (Carter et al., 1997; Dye et al., 1997, 1999; Longphre et al., 2000; Samet et al., 1996). In our study, lower doses of ROFA showed a significant dose-dependent inhibition of inducible TAP gene expression. Of note, 2.5 μg/cm2, the lowest dose studied, decreased inducible TAP expression by 30%, suggesting that even very low exposures of air pollution may be detrimental to the innate immune response.
The soluble component of ROFA is comprised primarily of transition metals (Dreher et al., 1997). In particular, instillation of nickel and vanadium sulfates in an in vivo rat model has been shown to cause an increase in inflammatory and stress processes (Nadadur and Kodavanti, 2002) as well as an increase in airway epithelial lesions comparable to that observed for ROFA (Kodavanti et al., 2001). In addition, this transition metal increases cell permeability, lung mucin production (Longphre et al., 2000), IL-8 (Samet et al., 1998) and IL-6 (Dye et al., 1999), and lactoferrin receptor gene expression (Ghio et al., 1999). Although NiSO4 has been demonstrated to increase IL-8 expression in human airway epithelial cells (Salnikow et al., 2004), it did not increase the expression of the lactoferrin receptor gene (Dye et al., 1999). In vitro, soluble vanadium has been shown to decrease tyrosine phosphatase activity, leading to an increase in protein phosphotyrosines, disrupting their homeostasis in the cell, which leads to increases in gene expression (Samet et al., 1997). These results imply a stimulatory role for both soluble nickel and vanadium.
The data show that inhibition of LPS-induced TAP upregulation by ROFA occurs in response to the soluble fraction of ROFA and that the major component of the leachate that inhibits this inducible β-defensin is vanadium. This was unexpected as vanadium, nickel, and iron are all transition metals, and all can have a 2+oxidation state. It is possible that iron-binding proteins such as ferritin and lactoferrin are capable of mitigating any harmful effects of iron, but not by other transition metals. It has been suggested that the effects of nickel are inhibited by the presence of iron in the soluble component of ROFA (Salnikow et al., 2004) and this may be one reason that the nickel effect on TAP gene expression is blunted compared with vanadium. While this result was surprising in the context of the previous investigations, it is consistent with the above findings using ROFA-leachate and the hypothesis that these soluble metals are mediating the response observed in BTE cells.
The current theory for the effects of particulate air pollution is proposed by Samet et al. 2002 whereby higher doses of fly ash cause an increase in oxidative stress, which leads to phosphorylation of IκB and activation of NF-κB, resulting in increased transcriptional activation of inflammatory mediators and subsequent lung injury. Our study suggests that immunity of the airway epithelium is compromised by air particulate pollution at low doses, leading to a decrease in the innate immune response. It is possible that at low doses of vanadium, there is an inhibitory effect on tyrosine phosphatases, leading to the inhibition of activation of NF-κB, and subsequently a suppression of the LPS- or cytokine-mediated induction of β-defensins.
The fact that TiO2 and SiO2 are inert with respect to suppressing TAP gene expression correlates with their presence in the washed-particle component of ROFA (Dreher et al., 1997). Reports by other investigators also show that TiO2 does not affect transcription of innate immune genes (Stringer and Kobzik, 1998). Furthermore, the inhibitory effects of the soluble components of ROFA mitigate the overall upregulatory effect of the insoluble components in the washed particle seen in Figure 3B.
Our study suggests that particles containing vanadium may alter susceptibility to infection by suppressing β-defensin induction by exposing airway cells prior to infection. We did not test suppression of β-defensin gene expression by giving ROFA or vanadium after infection, proinflammatory cytokine stimulation or LPS stimulation. Studies by Zelikoff et al. 2002, 2003 showed that concentrated air particles from New York City air concentrated ambient air PM2.5 [CAPS] given via inhalation before infecting rats with Streptococcus pneumonia had little effect, but when given after infection significantly increased bacterial burdens compared to those in infected filtered-air–exposed controls. It is interesting to note that CAPS contains no vanadium (Dr Judy Zelikoff, personal communication). However, other transition metals such as iron and nickel reduced bacterial clearance after inhalation exposure of previously infected rats (Zelikoff et al., 2002). In our model, iron and nickel had little effect but that does not rule out that they would not suppress β-defensin-2 if given after LPS stimulation.
