

Abstract
Codon usage bias in Drosophila melanogaster genes has been attributed to negative selection of those codons whose cellular tRNA abundance restricts rates of mRNA translation. Previous studies, which involved limited numbers of genes, can now be compared against analyses of the entire gene complements of 12 Drosophila species whose genome sequences have become available. Using large numbers (6138) of orthologs represented in all 12 species, we establish that the codon preferences of more closely related species are better correlated. Differences between codon usage biases are attributed, in part, to changes in mutational biases. These biases are apparent from the strong correlation (r = 0.92, P < 0.001) among these genomes' intronic G + C contents and exonic G + C contents at degenerate third codon positions. To perform a cross-species comparison of selection on codon usage, while accounting for changes in mutational biases, we calibrated each genome in turn using the codon usage bias indices of highly expressed ribosomal protein genes. The strength of translational selection was predicted to have varied between species largely according to their phylogeny, with the D. melanogaster group species exhibiting the strongest degree of selection.
CODON usage bias reflects a higher prevalence of particular, over other synonymous, codons. This phenomenon has been observed for bacteria (Sharp and Li 1986), yeast (Sharp et al. 1986), nematodes (Stenico et al. 1994), fruit flies (Shields et al. 1988), and mammals (Duret 2002). It varies between species, and between genes within a species, and has arisen from a complex interplay between mutation, selection, and drift (Bulmer 1991). Observations of codon usage bias provide insights into variations in selective strengths and into mutational biases over evolutionary distances separating distinct species. Conservation of codon usage is also of practical importance for phylogenetic methods, such as PAML (Goldman and Yang 1994), that use codon-based models to estimate phylogenetic distances among coding sequences. These methods generally assume that codons are chosen randomly from all available synonymous codons, subject to nucleic acid compositional biases and to selection. A negative correlation between the number of synonymous substitutions per synonymous site, dS, and the codon usage bias of a gene has been reported and, at times, refuted on a number of occasions using different methods (Sharp and Li 1989; Moriyama and Hartl 1993; Dunn et al. 2001; see Bierne and Eyre-Walker 2003 for a discussion). Recent studies have demonstrated the pitfalls of unequal codon usage for phylogeny estimation (Inagaki and Roger 2006) and for estimating the selection strength of codon usage bias (Aris-Brosou and Bielawski 2006).
Recently, the genome sequences of 12 Drosophila species have become available (Adams et al. 2000; Richards et al. 2005; Drosophila 12 Genomes Consortium 2007). The last common ancestor of these fruit flies is believed to have lived ∼63 MYA (Tamura et al. 2004). This species set contains (1) pairs of recently diverged species such as D. simulans/D. sechellia and D. pseudoobscura/D. persimilis; (2) species at increasing levels of divergence from D. melanogaster such as D. erecta, D. yakuba, D. ananassae, D. pseudoobscura, and D. willistoni; and (3) a set of more distantly related species such as D. mojavensis, D. virilis, and D. grimshawi (Figure 1).
Figure 1.—

Tree topology of the evolutionary relationships among the 12 fruit fly species. This reflects the topology of a tree based on median whole-genome dS values (see Heger and Ponting 2007, for details; branch lengths are not shown to scale).
Codon usage bias in Drosophila species in general, and in D. melanogaster in particular, is well established (Shields et al. 1988) and has been attributed both to mutational biases, as reflected by unequal A or T, over G or C, nucleotide composition within selectively neutral sequence, and to selection to improve translational efficiency (Bulmer 1991). Correlations have been observed between the codon usage bias of a gene and a variety of parameters (reviewed in Powell and Moriyama 1997), including gene length and amino acid substitution rates (Betancourt and Presgraves 2002). The two most persuasive determinants advanced so far for translational selection acting on Drosophila codon usage bias are tRNA abundance (Moriyama and Powell 1997) and gene expression level (Duret and Mouchiroud 1999), which are consistent with results found for many bacterial genomes (Sharp and Li 1986, reviewed in Kurland 1991).
Mutational biases and their contributions to codon usage bias are poorly understood. For reasons unknown, preferred codons in D. melanogaster tend to have a G or C in third position (Shields et al. 1988), raising the G + C content at third positions well above the G + C content in noncoding DNA. In contrast, mutational events in D. melanogaster are biased toward A + T base pairs (Petrov and Hartl 1999), perhaps because of recombination-driven biased gene conversion (Duret 2002). Mutational bias and codon usage are linked through a sizable and significant correlation between intronic G + C content (GCi) and the G + C content at synonymous third codon positions (GC3) (Kliman and Hey 1994; Kliman and Eyre-Walker 1998). Recombination rates have been linked to codon usage bias (Hey and Kliman 2002; Marais and Piganeau 2002), but the effect seems to be small compared to the effects of selection (Bierne and Eyre-Walker 2006).
Codon usage variation has been studied not only between genes from one species, but also between orthologs from among several species. In general, codon usage bias between orthologs has been found to be conserved even over long evolutionary distances, although some differences are apparent for individual genes (Powell and Moriyama 1997). Codon usage is reported to have shifted in D. willistoni compared to D. melanogaster (Powell et al. 2003), but it is not clear whether this change arose adaptively or else was a “frozen accident.” An excess of fixations of unpreferred vs. preferred codons in D. melanogaster has been interpreted as resulting from relaxed selection on codon usage bias (Akashi 1996; McVean and Vieira 2001). However, in D. simulans there are conflicting reports on whether constraint on codon usage similarly has undergone relaxation (Begun 2001; McVean and Vieira 2001), or has achieved mutation-selection-drift equilibrium (Dumont et al. 2004).
