

Abstract
Heritability is often used by plant breeders and geneticists as a measure of precision of a trial or a series of trials. Its main use is for computing the response to selection. Most formulas proposed for calculating heritability implicitly assume balanced data and independent genotypic effects. Both of these assumptions are often violated in plant breeding trials. This article proposes a simulation-based approach to tackle the problem. The key idea is to directly simulate the quantity of interest, e.g., response to selection, rather than trying to approximate it using some ad hoc measure of heritability. The approach is illustrated by three examples.
HERITABILITY is often used by plant breeders to quantify the precision of single field trials or of series of field trials. It is defined as the proportion of phenotypic variance among individuals in a population that is due to heritable genetic effects, also known as heritability in the narrow sense. Similarly, heritability in the broad sense is defined as the proportion of phenotypic variance that is attributable to an effect for the whole genotype, comprising the sum of additive, dominance, and epistatic effects (Nyquist 1991; Falconer and Mackay 1996). Heritability is a key parameter in quantitative genetics because it determines the response to selection. The original definitions of heritability were proposed in an animal breeding context, where the basic unit of observation and selection is usually the individual animal. By contrast, in plant breeding there are many different mating designs, and observational units are quite diverse, ranging from individual plants to means of a genotype tested across a wide range of environments in designed experiments. As pointed out by Holland et al. (2003), this complicates both the definition and the estimation of heritability. A particular difficulty is that virtually all equations for heritability assume balanced data, whereas the majority of trials exhibit some form of imbalance. Specifically, large sets of genotypes are usually tested in designs involving incomplete blocks, and common heritability definitions do not apply in such cases. Also, the standard definitions assume that trials are analyzed by models with independent random effects for blocks, plots, plants, etc., whereas analysis of field trials is often done by spatial models, which imply complex variance–covariance structures pertaining to observational units.
For a balanced series of m trials laid out in randomized complete blocks with r replicates, broad-sense heritability on an entry-mean basis is defined as
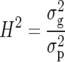 |
(1) |
(Falconer and Mackay 1996), where  is the genotypic variance and
is the genotypic variance and 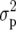 is the phenotypic variance. Several authors use the notation H in place of H2, e.g., Nyquist (1991). Also, H2 is sometimes termed “repeatability.” The “phenotype” is the mean of a genotype across m trials (environments) and r replicates per trial. This has variance
is the phenotypic variance. Several authors use the notation H in place of H2, e.g., Nyquist (1991). Also, H2 is sometimes termed “repeatability.” The “phenotype” is the mean of a genotype across m trials (environments) and r replicates per trial. This has variance
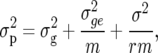 |
(2) |
where 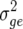 is the genotype–environment interaction variance and
is the genotype–environment interaction variance and  is the residual error variance. Similarly, the narrow-sense heritability for a mean is defined as
is the residual error variance. Similarly, the narrow-sense heritability for a mean is defined as
 |
(3) |
where 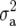 is the additive genetic variance and
is the additive genetic variance and 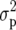 is as given in Equation 2. In the case of a balanced design, H2 and h2 have a number of simple and direct interpretations. For example, H2 is the fraction of phenotypic variation that can be explained by the genotype, where the phenotype is taken to be the arithmetic entry mean. Thus, H2 is equivalent to the coefficient of determination of a linear regression of the (latent) genotype on the observed phenotype. It is also the squared correlation between predicted (phenotypic) value and actual genetic or genotypic value. Most importantly, heritability (H2 and h2) can be used to predict the response to selection R as R = h2S (or H2S), where S is the selection differential (Falconer and Mackay 1996), and this is perhaps its main use for plant breeders.
is as given in Equation 2. In the case of a balanced design, H2 and h2 have a number of simple and direct interpretations. For example, H2 is the fraction of phenotypic variation that can be explained by the genotype, where the phenotype is taken to be the arithmetic entry mean. Thus, H2 is equivalent to the coefficient of determination of a linear regression of the (latent) genotype on the observed phenotype. It is also the squared correlation between predicted (phenotypic) value and actual genetic or genotypic value. Most importantly, heritability (H2 and h2) can be used to predict the response to selection R as R = h2S (or H2S), where S is the selection differential (Falconer and Mackay 1996), and this is perhaps its main use for plant breeders.