β-Defensins were initially discovered in BTE cells (Diamond et al., 1991), and subsequently, we and others have described their expression and activation in human tracheal (Becker et al., 2000) and alveolar epithelial cells (Rivas-Santiago et al., 2005), as well as bovine alveolar macrophages (Ryan et al., 1998). While bovine cells are quite sensitive to LPS (Diamond et al., 1996), human airway epithelium, both respiratory and alveolar, are less so (Tsutsumi-Ishii and Nagaoka, 2003). However, all cell types are responsive to stimulation with live bacteria (Legarda et al., 2005; Rivas-Santiago et al., 2005) and cytokines (Tsutsumi-Ishii and Nagaoka, 2003), and they all utilize similar pathways (reviewed in Laube et al., 2006). The inhibition of β-defensin 2 gene upregulation we observed occurred in both bovine and human cells, and with both LPS and cytokines, suggesting that the effect is at a later point in the signal transduction pathway, closer to NF-κB activation. Furthermore, these data support the use of the bovine cells as a model, and indicate that air pollutant particles containing soluble vanadium have a similar inhibitory effect on the human innate immune response in the airway.
Acknowledgments
The authors wish to thank Drs Allen Ledbetter, Kevin Dreher, and Daniel Costa, U.S. EPA for their generous gift of ROFA and for many helpful discussions. We also wish to thank Janice Rhodes for technical help with the initial studies, and Dr Sunghan Yim for helpful discussions and technical help. Statistical analysis was performed as advised by Dr Joan Skurnick, Department of Preventive Medicine and Community Health, UMDNJ-New Jersey Medical School, Newark, NJ. Mass spectrometry analysis was performed by Dr Hong Li, UMDNJ-New Jersey Medical School Center for Advanced Proteomics Research. This research was supported by U.S. Public Health Service grant NIH R01HL67871 (G.D.), a grant from the University of Medicine and Dentistry of New Jersey Foundation (L.K.R.) and a U.S. EPA Graduate STAR fellowship (M.E.K.).
References
- Antonini JM, Roberts JR, Jernigan MR, Yan HM, Ma JYC, Clarke RW. Residual oil fly ash increases the susceptibility to infection and severly damages the lungs after pulmonary challenge with a bacterial pathogen. Toxicol Sci. 2002;70:110–119. doi: 10.1093/toxsci/70.1.110. [DOI] [PubMed] [Google Scholar]
- Becker M, Diamond G, Verghese M, Randell SH. CD 14-dependent LPS-induced β-defensin expression in human tracheobronchial epithelium. J Biol Chem. 2000;275:29731–29736. doi: 10.1074/jbc.M000184200. [DOI] [PubMed] [Google Scholar]
- Carter JD, Ghio AJ, Samet JM, Devlin RB. Cytokine production by human airway epithelial cells after exposure to an air pollution particle is metal-dependent. Toxicol Appl Pharmacol. 1997;146:180–188. doi: 10.1006/taap.1997.8254. [DOI] [PubMed] [Google Scholar]
- Caverly JM, Diamond G, Gallup JM, Brogden KA, Dixon RA, Ackermann MR. Coordinated expression of tracheal antimicrobial peptide and inflammatory-response elements in the lungs of neonatal calves with acute bacterial pneumonia. Infect Immun. 2003;71:2950–2955. doi: 10.1128/IAI.71.5.2950-2955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa DL, Dreher KL. Bioavailable transition metals in particulate matter mediate cardiopulmonary injury in healthy and compromised animal models. Environ Health Perspect. 1997;105(Suppl 5):1053–1060. doi: 10.1289/ehp.97105s51053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond G, Jones DE, Bevins CL. Airway epithelial cells are the site of expression of a mammalian antimicrobial gene. Proc Natl Acad Sci USA. 1993;90:4596–4600. doi: 10.1073/pnas.90.10.