We have contributed predictions of protein-coding transcripts and genes, and their orthology and paralogy relations among the 12 Drosophila species, as described elsewhere (Heger and Ponting 2007). These have been made freely available via the AAA website (http://rana.lbl.gov/drosophila/wiki/index.php/Main_Page). In a separate article (Heger and Ponting 2007) we have considered the variations in selective pressures that operated on amino acid sequences for genes from each of the 12 genomes. Here, we sought first to investigate variations in selective pressures that acted upon codon use for these species, and thereafter to compare directly the strengths of these two selective processes for each Drosophila lineage in turn.
As expected, we observe codon usage bias for each of the 12 Drosophila species. Mutational biases and selective forces, however, contribute unequally to these species' codon usage biases. There is a strong correlation between the genomewide intronic G + C content and exonic G + C content of degenerate third codon positions (r = 0.92, P < 0.001). Thus, it is clear that variable mutational biases need to be appropriately accounted for if variable selective forces acting on codon usage are to be estimated accurately. We propose the set of ribosomal proteins as an internal calibration point when inferring the strength and type of codon usage bias within each genome. Following calibration, we examined codon usage across 6138 orthologs per genome. We find that codon usage bias due to translational selection has persisted between species, but that the strengths of selection have varied. While species in the melanogaster group and D. willistoni exhibit strong selection on codon bias, more relaxed selection is apparent for all remaining species.
MATERIALS AND METHODS
Data sets:
Chromosomes, transcripts, and translations for D. melanogaster (dmel) were obtained from ENSEMBL release 37 (Birney et al. 2006). The sequence data are based on BDGP assembly release 4, and annotations derive from FlyBase release 4.2.1 (Grumbling and Strelets 2006). This set contained 19,369 transcripts from 13,836 genes.
Genomic sequences for D. simulans (dsim), D. sechellia (dsec), D. yakuba (dyak), D. erecta (dere), D. ananassae (dana), D. pseudoobscura (dpse), D. persimilis (dper), D. willistoni (dwil), D. grimshawi (dgri), D. virilis (dvir), and D. mojavensis (dmoj) were obtained from the community server for the assembly/alignment/annotation project (http://rana.lbl.gov/drosophila/wiki/index.php/Main_Page), release comparative analysis freeze 1 (caf1).
Transcript and gene prediction:
Transcripts and genes were predicted by a pipeline developed around the alignment tool Exonerate (Slater and Birney 2005). Predictions have been submitted to the collaborative annotation effort headed by M. Eisen (Drosophila 12 Genomes Consortium 2007). Briefly, the pipeline predicts transcripts by homology using transcripts from D. melanogaster as templates. The pipeline assesses the quality of a prediction by checking if the intron positions of the template are conserved in the prediction. Further details on the gene prediction process can be found in a companion article (Heger and Ponting 2007). For this analysis, only transcripts with conserved gene structure were considered. The numbers of genes analyzed are provided in Table 1.
TABLE 1.
G + C content in introns (GCi), G + C content in degenerate third codon positions (GC3D), and strength of selection on codon bias (ΔL) in 12 Drosophila genomes
| Species | Genes | GCi (%) | GC3D (%) | Correlation GCi ∼ GC3D | 〈Lc〉R | 〈Lc〉B | ΔL | 〈ENC〉R+B |
|---|---|---|---|---|---|---|---|---|
| D. melanogaster | 13,836 | 39.0 (8) | 64.5 (5) | 0.35 | 0.75 | 1.27 | 100.0 (2) | 100.0 (8) |
| D. simulans | 9,092 | 39.6 (5) | 65.9 (6) | 0.39 | 0.74 | 1.27 | 101.9 (1) | 98.2 (4) |
| D. sechellia | 10,527 | 39.6 (6) | 65.7 (7) | 0.39 | 0.75 | 1.23 | 92.3 (5) | 98.5 (6) |
| D. yakuba | 11,900 | 39.5 (6) | 65.9 (8) | 0.36 | 0.74 | 1.25 | 96.2 (3) | 98.3 (5) |
| D. erecta | 11,483 | 40.1 (3) | 66.4 (3) | 0.41 | 0.75 | 1.22 | 90.4 (6) | 97.9 (3) |
| D. ananassae | 11,158 | 39.4 (7) | 66.0 (4) | 0.40 | 0.77 | 1.27 | 96.2 (4) | 101.0 (9) |
| D. pseudoobscura | 10,039 | 43.4 (1) | 68.4 (1) | 0.42 | 0.86 | 1.17 | 59.6 (9) | 97.2 (1) |
| D. persimilis | 8,338 | 43.1 (2) | 68.3 (2) | 0.41 | 0.86 | 1.18 | 61.5 (8) | 97.3 (2) |
| D. willistoni | 9,976 | 34.8 (12) | 45.7 (12) | 0.28 | 0.90 | 1.33 | 80.8 (7) | 108.6 (12) |
| D. virilis | 9,470 | 38.1 (9) | 61.4 (10) | 0.33 | 0.91 | 1.19 | 55.8 (11) | 99.7 (7) |
| D. mojavensis | 9,192 | 36.9 (10) | 61.6 (9) | 0.40 | 0.92 | 1.21 | 55.8 (10) | 101.2 (10) |
| D. grimshawi | 9,422 | 35.8 (11) | 58.9 (11) | 0.20 | 0.91 | 1.18 | 50.0 (12) | 103.3 (11) |
Ranks are in parentheses. Selection strength (ΔL) is considered to be the average message length difference between ribosomal sequences and all sequences. 〈Lc〉 is the average message length per codon for the set of ribosomal protein genes (R) or for the bulk of genes excluding ribosomal protein genes (B). 〈ENC〉R+B is the average ENC value calculated for all transcripts per genome. ΔL and ENC are given as percentages, relative to values for D. melanogaster. G + C contents in exons and introns are computed over all predicted transcripts, whereas the comparison of selection strengths considered only ortholog sets with representatives in all 12 species (see materials and methods for details). Correlations between GC3D and GCi are all significant at P < 0.001.