When data are unbalanced or when genetic effects are correlated or heteroscedastic, Equation 2 no longer holds, and no simple equation is available for computing response to selection. Essentially this occurs because the correlation between phenotype and response to selection differs among genotypes, and thus there is no simple linear relationship between response to selection (R) and selection differential (S), as in the balanced case. The phenotype might be taken to be the adjusted entry mean, i.e., the best linear unbiased estimator (BLUE) assuming fixed genotypic effects. Analytical computation of the response to selection is then hampered not only by heterogeneity of the variance of an adjusted mean, but also by correlations among means induced by the least-squares adjustment for blocking. Also, with unbalanced data and a large number of genotypes, it is desirable to estimate genetic effects by best linear unbiased prediction (BLUP) rather than by BLUE, and in the computation of BLUP, an adjusted entry mean, conventionally considered as the phenotype, is usually not directly involved. A further complication arises when genetic effects are correlated, as is the case, e.g., when pedigree information is exploited.
This raises the question of how heritability should be computed in the more general case of unbalanced data and correlated genetic effects. Various approaches can be found in the literature, most of which propose a generalized definition of heritability accounting for unbalanced data and/or correlated genetic effects (Holland et al. 2003; Cullis et al. 2006; Helms and Hammond 2006; Oakey et al. 2006; K. Emrich, unpublished results). We believe that alternative definitions of heritability are sometimes problematic, because they do not always share the same straightforward interpretations as their balanced-data counterparts. Also, it is often not the heritability itself that is of interest, but some related quantity, which in the balanced case can be directly computed from it by a simple equation, such as that for the response to selection. We think that it is important to carefully think about the specific notion or definition of heritability one has in mind or which quantity derived from it in the balanced case is of immediate interest. It is usually possible to compute such quantities directly, and in complex settings this may be more straightforward than trying to compute a meaningful measure of heritability. For example, in the case of unbalanced data or correlated genetic effects, it is more accurate to compute the response to selection directly rather than plugging some ad hoc estimate of heritability into the standard equation for response to selection that assumes balanced data and independent genetic effects. In fact, Hanson (1963, p. 127) proposed to define heritability as “the fraction of the selection differential expected to be gained when selection is practiced on a defined reference unit.” As emphasized by Holland et al. (2003, p. 11), “heritability has meaning only in reference to defined selection units and response units, and these can vary among breeding schemes.” In this article, we take a similar view, stressing the importance of identifying both the statistic on which selection is to be based, denoted here as “selection statistic,” and the genetic effect of interest, denoted here as “target effect.” For example, we may choose to make selections on the basis of BLUP of the sum of additive genetic value of two parents (selection statistic), thus aiming to select the offspring with the best genotypic value (target effect). Generally, the notion of heritability is related to the correlation of selection statistic and target effect.
In this article we propose a general method to compute heritability and related quantities, such as response to selection. To be generally applicable, the proposed computational method employs simulation.
SIMULATING HERITABILITY AND RELATED QUANTITIES
We assume a standard linear mixed model for the observed data y, which is of the form
 |
(4) |
where  and
and 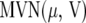 denotes the multivariate normal distribution with mean vector μ and variance–covariance matrix V. The random vector u contains the genetic or genotypic effects of the tested genotypes, as well as nongenetic effects and genotype-by-environment interaction effects. The solution to the mixed-model equations (MME), yielding the BLUE of β and the BLUP of u, is given by
denotes the multivariate normal distribution with mean vector μ and variance–covariance matrix V. The random vector u contains the genetic or genotypic effects of the tested genotypes, as well as nongenetic effects and genotype-by-environment interaction effects. The solution to the mixed-model equations (MME), yielding the BLUE of β and the BLUP of u, is given by
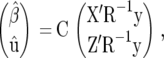 |
(5) |
where
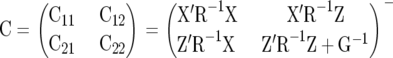 |
(6) |
is a g-inverse of the coefficient matrix of the MME (McLean et al. 1991) and C11 has the same dimension as 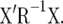
Let g be the genetic effects of interest, i.e., the target effect, and assume that g is correlated with u. A special case occurs when g is a linear function of u. For example, when a clonal crop is tested and interest is in the genotypic effect, g is simply a subset of u, which constitutes a simple instance of a linear function of u. When the mating design allows dissection of additive and dominance effects, and one is interested in estimating genotypic values of tested genotypes, elements of g are linear combinations of elements in u. As a result of the linear dependence, g and u are also correlated. Finally, when one is interested in predicting the breeding value of unobserved progeny, g is a vector that is correlated with u through the pedigree, but it is not necessarily a linear function of u in this case (Henderson 1977).