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond G, Kaiser V, Rhodes J, Russell JP, Bevins CL. Transcriptional regulation of β-defensin gene expression in tracheal epithelial cells. Infect Immun. 2000b;68:113–119. doi: 10.1128/iai.68.1.113-119.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond G, Legarda D, Ryan LK. The innate immune response of the respiratory epithelium. Immunol Rev. 2000a;173:27–38. doi: 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- Diamond G, Russell JP, Bevins CL. Inducible expression of an antibiotic peptide gene in lipopolysaccharide-challenged tracheal epithelial cells. Proc Natl Acad Sci USA. 1996;93:5156–5160. doi: 10.1073/pnas.93.10.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond G, Zasloff M, Eck H, Brasseur M, Maloy WL, Bevins CL. Tracheal antimicrobial peptide, a novel cysteine-rich peptide from mammalian tracheal mucosa: Peptide isolation and cloning of a cDNA. Proc Natl Acad Sci USA. 1991;88:3952–3956. doi: 10.1073/pnas.88.9.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher KL, Jaskot RH, Lehmann JR, Richards JH, McGee JK. Soluble transition metals mediate residual oil fly ash induced lung injury. J Toxicol Environ Health. 1997;50:285–305. [PubMed] [Google Scholar]
- Dye JA, Adler KB, Richards JH, Dreher KL. Epithelial injury induced by exposure to residual oil fly-ash particles: Role of reactive oxygen species? Am J Respir Cell Mol Biol. 1997;17:625–633. doi: 10.1165/ajrcmb.17.5.2749. [DOI] [PubMed] [Google Scholar]
- Dye JA, Adler KB, Richards JH, Dreher KL. Role of soluble metals in oil fly ash-induced airway epithelial injury and cytokine gene expression. Am J Physiol Lung Cell Mol Physiol. 1999;21:L498–L510. doi: 10.1152/ajplung.1999.277.3.L498. [DOI] [PubMed] [Google Scholar]
- Ghio AJ, Carter JD, Dailey LA, Devlin RB, Samet JM. Respiratory epithelial cells demonstrate lactoferrin receptors that increase after metal exposure. Am J Physiol Lung Cell Mol Physiol. 1999;20:L933–L940. doi: 10.1152/ajplung.1999.276.6.L933. [DOI] [PubMed] [Google Scholar]
- Ghio AJ, Carter JD, Samet JM, Reed W, Quay J, Dailey LA, Richards JH, Devlin RB. Metal-dependent expression of ferritin and lactoferrin by respiratory epithelial cells. Am J Physiol Lung Cell Mol Physiol. 1998;18:L728–L736. doi: 10.1152/ajplung.1998.274.5.L728. [DOI] [PubMed] [Google Scholar]
- Hatch GE, Boykin E, Graham JA, Lewtas J, Pott F, Loud K, Mumford JA. Inhalable particles and pulmonary host defense: In vivo and in vitro effects of ambient air and combustion particles. Environ Res. 1985;36:67–80. doi: 10.1016/0013-9351(85)90008-8. [DOI] [PubMed] [Google Scholar]
- Kaiser V, Diamond G. Expression of mammalian defensin genes. J Leukoc Biol. 2000;68:779–784. [PubMed] [Google Scholar]
- Kennedy T, Ghio AJ, Reed W, Samet J, Zagorski J, Quay J, Carter J, Dailey L, Hoidal JR, Devlin RB. Copper-dependent inflammation and nuclear factor-κB activation by particulate air pollution. Am J Respir Cell Mol Biol. 1998;19:366–378. doi: 10.1165/ajrcmb.19.3.3042. [DOI] [PubMed] [Google Scholar]
- Kodavanti UP, Schladweiler MC, Richards JH, Costa DL. Acute lung injury from intratracheal exposure to fugitive residual oil fly ash and its constituent metals in normo- and spontaneously hypertensive rats. Inhal Toxicol. 2001;13:37–54. doi: 10.1080/089583701459056. [DOI] [PubMed] [Google Scholar]
- Laube DM, Yim S, Ryan LK, Kisich KO, Diamond G. Antimicrobial peptides in the airway. Curr Top Microbiol Immunol. 2006;306:155–184. doi: 10.1007/3-540-29916-5_6. [DOI] [PubMed] [Google Scholar]