Ortholog sets:
Orthology prediction between D. melanogaster genes and the gene set of each of the 11 other species was performed using PhyOP, essentially as described previously (Goodstadt and Ponting 2006), but with modifications as described elsewhere (Heger and Ponting 2007). Ortholog sets were built around each D. melanogaster gene by collecting ortholog transcripts in each of the other 11 Drosophila species. Gene lengths and codon bias indices, such as codon adaptation index (CAI) or effective number of codons (ENC), were averaged over multiple transcripts, when present, and over multiple orthologs for cases of lineage-specific duplications. Ortholog sets lacking genes from 1 or more species were discarded, resulting in 6138 ortholog sets with representatives from all 12 species.
Annotated ribosomal proteins were obtained from FlyBase (Release 4.3, March 2006, Grumbling and Strelets 2006), and their orthologs were collected for each newly sequenced genome. This resulted in between 67–75 ribosomal protein genes per species, depending on the incompleteness of the genome assembly and the presence or absence of lineage-specific gene duplicates, and 57 ribosomal protein genes with orthologs in each species.
G + C content:
We tested for a correlation between the nucleotide compositions for introns and those for the third codon positions of coding exons. For this, it was paramount to exclude introns containing exons from, for example, alternative transcripts and mispredictions. Consequently, we removed all introns that overlapped with an exon from any other transcript on either strand. To be as comprehensive as possible, fragmentary predictions and predictions with in-frame stop codons or frameshifts were considered as part of this filtering procedure. This step removed 4% of all introns in D. melanogaster and between 13–19% of introns in the newly sequenced genomes.
The G + C content of a gene's introns (GCi) was defined as the G + C content of its concatenated intronic sequences. Ten bases at either end of each intron were discarded to exclude splice site motifs. The G + C content (GC3) for third codon positions of a gene's coding sequence, and the G + C content (GC3D) of such positions that are degenerate, were also calculated using concatenated sequences.
Measurement of codon usage bias:
We employed three measures to assess codon usage biases among species. First, we calculated the deviation from uniform codon usage, as measured by the ENC (Wright 1990) and implemented by codonW (http://codonw.sourceforge.net). ENC ranges from values of 20 for genes with an invariable preference for a single codon for each amino acid to 61 for genes exhibiting no codon preferences.
Second, we applied the CAI (Sharp and Li 1987) as a measure of the departure of a sequence from its optimal codon usage. Optimal codon usage has often been defined by a set of highly expressed genes (for D. melanogaster, see Shields et al. 1988). We were unable to employ this definition uniformly due to the lack of expression data for all 12 species. Instead, for each species we used a common set of ribosomal protein genes as a proxy for such a set of highly expressed genes. Codon frequencies for ribosomal protein genes provided the codon weights used subsequently for computing values of the CAI of other genes. Importantly, using our set of D. melanogaster ribosomal protein genes, we were able to reproduce the codon usage and the previously described preferred codons for each amino acid type (Shields et al. 1988). The preferred codon for each amino acid was unchanged and the correlation coefficient between the remaining weights was high (r = 0.96; P < 0.001). This CAI and ribosomal protein set strategy avoids the pitfalls of parameter fluctuations between species (Akashi et al. 2006).
Third, we use the average message length per codon as a measure of codon usage bias. Indices derived from information theory have been used previously to estimate codon usage bias and are based on the computation of relative entropies (Zeeberg 2002; Wan et al. 2003). Here, we compute the total message length ML of a transcript of n codons, amino acid frequencies na and codon frequencies nc, given codon usage table P, as
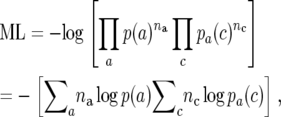 |
where p(a) is the probability of observing amino acid a and pa(c) is the probability of observing codon c for amino acid a. The message length is thus dependent on the amino acid sequence of the transcript as well as the codon usage. In our analysis, we use only the contribution of the codon usage to ML. The message length is sequence-length dependent and can be normalized by dividing by the sequence length n giving the message length per codon
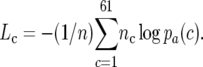 |
To allow comparisons between selection strengths on codon usage bias among genomes, we calculate ΔL, the difference between the message length per codon averaged over a reference set of highly biased genes (R, here the set of ribosomal protein genes) and the message length per codon averaged over all other sequences (B, “bulk”): 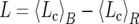 . We use only sequences with orthologs in all species because this permits comparison of ΔL among all genomes.