To allow a joint treatment of all cases, we cast the estimation problem in terms of correlated random target effects. Let the variance–covariance matrix of the random effects u and the target effects g be given as
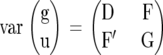 |
(7) |
We then have
 |
(8) |
(Henderson 1977, p. 784), where
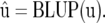 |
(9) |
The joint distribution of g and 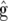 is multivariate normal with zero mean and variance–covariance matrix
is multivariate normal with zero mean and variance–covariance matrix
 |
(10) |
(see the appendix), where 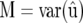 is the unconditional variance of
is the unconditional variance of 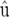 (Searle et al. 1992, p. 287; also see the appendix). In many cases, for example, when g is a linear function or a subset of u, a large block of F equals zero, which may be exploited to efficiently evaluate (10). To illustrate, consider the case where
(Searle et al. 1992, p. 287; also see the appendix). In many cases, for example, when g is a linear function or a subset of u, a large block of F equals zero, which may be exploited to efficiently evaluate (10). To illustrate, consider the case where  and g is independent of
and g is independent of  such that
such that
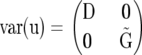 |
(11) |
We then have 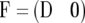 and
and 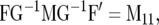 where M11 is the part of M pertaining to
where M11 is the part of M pertaining to 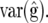 This result makes evaluation of (10) computationally efficient.
This result makes evaluation of (10) computationally efficient.
To obtain the REML estimate of M from a mixed-model package, we may exploit that
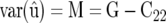 |
(12) |
(McLean et al. 1991, p. 55), where C22 can be extracted from the mixed-model equations (Equations 5 and 6).
Now let a decomposition of Ω be given by
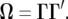 |
(13) |
This can be obtained, e.g., by a singular value decomposition or, in the case of a positive-definite Ω, by a Cholesky decomposition. Note that Ω is not necessarily positive definite. We may simulate values of  for an experiment with the same design as that underlying the actual data y from
for an experiment with the same design as that underlying the actual data y from
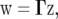 |
(14) |
where z is a 2I random vector of independent standard normal deviates and I is the size of g. For one random draw of w, a vector z of independent standard normal deviates is generated using a random number generator, e.g., based on the Box–Muller method [function normal() in SAS]. The simulated z is then plugged into (14) to yield a random draw of w. A large number (Q, say) of draws of w can thus be simulated and quantities of interest computed. The approach suggested here is akin to the parametric bootstrap (Efron and Tibshirani 1993). It has the advantage that the underlying experimental design (blocking and treatment structure) is implicitly accounted for through the variance–covariance matrix Ω. Quantities of interest can thus be computed for virtually any experimental design and genetic structure. The procedure is now exemplified for two cases.
Response to selection:
Assume that n entries are selected from the collection of I entries in the trial, so the selection fraction p = n/I. Let gi and  denote the true genotypic value of the ith entry and its BLUP, respectively. In each simulation run, select the p100% entries with the best values of
denote the true genotypic value of the ith entry and its BLUP, respectively. In each simulation run, select the p100% entries with the best values of 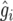 and compute
and compute
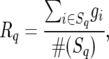 |
(15) |
where Sq is the selected subset in the qth simulation run and #(Sq) is the size of Sq. The simulated expected response to selection is then computed as
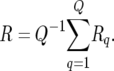 |
(16) |
Squared correlation of predicted and true genotypic values:
In each simulation run, compute the sample correlation (rq) of  and
and 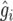 . The simulated expected correlation of predicted and true genotypic value is computed as
. The simulated expected correlation of predicted and true genotypic value is computed as
 |
(17) |
The method is not exactly identical to the standard procedure of computing the heritability in the balanced case, but it is asymptotically equivalent for an increasing number of entries. For example, in a balanced completely randomized design and with independent genotypic effects, we have asymptotically that
 |
(18) |
Inference:
Computation of standard errors for simulated quantities such as R and r2 can be done by a parametric bootstrap procedure (Efron and Tibshirani 1993). A bootstrap replicate involves simulating data according to the fitted model (4), estimating model parameters, computing Ω from the simulated variance parameter estimates, and then simulating the quantity of interest. Standard errors, confidence intervals, etc., can be obtained from a large number of parametric bootstrap samples. While the procedure is straightforward, it may be computationally demanding with large problems.