- Legarda D, Klein-Patel ME, Yim S, Yuk MH, Diamond G. Suppression of NF-κB-mediated β-defensin gene expression in the mammalian airway by the Bordetella type III secretion system. Cell Microbiol. 2005;7:489–497. doi: 10.1111/j.1462-5822.2004.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longphre M, Li D, Li J, Matovinovic E, Gallup M, Samet JM, Basbaum C. Lung mucin production is stimulated by the air pollutant residual oil fly ash. Toxicol Appl Pharmacol. 2000;162:86–92. doi: 10.1006/taap.1999.8838. [DOI] [PubMed] [Google Scholar]
- Nadadur SS, Kodavanti UP. Altered gene expression profiles of rat lung in response to an emission particulate and its metal constituents. J Toxicol Environ Health A. 2002;65:1333–1350. doi: 10.1080/00984100290071559. [DOI] [PubMed] [Google Scholar]
- Peel JL, Tolbert PE, Klein M, Metzger KB, Flanders WD, Todd K, Mulholland JA, Ryan PB, Frumkin H. Ambient air pollution and respiratory emergency department visits. Epidemiology. 2005;16:164–174. doi: 10.1097/01.ede.0000152905.42113.db. [DOI] [PubMed] [Google Scholar]
- Pope CA, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K, Thurston GD. Lung cancer, cardiopulmonary mortality and long-term exposure to fine particulate air pollution. JAMA. 2002;287:1132–1141. doi: 10.1001/jama.287.9.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CA, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ. Cardiovascular mortality and long-term exposure to particulate air pollution. Circulation. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
- Prahalad AK, Inmon J, Dailey LA, Madden MC, Ghio AJ, Gallagher JE. Air pollution particles mediated oxidative DNA base damage in a cell free system and in human airway epithelial cells in relation to particulate metal content and bioreactivity. Chem Res Toxicol. 2001;14:879–887. doi: 10.1021/tx010022e. [DOI] [PubMed] [Google Scholar]
- Quay JL, Reed W, Samet J, Devlin RB. Air pollution particles induce IL-6 gene expression in human airway epithelial cells via NF-κB activation. Am J Respir Cell Mol Biol. 1998;19:98–106. doi: 10.1165/ajrcmb.19.1.3132. [DOI] [PubMed] [Google Scholar]
- Rivas-Santiago B, Schwander SK, Sarabia C, Diamond G, Klein-Patel ME, Hernandez-Pando R, Ellner JJ, Sada E. Human {beta}-defensin 2 is expressed and associated with mycobacterium tuberculosis during infection of human alveolar epithelial cells. Infect Immun. 2005;73:4505–4511. doi: 10.1128/IAI.73.8.4505-4511.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JR, Taylor MD, Castranova V, Clarke RW, Antonini JM. Soluble metals associated with residual oil fly ash increase morbidity and lung injury after bacterial infection in rats. J Toxicol Environ Health A. 2004;67:251–263. doi: 10.1080/15287390490266927. [DOI] [PubMed] [Google Scholar]
- Ryan LK, Rhodes J, Bhat M, Diamond G. Expression of beta-defensin genes in bovine alveolar macrophages. Infect Immun. 1998;66:878–881. doi: 10.1128/iai.66.2.878-881.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikow K, Li X, Lippman M. Effect of nickel and iron co-exposure on human lung cells. Toxicol Appl Pharmacol. 2004;196:258–265. doi: 10.1016/j.taap.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Samet JM, Dominici F, Curriero FC, Coursac I, Zeger SL. Fine particulate air pollution and mortality in 20 U.S. cities. N Engl J Med. 2000;343:1742–1749. doi: 10.1056/NEJM200012143432401. [DOI] [PubMed] [Google Scholar]