. We use only sequences with orthologs in all species because this permits comparison of ΔL among all genomes.
Due to the strong phylogenetic signal in ΔL and other indices we confirmed that all correlations reported in the article remained significant after applying the phylogenetic-contrasts method (Felsenstein 1985) (see supplemental Table S1 at http://www.genetics.org/supplemental/).
Statistical analyses were performed with the R software package (http://www.r-project.org). Phylogenetic contrasts (Felsenstein 1985) were computed with the CONTRAST program of the PHYLIP package (Felsenstein 1989). Phylogenetic eigenvector regression (Diniz-Filho et al. 1998) was performed with the ade4 (http://pbil.univ-lyon1.fr/ADE-4) package. In these analyses, the species' relationships were given by their divergence in terms of synonymous substitutions per site.
RESULTS
Mutational biases:
Codon usage bias can arise simply from nucleotide substitution biases favoring the inclusion, for example, of A or T, over G or C, or it can arise because of translational selection. Discriminating selective from mutational sequence biases is challenging for Drosophila species if only because much of their intronic sequence is under greater constraint than are synonymous sites (Andolfatto 2005). Nevertheless, if it is assumed that selection greatly affects neither GCi (the G + C composition of introns) nor GC3D (the G + C content of degenerate third codon positions), then the changes in mutational bias along Drosophila lineages can be inferred from the variations of these two quantities.
We first investigated whether G + C content has changed among the genes from each of the 12 Drosophila species' genomes. As might be expected, average GCi or GC3D values are most similar between the more closely related species, with values in the melanogaster group varying by only 1.4% in GC3D or 1.1% in GCi (Table 1). For D. melanogaster genes, GCi and GC3D were known previously to be correlated strongly (Kliman and Hey 1994). We observed significant covariation between genes' GCi and GC3D values for each of the 12 Drosophila species, albeit at correlation coefficients between 0.2 and 0.4 (Table 1). Furthermore, the GCi and GC3D values calculated by concatenating intronic and coding sequences for each of these species are also correlated and exhibit a strong linear dependence (Figure 2A). We find that GCi, averaged across each genome, can explain 10.6% of the variance in GC3D between genes (P < 10−5).
Figure 2.—

Correlation between G + C content in introns and in degenerate third positions in codons (regression computed without D. willistoni: R = 0.92, P < 0.001).
Only D. willistoni fails to follow this linear relationship with an extraordinarily low GC3D value for its intronic G + C content (Table 1; Figure 2A). Rodríguez-Trelles et al. (2000) previously observed this unusual characteristic of D. willistoni sequence within a small set of eight genes. These authors also concluded that the common ancestor of the Sophophora (which include both melanogaster–obscura and saltans–willistoni lineages) possessed a genome with an elevated G + C content (Rodriguez-Trelles et al. 2000). This is consistent with the most parsimonious interpretation of G + C content evolution among the 12 genomes since D. willistoni exhibits the lowest of all GCi and GC3D contents. Assuming, once more, that selection has not contributed to this dramatic base composition change, then mutational biases must have altered substantially on the D. willistoni lineage since its last common ancestor with D. melanogaster. This alteration in mutational preferences, and more minor changes on other Drosophila lineages, will contribute to changes in codon usage even in the absence of selection.
Ribosomal proteins as a calibration to infer translational selection variation among genomes:
We sought evidence that translational selection has affected codon usage biases differently among the 12 Drosophila species. To achieve this, we needed to account for the changes in codon usage arising from mutational biases that, for example, led to the unusually low G + C content seen for D. willistoni. Codon usage indices such as the ENC (Wright 1990) compare the observed codon usage for a gene against uniform codon usage and, as such, do not compensate for the influence of mutational biases on codon usage. By way of contrast, indices such as the CAI (Sharp and Li 1987) estimate codon usage bias relative to a reference gene set, usually a set of highly biased genes. Consequently, these methods can, in principle, take account of mutational biases by assuming that such biases affect genes subject to translational selection equivalently to those that are not. Although reference sets of highly biased genes are readily available for D. melanogaster (Shields et al. 1988), similar sets have not been compiled for the other Drosophila species. Consequently, if we are to understand the contributions of translational selection to Drosophila species' codon usage biases, we required an internal calibration point that might allow us to compare CAI values between species.
For this, we propose the use of a set of ribosomal protein genes as an appropriate reference gene set, all assumed to be highly expressed with strong codon usage bias in each of the additional 11 Drosophila species. This use is owing to their strong conservation, which results in gene prediction and orthology assignment being relatively straightforward, and because the majority of ribosomal protein genes are found among genes with strong codon usage biases in D. melanogaster (Shields et al. 1988; Moriyama and Powell 1997), in Escherichia coli (Jia and Li 2005) and in Saccharomyces cerevisiae (Sharp et al. 1986). Finally, we use ribosomal protein genes since they are ubiquitous and continuously expressed, and thus their codon usage is unlikely to have been optimized for certain tissues or developmental stages.