OTHER PROPOSALS TO COMPUTE HERITABILITY
As an ad hoc measure of heritability, one may compute
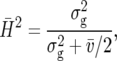 |
(19) |
where  is the mean variance of a difference of two adjusted treatment means (BLUE) (Holland et al. 2003, p. 64; K. Emrich, unpublished results). This measure is closely related to the concept of “effective error variance” (Cochran and Cox 1957). A similar measure proposed by Cullis et al. (2006) is based on BLUP rather than BLUE. It is computed as
is the mean variance of a difference of two adjusted treatment means (BLUE) (Holland et al. 2003, p. 64; K. Emrich, unpublished results). This measure is closely related to the concept of “effective error variance” (Cochran and Cox 1957). A similar measure proposed by Cullis et al. (2006) is based on BLUP rather than BLUE. It is computed as
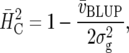 |
(20) |
where 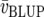 is the mean variance of a difference of two BLUP. Cullis et al. (2006) proposed to use this measure to approximate the response to selection as
is the mean variance of a difference of two BLUP. Cullis et al. (2006) proposed to use this measure to approximate the response to selection as  where i denotes the selection intensity. Both
where i denotes the selection intensity. Both  and
and 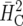 coincide with H2 in Equation 1 in the balanced case. Their use is restricted mainly to settings where the genotypic effects are assumed to be independently distributed and the design is nearly variance balanced, i.e., when the pairwise variances are rather homogeneous, as is the case in well-designed experiments (Cullis et al. 2006). The measures are expected to perform less well in the case of strong unbalancedness and in the case of correlation due to the pedigree, in particular when response to selection is computed from them. This is illustrated in the examples.
coincide with H2 in Equation 1 in the balanced case. Their use is restricted mainly to settings where the genotypic effects are assumed to be independently distributed and the design is nearly variance balanced, i.e., when the pairwise variances are rather homogeneous, as is the case in well-designed experiments (Cullis et al. 2006). The measures are expected to perform less well in the case of strong unbalancedness and in the case of correlation due to the pedigree, in particular when response to selection is computed from them. This is illustrated in the examples.
Several generalized heritability measures, which are also applicable when genotypic effects are modeled using the pedigree via the numerator relationship matrix, were recently proposed by Oakey et al. (2006). These measures involve eigenvalue analyses, and their interpretation is somewhat abstract.
EXAMPLES
We exemplify the simulation-based approach proposed in this article using three examples. In the first example, the result obtained by the new method is virtually identical to the approaches based on 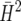 and
and 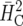 described in the preceding section. The other two examples show that
described in the preceding section. The other two examples show that  and
and 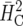 can yield misleading results or may not even be feasible. All analyses were done by REML as implemented in the MIXED procedure of the SAS system. Simulations are based on Q = 100,000 data sets. SAS code for example 1 is available at http://www.uni-hohenheim.de/bioinformatik/.
can yield misleading results or may not even be feasible. All analyses were done by REML as implemented in the MIXED procedure of the SAS system. Simulations are based on Q = 100,000 data sets. SAS code for example 1 is available at http://www.uni-hohenheim.de/bioinformatik/.
Example 1:
John and Williams (1995, p. 146) report results from a yield trial with oats laid out as an α-design. The trial had 24 genotypes, three complete replications, and six incomplete blocks within each replication. The block size was four. The data were analyzed by a linear mixed model with effects for genotypes, replicates, and incomplete blocks. Blocks were modeled as independent random effects to recover interblock information. Genotypic effects (gi) were also assumed to be independent among genotypes. The standard errors of a difference among adjusted means were rather homogeneous (Table 1), as is to be expected for a well-designed experiment.
TABLE 1.
Quantiles of standard errors of a difference among adjusted means (SED) for example 1
| Quantile (%) | SED |
|---|---|
| 100 (maximum) | 0.2699 |
| 90 | 0.2698 |
| 75 | 0.2690 |
| 50 (median) | 0.2681 |
| 25 | 0.2584 |
| 10 | 0.2577 |
| 0 (minimum) | 0.2575 |
We found  and
and 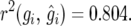 The parametric bootstrap standard error of
The parametric bootstrap standard error of 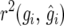 based on 1000 bootstrap samples was 0.0876. The very similar results for the simulated and the other procedures (
based on 1000 bootstrap samples was 0.0876. The very similar results for the simulated and the other procedures ( and
and  ) are expected due to the near constancy of the standard error of a difference (Table 1). By simulation based on (16), we computed response to selection in the selected fraction for different fractions p = n/I, where I is the total number of genotypes and n the number of selected genotypes. For comparison, a naïve estimate of response to selection based on
) are expected due to the near constancy of the standard error of a difference (Table 1). By simulation based on (16), we computed response to selection in the selected fraction for different fractions p = n/I, where I is the total number of genotypes and n the number of selected genotypes. For comparison, a naïve estimate of response to selection based on  and
and  was computed also by simulation, assuming that genotypic means and true genetic values gi are bivariate normal with variances
was computed also by simulation, assuming that genotypic means and true genetic values gi are bivariate normal with variances  or
or 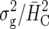 and
and  respectively, and with covariance
respectively, and with covariance 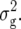 The results by all three methods are very similar (Table 2).