- Samet JM, Graves LM, Quay J, Dailey LA, Devlin RB, Ghio AJ, Wu W, Bromberg PA, Reed W. Activation of MAPKs in human bronchial epithelial cells exposed to metals. Am J Physiol Lung Cell Mol Physiol. 1998;19:L551–L558. doi: 10.1152/ajplung.1998.275.3.L551. [DOI] [PubMed] [Google Scholar]
- Samet JM, Reed W, Ghio AJ, Devlin RB, Carter JD, Dailey LA, Bromberg PA, Madden MC. Induction of prostaglandin H synthase 2 in human airway epithelial cells exposed to residual oil fly ash. Toxicol Appl Pharmacol. 1996;141:159–168. doi: 10.1006/taap.1996.0272. [DOI] [PubMed] [Google Scholar]
- Samet JM, Silbajoris R, Huang T, Jaspers I. Transcription factor activation following exposure of an intact lung preparation to metallic particulate matter. Environ Health Perspect. 2002;110:985–990. doi: 10.1289/ehp.02110985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samet JM, Stonehuerner J, Reed W, Devlin RB, Dailey LA, Kennedy TP, Bromberg PA, Ghio AJ. Disruption of protein tyrosine phosphate homeostasis in bronchial epithelial cells exposed to oil fly ash. Am J Physiol Lung Cell Mol Physiol. 1997;272:L426–L432. doi: 10.1152/ajplung.1997.272.3.L426. [DOI] [PubMed] [Google Scholar]
- Schonwetter BS, Stolzenberg ED, Zasloff MA. Epithelial antibiotics induced at sites of inflammation. Science. 1995;267:1645–1648. doi: 10.1126/science.7886453. [DOI] [PubMed] [Google Scholar]
- Schutte BC, McCray PB., Jr β-Defensins in lung host defense. Annu Rev Physiol. 2002;64:709–748. doi: 10.1146/annurev.physiol.64.081501.134340. [DOI] [PubMed] [Google Scholar]
- Schwartz J. Total suspended particulate matter and daily mortality in Cincinnati, Ohio. Environ Health Perspect. 1994a;102:186–189. doi: 10.1289/ehp.94102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz J. Air pollution and daily mortality: A review and meta-analysis. Environ Res. 1994b;64:35–52. doi: 10.1006/enrs.1994.1005. [DOI] [PubMed] [Google Scholar]
- Stringer B, Kobzik L. Environmental particulate-mediated cytokine production in lung epithelial cells (A549): Role of preexisting inflammation and oxidant stress. J Toxicol Environ Health A. 1998;55:31–44. doi: 10.1080/009841098158601. [DOI] [PubMed] [Google Scholar]
- Tsutsumi-Ishii Y, Nagaoka I. Modulation of human beta-defensin-2 transcription in pulmonary epithelial cells by lipopolysaccharide-stimulated mononuclear phagocytes via proinflammatory cytokine production. J Immunol. 2003;170:4226–4236. doi: 10.4049/jimmunol.170.8.4226. [DOI] [PubMed] [Google Scholar]
- Veronesi B, Oortgiesen M, Carter JD, Devlin RB. Particulate matter initiates inflammatory cytokine release by activation of capsaicin and acid receptors. Toxicol Appl Pharmacol. 1999;154:106–115. doi: 10.1006/taap.1998.8567. [DOI] [PubMed] [Google Scholar]
- Ware JH, Ferris BG, Dockery DW, Spengler JD, Stram DO. Effects of ambient sulfur oxides and suspended particles on respiratory health of pre-adolescent children. Am Rev Respir Dis. 1986;133:824–842. [PubMed] [Google Scholar]
- Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OMZ, et al. β-Defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–528. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- Zelikoff JT, Chen LC, Cohen MD, Fang K, Gorden T, Li Y, Nadziejko C, Schlesinger RB. Effects of inhaled ambient particulate matter on pulmonary antimicrobial immune defense. Inhal Toxicol. 2003;15:131–150. doi: 10.1080/08958370304478. [DOI] [PubMed] [Google Scholar]
- Zelikoff JT, Schermerhorn KR, Fang K, Cohen MD, Schlesinger RB. A role for associated transition metals in the immunotoxicity of inhaled ambient particulate matter. Environ Health Perspect. 2002;110(Suppl 5):871–875. doi: 10.1289/ehp.02110s5871. [DOI] [PMC free article] [PubMed] [Google Scholar]