Of the 69 ribosomal protein genes present in D. melanogaster, we were able to assign between 67 and 75 orthologous genes in the 11 other sequenced genomes. Six ribosomal protein genes were not found in 1 of the 11 species but were present in all others, likely reflecting gaps among the genome sequence assemblies. Another 6 D. melanogaster ribosomal protein genes were absent from >2 other genomes, but reflect likely duplication events in the D. melanogaster lineage. Similarly, some ribosomal protein genes have been duplicated in other lineages, leading to some Drosophila species exhibiting higher counts of ribosomal protein genes than D. melanogaster.
CAI codon usage preferences for these ribosomal protein genes were found to have remained relatively constant across all Drosophila species (Figure 3A), with the notable exception of D. willistoni (see below). The preferred codons, those that have been used most frequently in ribosomal protein genes, are identical between the major subgroups (D. melanogaster to D. pseudoobscura and D. virilis to D. grimshawi), with the exception of aspartic acid, whose most frequently used codon has changed from GAC (58% frequency in D. melanogaster) to GAT (61% in D. virilis). Nevertheless, this represents only a minor change and is also likely to be unimportant since aspartic acid is thought to provide the least contribution to codon usage bias of all amino acids (Powell and Moriyama 1997; McVean and Vieira 2001). Change among the preferred codons for the ribosomal protein genes has thus been minor, and it groups species together according to their phylogenetic relationships.
Figure 3.—

Codon usage has not varied greatly across the 12 species. Variations in codon preferences for (A) ribosomal protein genes and (B) all genes across 12 genomes. Codon usage was computed as the percentage difference with respect to D. melanogaster. Codons are colored according to this difference (see adjacent color bar). The species' phylogeny is provided at the bottom of each chart for ease of reference.
D. willistoni:
The codon usage of D. willistoni genes contrasts greatly with those of the other 11 species, as has been observed previously (Powell et al. 2003). The unusually low GC3D values for D. willistoni genes result in the correlation (r = 0.71, P < 0.001) between GC3D and codon usage bias (CAI) being lower than for the other species (r > 0.80, P < 0.001). Consequently, the correlation between ENC and CAI for D. willistoni genes is considerably weaker (r = 0.41, P < 0.001) than for the other species (r = 0.66–0.83; P < 0.001).
A consequence of the proposed shift in base composition within the D. willistoni lineage is the change of preferred codons among the ribosomal protein genes for arginine, valine, glycine, and aspartic acid (arginine: CGC to CGT in D. willistoni, valine: GTG to GTC in D. willistoni, glycine: GGC to GGT in D. willistoni, aspartic acid: GAC to CGT in D. willistoni), with respect to the melanogaster group and pseudoobscura subgroup. These are significant changes since the former three amino acids are known to contribute greatly to selection on codon usage for other Drosophila species (McVean and Vieira 2001).
Codon usage bias is better conserved between more closely related orthologs:
Codon usage bias (CAI) values calculated among all 6138 orthologs were found to be correlated among all species pairs, but better conserved among more closely related, than more distantly related, Drosophila species (Figure 4). Correlation coefficients rank from 0.98 for pairs of orthologous transcripts in very closely related species, such as D. simulans and D. sechellia, to 0.52 for pairs of orthologous transcripts in the distantly related pair D. mojavensis and D. sechellia. Like ribosomal protein genes' codon usage, the strength of the correlation between orthologs' CAI values has been determined, in large part, by the evolutionary distance between species.
Figure 4.—

Variations in codon usage bias are consistent with the species' phylogeny. Correlations between codon usage bias indices across 6138 orthologs for each pairwise species comparison, shaded according to correlation strengths. Top diagonal, CAI based on ribosomal protein gene weights; bottom diagonal, ENC (see adjacent bar for scale). Note that the correlation between species' codon usage biases is stronger when CAI is used as a measure. All correlations are highly significant (P < 0.001). The species' phylogeny is provided at the bottom of the chart for ease of reference.
Figure 4 also illustrates the benefit, for between-species comparisons, of using CAI values, normalized using ribosomal protein genes, to estimate codon usage bias. Even though correlations remain highly significant between orthologs across species when using the ENC index, correlations are much weaker than those when CAI values are used, particularly for the more distantly related species. For example, the correlation between the species pair D. sechellia and D. mojavensis is 0.52 for CAI and 0.29 for ENC. The mean ENC value per genome is strongly influenced by mutational biases as it is negatively correlated with intronic G + C composition (r = −0.84, P < 0.001); no such correlation is seen between CAI and intronic G + C content (see below). This demonstrates that nucleotide composition evolution as well as translational selection contribute greatly to codon usage bias, as measured by ENC, but only translational selection contributes to our version of CAI.
A complementary approach to this might have been the application of codon usage preferences for D. melanogaster genes to their orthologs in other Drosophila species. This approach would have merit since we observe little change in the correlation between orthologs' CAI values, if these are instead calculated on the basis of published D. melanogaster preferences (Shields et al. 1988). This indicates that the relative ranking of codon bias strength remains even for considerable phylogenetic divergences. Nevertheless, CAI values are affected by G + C compositional variation between genomes, which results in increasing incompatibility of D. melanogaster codon usage preferences for the more distantly related Drosophila species. The average CAI value per genome based on codon usage preferences of D. melanogaster correlates strongly with intronic G + C (r = 0.81, P < 0.002) whereas when the mean CAI is calculated for each species using weights from its own ribosomal protein gene set, no such correlation is apparent (r = 0.17, P = 0.6). Consequently, to differentiate between mutational and selective effects on codon usage biases it is best to estimate CAI values for each genome in turn using ribosomal protein genes for calibration.