The results by all three methods are very similar (Table 2).
TABLE 2.
Response to selection for example 1 computed by simulation based on (16), based on an ad hoc approach using  , and based on
, and based on 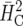 (Cullis et al. 2006) for different selection fractions p = n/I with I = 24
(Cullis et al. 2006) for different selection fractions p = n/I with I = 24
| Response to selection
|
|||
|---|---|---|---|
| n | Based on (16) |
Ad hoc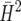
|
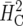 |
| 1 | 0.6625 | 0.6602 | 0.6627 |
| 2 | 0.5868 | 0.5843 | 0.5866 |
| 3 | 0.5319 | 0.5293 | 0.5313 |
| 4 | 0.4875 | 0.4854 | 0.4872 |
| 5 | 0.4499 | 0.4476 | 0.4493 |
| 10 | 0.3086 | 0.3067 | 0.3079 |
| 15 | 0.1999 | 0.1985 | 0.1993 |
Model with random effects for incomplete blocks.
For comparison, we analyzed the same data dropping the incomplete block effect. This is equivalent to analysis as a randomized complete block design, for which variance balance holds and the usual heritability definition is adequate. The estimates were 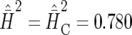 and
and 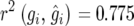 (SE = 0.0881). The slight discrepancy can be explained by the approximation in Equation 18. By comparison, the simulated value of the right-hand side of inequality (18) is 0.781 (SE = 0.0889), which is very close to
(SE = 0.0881). The slight discrepancy can be explained by the approximation in Equation 18. By comparison, the simulated value of the right-hand side of inequality (18) is 0.781 (SE = 0.0889), which is very close to 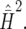
Example 2:
We analyzed a series of official rapeseed variety trials in Germany. The data set comprised 120 cultivars tested in 4 years and at four locations. At some locations, several trials were performed. The series was rather unbalanced, because old cultivars were dropped, while new cultivars entered the trials. Trials were laid out in complete blocks. The data are a subset of a data set that is more fully described in Piepho and Michel (2000). Trials' means were analyzed by the variance-components model
 |
(21) |
where L, Y, T, and G denote sites, years, trials, and cultivars, respectively, and the model is written using the syntax of Patterson (1997). Fixed effects appear before the colon and random effects after the colon. The residual was assumed to have variance equal to the variance of a mean computed for each trial. The variance-component estimates are shown in Table 3. Notably, the genetic variance was relatively small, presumably because released varieties result from intensive selection so that genetic variance is narrowed down compared to that in breeding trials. The standard errors of a difference among adjusted means were rather heterogeneous (Table 4), reflecting the unbalanced data structure. We found  and
and  Obviously, the first estimate is rather different from the other two. The result shows that the ad hoc measure based on adjusted means (
Obviously, the first estimate is rather different from the other two. The result shows that the ad hoc measure based on adjusted means ( ) cannot be recommended for very unbalanced data, while the
) cannot be recommended for very unbalanced data, while the  which is based on BLUP, works remarkably well. This is in agreement with the results of Cullis et al. (2006).
which is based on BLUP, works remarkably well. This is in agreement with the results of Cullis et al. (2006).
TABLE 3.
Variance component estimates (REML) for example 2 based on model (21)
| Term | Variance |
|---|---|
| G | 3.8299 |
| G · L | 0.2094 |
| G · Y | 1.9245 |
| G · L · Y | 5.8699 |
| G · L · Y · T | 1.8617 |
TABLE 4.