Translational selection among ribosomal protein genes:
A set of genes is appropriate for calibrating codon bias measures if it fulfills two requirements: (1) selection on codon usage bias has acted nonuniformly among all genes, and (2) genes in the set exhibit strong and consistent codon usage biases.
First, we show that selection on codon usage has acted nonuniformly among genes. To test the null hypothesis of equal codon sampling, we randomized transcripts for each species according to the codon usage in their entire gene sets. We find, for all 12 species, that the observed distributions of CAI values are considerably more broad than the simulated distributions, because of greater than expected numbers of sequences with low and with high codon usage biases (Figure 5 inset). Thus, codon usage bias occurs nonuniformly among genes in each of these species.
Figure 5.—

CAI value histograms for genes from 12 Drosophila species. These distributions are broader and are increasingly left shifted with increasing strengths of codon usage bias. CAI values of each gene were computed on the basis of codon usage preferences in ribosomal protein genes. The inset shows the effect of selection for codon usage bias on the distribution of CAI values. In the simulations, codons were chosen randomly for all 13,831 D. melanogaster genes according to the bulk codon usage in D. melanogaster. The observed distribution of CAI values is broader than the distribution of simulated CAI values, showing that a disproportionate number of genes have higher or lower codon usage than expected.
Second, we show that codon usage bias among ribosomal protein-coding genes is strong and consistent. CAI values or ribosomal protein-coding genes are, on average, 2.3 standard deviations above the mean for all genes (Figure 6). We repeated the analysis using randomly selected gene sets for calibration. In these simulations, the average CAI values did not exceed 0.42 standard deviations above the mean for any of the 12 species (100 replications per species). The ribosomal protein genes thus form a distinct set of proteins with a characteristic codon usage bias (P < 0.001, one-tailed Monte Carlo test).
Figure 6.—

Ribosomal protein genes exhibit codon usage bias for each of the 12 Drosophila species. For each ribosomal protein gene, we computed the z-score (number of standard deviations above the mean) of its codon usage bias (CAI value) compared to the bulk of all proteins (excluding ribosomal protein genes). Shown, for each species, is the distribution of z-scores of ribosomal proteins from the bulk of sequences. The box indicates first, second, and third quartiles, the whiskers extend to the 5 and 95 percentiles. Outliers are also shown.
Our observations are consistent with selection acting on ribosomal protein-coding genes and the remaining majority of sequences sampling codons simply according to mutational biases of nucleotide substitution.
Measuring the strength of selection for codon usage:
If translational selection has resulted in codon usage bias, then a biased transcript must have conferred a benefit to the organism. We assume no positional effect on codon choice: a biased transcript will simply use more preferred codons than an unbiased transcript, if both translate into the same amino acid sequence. With this simple model, the information theoretical message length can be used to quantify this benefit.
To assess selection strength on codon usage bias, we compare the mean message lengths per codon Lc (see materials and methods) of ribosomal protein genes, with the mean message lengths per codon of all other proteins, reasoning that the larger the difference between them, the stronger the selection on codon usage bias. The relative selection strength is given by ΔL. The use of ΔL, rather than any other index, is advantageous in that it can be considered as an extra cost per codon incurred for translating a typical transcript compared to the translation of a transcript of a ribosomal gene. Such a straightforward interpretation is less easily obtained for the other indices.
We observe no significant correlation (r = −0.16, P = 0.61) between our measure of relative selection strength ΔL and intronic G + C for the 12 species. This indicator of selection strength thus appropriately appears to be independent of background G + C content and thus mutational biases.
Using this method for inferring the strength of translational selection, we find that species in the D. melanogaster group, as well as D. willistoni despite its striking reductions in GCi and GC3D values, exhibit similar levels of codon usage selection strengths, ΔL (Table 1). Compared to these, species of the obscura group, together with D. virilis, D. mojavensis and D. grimshawi, exhibit smaller ΔL values which we interpret as indicating weaker codon usage selection strengths (Table 1).
The same subdivision into two groups is obtained using other indices (Figure 7) once they are calibrated with respect to ribosomal protein genes' codon usage. Even ENC, the deviation from uniform codon usage, shows this species subdivision if it is employed as ΔENC. It is notable that average ENC (〈ENC〉), an estimator of the location of the ENC distribution and a common indicator of codon usage bias, shows only marginal differences between the two groups and underestimates selection strength in D. willistoni. It is thus not an appropriate proxy for selection strength acting on codon usage bias.
Figure 7.—

After correction for mutational bias, codon usage indices are consistent and likely reflect selection strength. The top section shows the intronic GC content (GCi) and indices uncorrected for mutational bias while the bottom section shows measures corrected for mutational bias. The indices are L, message length; CAI, codon adaptive index; and ENC, effective number of codons. < > values are the average over all genes while Δ-values are computed as the difference of a particular index averaged over ribosomal protein genes vs. the index averaged over all others. Uncorrected CAI values use D. melanogaster weights for all genomes. All measures are relative to D. melanogaster and oriented such that higher values correspond to higher codon usage bias. The species' phylogeny is provided at the bottom of the chart for ease of reference.
Changes of selection strength on codon usage bias:
We conclude that selective forces acting to generate codon usage bias in all 12 Drosophila species examined and the strength of codon usage bias are well reflected by the codons of ribosomal protein genes. The selection strength on codon usage bias (ΔL) has not been constant among these species, with the melanogaster group showing significantly greater selection strengths on codon usage bias than the remaining species (P < 10−4, discounting D. willistoni; P < 10−5, retaining D. willistoni). As differences in ΔL readily map onto the Drosophila species' phylogeny, it might thus appear that the degree of selection strength on codon usage bias represents an inherited trait.
Nevertheless, differences in ΔL (Table 1) might simply reflect these species' phylogenetic heritage: ΔL values for the melanogaster group, for example, may simply reflect their common inheritance of codon usage from their last common ancestor. To investigate whether the differences in species' ΔL values are significant, or whether they simply reflect the influence of ancestor on descendant, we applied the phylogenetic eigenvector regression method (Diniz-Filho et al. 1998). Specifically, we compared ΔL values for melanogaster group species (with or without D. willistoni) against those of all other species (excepting D. willistoni) using this method. We find that these ΔL differences are not significant given their phylogenetic heritage (P = 0.08 or 0.37, with or without D. willistoni, respectively) implying that ΔL differences can be explained simply by the influence of ancestor on descendant (“phylogenetic inertia” Harvey and Purvis 1991).
DISCUSSION
We provide a first comparative genomic view on the mutational and selective effects on codon usage bias among 12 Drosophila species. Although population data provide the most detailed insights into the recent evolution of codon usage bias strength (Akashi 1996, 1999; Akashi et al. 1998; McVean and Vieira 1999, 2001; Dumont et al. 2004; Maside et al. 2004), such data, for multiple orthologous loci for all species, are, as yet, not available. Instead, we have exploited the 12 newly sequenced Drosophila genomes, and their predicted orthologous protein-coding genes, to investigate the relative contributions to codon usage of nucleotide composition and translational selection.
Here, we have provided evidence for translational selection on codon usage in each of these Drosophila species. Preferred codons are, in the main, preserved between ribosomal protein orthologs among these genomes. We have demonstrated that translational selection strength is highest within the melanogaster group, and for D. willistoni, and weaker in the remaining five species. D. willistoni is exceptional among these species for its large decrease in G + C content along its lineage, and thus for its large corresponding change in codon usage.
We have described how, by using an internal calibration, measuring codon usage bias using CAI is appropriate since this quantity varies independently of intronic G + C content. The approach rests on the assumption that intronic G + C content is unaffected by selection on codon usage. However, we acknowledge that this assumption may fail if selection has acted upon mRNA properties such as its stability or structure. Given the known correlation between tRNA pool size and codon usage bias (Moriyama and Powell 1997), we assume that the selection we are observing has acted primarily on rates of translation at the ribosome.
The degree of codon usage variation across genomes was observed to be greater for ribosomal protein genes than for the bulk of genes (Figure 3B). This was surprising since we had expected essentially stationary codon usage in ribosomal protein genes due to the constraints imposed by tRNAs' concentrations, compared with more variable codon usage, in concert with changes in mutational biases, among genes less susceptible to translational selection. One interpretation of these data is that tRNA concentrations have fluctuated over time. This would result in adaptive changes of codon bias among ribosomal protein genes that might exceed mutational changes. This hypothesis, which remains to be tested, implies that tRNA concentrations, themselves, would be subject to adaptive evolution.
Choice of method for estimating strengths of selection for codon usage:
We sought to compare the strengths of translational selection for codon usage among the 12 Drosophila species. To do so, we had considered whether we could apply the suppression of synonymous substitution rate (dS) as a proxy for selection strength (Powell and Moriyama 1997). However, accurate estimation of dS in the face of changeable mutational biases and nucleotide compositions would have been problematic (Singh et al. 2005; Aris-Brosou and Bielawski 2006). We also considered applying the dominant bias (DB) method (Carbone et al. 2003), which derives a set of the most biased transcripts using an iterative process. The DB method, however, is less effective when the codon usage of highly biased transcripts is not easily separable from the usage for the remaining bulk of sequences. Indeed, for strongly biased species (the melanogaster subgroup and D. willistoni), the DB method succeeded in reproducing the codon usage bias obtained using CAI and ribosomal protein genes for calibration, whereas for the more weakly biased species (the pseudoobscura group, D. mojavensis and D. virilis) proteins found to be biased by the DB method were of markedly different types and contained few ribosomal proteins. (By contrast, for as yet unknown reasons, for D. grimshawi, the species with the smallest selection strength, the dominant bias method again correctly produced ribosomal proteins in the set of highly biased genes.) For the pseudoobscura group, D. mojavensis and D. virilis, we considered the possibility that selection on codon usage bias is truly acting on a set of proteins distinct from the set of ribosomal proteins. Nevertheless, we discounted this possibility because of the lack of a clear functional bias (with respect to gene ontology terms, Ashburner et al. 2000) in the DB-derived set of biased proteins, and because in our analysis employing CAI we found that codon usage of ribosomal protein genes is indeed distinct from that for the remaining sequences. We conclude that application of the DB method may not be appropriate for all species.