Quantiles of standard errors of a difference among adjusted means (SED) for example 2
| Quantile (%) | SED |
|---|---|
| 100 (maximum) | 5.718 |
| 90 | 5.416 |
| 75 | 5.284 |
| 50 (median) | 4.431 |
| 25 | 3.972 |
| 0 | 3.007 |
| 0 (minimum) | 1.404 |
Recommendation of cultivars based on official trials is similar to selection in breeding trials. It therefore seems reasonable to compute a response to selection. An estimate of response to selection based on  or
or 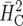 would not be appropriate due to the large unbalancedness. The results in Table 5 show that the naïve approach does not properly estimate the response to selection. It is important to point out that there is also a discrepancy for
would not be appropriate due to the large unbalancedness. The results in Table 5 show that the naïve approach does not properly estimate the response to selection. It is important to point out that there is also a discrepancy for 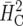 in terms of response to selection, most notably for small values of n, although the estimate of
in terms of response to selection, most notably for small values of n, although the estimate of  agreed very well with
agreed very well with 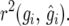
TABLE 5.
Response to selection for example 2 computed by simulation based on (16), based on an ad hoc approach using  , and based on
, and based on 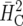 (Cullis et al. 2006) for different selection fractions p = n/I with I = 120
(Cullis et al. 2006) for different selection fractions p = n/I with I = 120
| Response to selection
|
|||
|---|---|---|---|
| n | Based on (16) |
Ad hoc
|
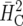 |
| 1 | 3.364 | 2.632 | 2.979 |
| 2 | 3.056 | 2.455 | 2.777 |
| 3 | 2.850 | 2.327 | 2.635 |
| 4 | 2.693 | 2.227 | 2.522 |
| 5 | 2.565 | 2.145 | 2.428 |
| 10 | 2.153 | 1.858 | 2.103 |
| 15 | 1.899 | 1.669 | 1.887 |
| 20 | 1.711 | 1.520 | 1.720 |
| 30 | 1.433 | 1.291 | 1.461 |
| 40 | 1.221 | 1.108 | 1.254 |
| 50 | 1.045 | 0.952 | 1.077 |
| 60 | 0.889 | 0.811 | 0.918 |
Example 3:
This data set comprises a series of 26 breeding trials with sugar beet conducted in a single location and year. The trials involved 831 entries. Each trial was laid out as a 6 × 6 lattice design with two replicates. The lattices were connected by four checks. Thus, apart from the four checks, the overall design accommodated 26 × 32 = 832 treatments. There were an additional 2 entries, which were treated as checks in the analysis. Also, 5 entries could not be sown and so had to be replaced by some of the other entries. The remaining 825 entries were testcrosses of S1 and S2 lines, derived from 33 crosses among 31 different female parents and 33 male parents, with two different testers. S1 lines were available for all crosses, while S2 lines were present for only some crosses. The parents were related by pedigree, i.e., some of them were half sibs or full sibs. Pedigree information was available for up to eight generations back from the tested entries. Definition of heritability accounting for pedigree information is difficult, although some proposals have been made (Holland et al. 2003, p. 46; Oakey et al. 2006). The measures 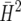 and
and 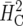 are not applicable here, because they assume independent genotypes. The present example shows that estimation of heritability and related quantities in the presence of pedigree information is straightforward with our approach.
are not applicable here, because they assume independent genotypes. The present example shows that estimation of heritability and related quantities in the presence of pedigree information is straightforward with our approach.
The linear model used for analysis was
 |
(22) |
where T denotes trial, R denotes replicate, B denotes incomplete block, C denotes check (C = 0 for noncheck entries), Ts denotes tester (two levels), G denotes genotype, and X is a dummy variable with X = 0 for checks and X = 1 for entries. Coding of the variables C was such that each check and the group of nonchecks had a separate mean. Jointly, the coding for C and X ensured that checks were blocked from the genotypic variance–covariance structure (Piepho et al. 2006). The factor G was defined such that each entry had a separate level and checks were assigned to one of the levels for entries. This made sure that the dimension of the matrix G equaled the number of entries. Which particular level of the entries was chosen for this purpose was immaterial, because entries were blocked from the genotypic variance–covariance structure. The genotypic effect X · G was modeled in two ways:
Independent genotypic variance, i.e.,
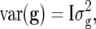 where I denotes an identity matrix: This model ignores pedigree information.
where I denotes an identity matrix: This model ignores pedigree information.Pedigree-based genotypic variance, i.e.,
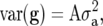 where A denotes the numerator relationship matrix computed from the pedigree (Mrode 2000): Under this model, genotypic effects are correlated, the covariance being proportional to the coefficient of coancestry. This type of model allows exploiting resemblance between relatives in the estimation of g.
where A denotes the numerator relationship matrix computed from the pedigree (Mrode 2000): Under this model, genotypic effects are correlated, the covariance being proportional to the coefficient of coancestry. This type of model allows exploiting resemblance between relatives in the estimation of g.
The model with pedigree-based variance–covariance structure yielded a poorer fit than the independent model in terms of minus twice the restricted log-likelihood (Table 6). We also fitted a model with the genetic variance–covariance structure 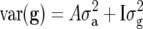 (Oakey et al. 2006). This converged to the model with
(Oakey et al. 2006). This converged to the model with 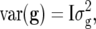 further indicating that the independent model was preferable. A possible reason for the better fit of the independent model is that selection has taken place and not all information on which selection has been based is included in the analysis. It is also not obvious how one should define a base population in this case (Piepho and Möhring 2006; Piepho et al. 2007).
further indicating that the independent model was preferable. A possible reason for the better fit of the independent model is that selection has taken place and not all information on which selection has been based is included in the analysis. It is also not obvious how one should define a base population in this case (Piepho and Möhring 2006; Piepho et al. 2007).
TABLE 6.
Model fit for two genotypic variance–covariance structures (example 3)
Form of 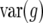
|
||
|---|---|---|
| Term | 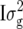 |
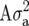 |
| T · R · B | 901.90 | 858.88 |
| X · G (independent) | 2,745.84 | — |
| X · G (pedigree) | — | 1,010.46 |
| Residual | 3,134.02 | 3,626.05 |
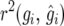 |
0.5970a | 0.5328 |
| −2 restricted log-likelihood | 20,969.4 | 20,997.2 |
Up to the fourth decimal place, the same estimate was obtained for 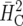 (Cullis et al. 2006).
(Cullis et al. 2006).
The response to selection was computed for all possible selection intensities and both models i and ii. Figure 1 shows a plot of the two selection responses. It is seen that pedigree-based BLUP performs worse than BLUP based on a model with independent genotypic effects, which agrees well with the model fit as assessed by the log-likelihood.
Figure 1.—

Plot of simulated response to selection under pedigree-based [R(pedigree)] and independent [R(independent)] models for genotypic variance and for all selection fractions. The dashed diagonal indicates equal response to selection (example 3).
CONCLUDING REMARKS
In estimating M, which is needed to obtain Ω, it is also possible to account for errors of estimation in the variance of u (Kackar and Harville 1984). We did not consider this option, as is common practice (Cullis et al. 2006), but an extension along the lines of Kackar and Harville (1984) seems straightforward. In large breeding trials, the effect of this refinement is expected to be marginal.
Our simulation-based approach can also be used to compare the efficiency of different genotypic variance–covariance models, as was illustrated in example 3. It therefore provides an alternative to cross-validation methods, which have been used to assess the performance of pedigree-based BLUP in plant breeding (Bernardo 1994). A problem with cross-validation is that it is not obvious how the data should be split into estimation and validation sets and that the complex correlation structure in many breeding trials violates the standard assumptions underlying the optimality theory for cross-validation (Efron and Tibshirani 1993). Our simulation approach provides a simple alternative that circumvents these problems.
The key issue in the case of unbalanced data and pedigree-based models is that any measure of heritability loses the standard interpretations that it allows for the balanced case with independent genotypes. A prominent example occurs in animal breeding, when the mixed model involves only a random residual and an additive genetic effect (breeding value) modeled by the numerator relationship matrix A. It is customary in this case to express the mixed-model equations in a form involving the term  where
where 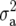 is the additive genetic variance and
is the additive genetic variance and  the residual (Mrode 2000, p. 39). While this expression is useful,
the residual (Mrode 2000, p. 39). While this expression is useful,  does not lend itself to the computation of a response to selection by standard equations, because breeding values in a population will be correlated and heteroscedastic due to the pedigree. Our proposed solution to this problem is to directly simulate the quantity of interest (response to selection, correlation between true and predicted genotypic value, etc.) rather than trying to compute it from some measure of heritability. The approach is straightforward to implement. The only potential downside is computing time in the case of very large problems.
does not lend itself to the computation of a response to selection by standard equations, because breeding values in a population will be correlated and heteroscedastic due to the pedigree. Our proposed solution to this problem is to directly simulate the quantity of interest (response to selection, correlation between true and predicted genotypic value, etc.) rather than trying to compute it from some measure of heritability. The approach is straightforward to implement. The only potential downside is computing time in the case of very large problems.
Acknowledgments
We thank Volker Michel (Landesforschungsanstalt für Landwirtschaft und Fischerei Mecklenburg-Vorpommern, Gülzow, Germany) for providing the rapeseed data. We also thank Axel Schechert (FR. STRUBE Saatzucht GmbH & Co. KG, Söllingen, Germany) for making available the sugar beet data. This research was conducted within the Breeding and Informatics project of the Genome Analysis of the Plant Biological System initiative (http://www.gabi.de).
APPENDIX
We here derive the variance of 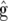 and the covariance of
and the covariance of  and g. The BLUP of u takes the form
and g. The BLUP of u takes the form
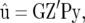 |
(Searle et al. 1992, p. 274), where
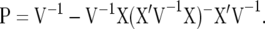 |
Thus,
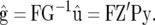 |
From this we find
 |
Exploiting that the variance of the BLUP of u is
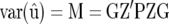 |
(Searle et al. 1992, p. 287), we find
 |
thus establishing the covariance given in Equation 10. Also,
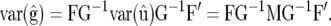 |
References
- Bernardo, R., 1994. Prediction of maize single-cross performance using RFLPs and information from related hybrids. Crop Sci. 34: 20–25. [Google Scholar]
- Cochran, W. G., and G. M. Cox, 1957. Experimental Designs, Ed. 2. Wiley, New York.
- Cullis, B. R., A. Smith and N. Coombes, 2006. On the design of early generation variety trials with correlated data. J. Agric. Biol. Environ. Stat. 11: 381–393. [Google Scholar]
- Efron, B., and R. Tibshirani, 1993. An Introduction to the Bootstrap. Chapman & Hall, London.
- Falconer, D. S., and T. F. C. Mackay, 1996. An Introduction to Quantitative Genetics, Ed. 4. Prentice Hall, London.
- Hanson, W. D., 1963. Heritability, pp. 125–139 in Statistical Genetics and Plant Breeding, edited by W. D. Hanson and H. F. Robinson. Pub. 982, National Academy of Science-National Research Council, Washington, DC.
- Helms, T. C., and J. J. Hammond, 2006. Genetic gain equation with correlated genotype x environment effects. Crop Sci. 46: 1137–1142. [Google Scholar]
- Henderson, C. R., 1977. Best linear unbiased prediction of breeding values not in the model for records. J. Dairy Sci. 60: 783–787. [Google Scholar]
- Holland, J. B., W. E. Nyquist and C. T. Cervantes-Martinez, 2003. Estimating and interpreting heritability for plant breeding: an update. Plant Breed. Rev. 22: 9–112. [Google Scholar]
- John, J. A., and E. R. Williams, 1995. Cyclic and Computer Generated Designs. Chapman & Hall, London.
- Kackar, A. N., and D. A. Harville, 1984. Approximation for standard errors of estimators of fixed and random effects in mixed linear models. J. Am. Stat. Assoc. 79: 853–861. [Google Scholar]
- McLean, R. A., W. L. Sanders and W. W. Stroup, 1991. A unified approach to mixed linear models. Am. Stat. 45: 54–64. [Google Scholar]
- Mrode, R. A., 2000. Linear Models for the Prediction of Animal Breeding Values. CAB International, Wallingford, UK.
- Nyquist, W. E., 1991. Estimation of heritability and prediction of selection response in plant populations. Crit. Rev. Plant Sci. 10: 235–322. [Google Scholar]
- Oakey, H., A. Verbyla, W. Pitchford, B. Cullis and H. Kuchel, 2006. Joint modelling of additive and non-additive genetic line effects in single field trials. Theor. Appl. Genet. 113: 809–819. [DOI] [PubMed] [Google Scholar]
- Patterson, H. D., 1997. Analysis of series of variety trials, pp. 139–161 in Statistical Methods for Plant Variety Evaluation, edited by R. A. Kempton and P. N. Fox. Chapman & Hall, London.
- Piepho, H. P., and V. Michel, 2000. Überlegungen zur regionalen Auswertung von Landessortenversuchen. Informatik. Biomet. Epidemiol. Med. Biol. 31: 123–136. [Google Scholar]
- Piepho, H. P., and J. Möhring, 2006. Selection in cultivar trials—Is it ignorable? Crop Sci. 146: 193–202. [Google Scholar]
- Piepho, H. P., E. R. Williams and M. Fleck, 2006. A note on the analysis of designed experiments with complex treatment structure. HortScience 41: 446–452. [Google Scholar]
- Piepho, H. P., J. Möhring, A. E. Melchinger and A. Büchse, 2007. BLUP for phenotypic selection in plant breeding and variety testing. Euphytica (in press).
- Searle, S. R., G. Casella and C. E. McCulloch, 1992. Variance Components. Wiley, New York.