Confounding effects on measure of selection strength:
Our results show a strong phylogenetic division between strong selective strength on codon usage bias in the melanogaster subgroup and D. willistoni and weaker selective strength for the remaining species. Such a phylogenetic distribution can arise if all species are not considered equally in analyses. We considered whether a bias could have arisen from the gene prediction process because transcripts in the target genome were predicted according to template transcripts from D. melanogaster only: the likelihood of mispredicted exons increases with increasing divergence between template and target genome (Heger and Ponting 2007). Mispredicted exons could act to homogenize codon usage bias between ribosomal protein genes and bulk genes and thus reduce ΔL. However, we confirmed our previous results by using an extensively cleaned set of sequences that included only codons that were aligned in conserved blocks across 1:1 orthologs in all species. We observed no significant changes in ΔL values from the extensively cleaned data set (data not shown).
We considered that the strength of selection on amino acid sequence and the strength of selection on codon usage might be tightly coupled, with each affected only by a lineage's effective population size history. Nevertheless, maximum-likelihood estimates of the dN/dS ratio (Heger and Ponting 2007) showed no significant correlation (r = −0.14, P = 0.72) with our measure of selection strength, ΔL. [Nor is the dN/dS ratio correlated with mean ENC per genome (r = −0.38, P = 0.31.)]
Despite ΔL values differing between Drosophila species, these differences, in large part, can be explained by phylogenetic inertia. We do not, therefore, observe significant differences between the species, with respect to the selection strength on codon usage bias, that could be interpreted as changes in the selection gradient within internal branches of the Drosophila phylogeny. The differences in ΔL values thus are explained best as “frozen accidents” occurring at speciation nodes.
Comparisons with previous, smaller-scale, studies:
Results from our whole-genome approach appear, on the whole, to be consistent with those from previous smaller-scale analyses. It has been reported that D. simulans and D. virilis, compared with D. melanogaster, each exhibits a different codon usage bias and a stronger (D. simulans) or weaker (D. virilis) selection strength, presumably resulting from their proposed larger or smaller effective population sizes, respectively (Aquadro et al. 1988; Akashi 1996; McVean and Vieira 2001). In each case, our findings concur with these previous observations. The difference, however, in selection strengths we observe between D. melanogaster, D. simulans, and D. sechellia are slight (Table 1) and well within the range of measurement error.
The impact of codon usage for comparative analysis of the fly genomes:
The impact of mutational bias and translational selection on codon usage bias for Drosophila genes, and the variations in selective strengths between species, have implications for the accuracy of neutral rate estimations from synonymous substitution rates. Neutral rates will be considerably underestimated for genes or species exhibiting strong bias, which can be corrected for empirically (Hirsh et al. 2005). The confounding effects of codon bias on synonymous substitution rate dS will thus depend on the species pairs under study. In the D. melanogaster subgroup, both strength and type of codon bias appear to have remained relatively constant, and maximum-likelihood estimates of synonymous substitution rates are unlikely to be affected by codon usage bias. For the further diverged species, this may not necessarily be true, but here the effect of codon usage bias on dS estimates might be negligible when compared to the variance arising when multiple substitutions are accounted for.
Acknowledgments
We gratefully acknowledge the Drosophila Genomes Sequencing Consortium, in particular the various sequencing centers for the data and Mike Eisen (Lawrence Berkeley National Lab) for hosting the AAA web site. We thank two anonymous referees for their helpful comments. This work was funded by the United Kingdom Medical Research Council.
References
- Adams, M. D., S. E. Celniker, R. A. Holt, C. A. Evans, J. D. Gocayne et al., 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185–2195. [DOI] [PubMed] [Google Scholar]
- Akashi, H., 1996. Molecular evolution between Drosophila melanogaster and D. simulans: reduced codon bias, faster rates of amino acid substitution, and larger proteins in D. melanogaster. Genetics 144: 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., 1999. Inferring the fitness effects of DNA mutations from polymorphism and divergence data: statistical power to detect directional selection under stationarity and free recombination. Genetics 151: 221–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., R. M. Kliman and A. Eyre-Walker, 1998. Mutation pressure, natural selection, and the evolution of base composition in Drosophila. Genetica 102–103: 49–60. [PubMed] [Google Scholar]
- Akashi, H., W. Ko, S. Piao, A. John, P. Goel et al., 2006. Molecular evolution in the Drosophila melanogaster species subgroup: frequent parameter fluctuations on the timescale of molecular divergence. Genetics 172: 1711–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437: 1149–1152. [DOI] [PubMed] [Google Scholar]
- Aquadro, C. F., K. M. Lado and W. A. Noon, 1988. The rosy region of Drosophila melanogaster and Drosophila simulans. I. Contrasting levels of naturally occurring DNA restriction map variation and divergence. Genetics 119: 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aris-Brosou, S., and J. Bielawski, 2006. Large-scale analyses of synonymous substitution rates can be sensitive to assumptions about the process of mutation. Gene 378: 58–64. [DOI] [PubMed] [Google Scholar]
- Ashburner, M., C. A. Ball, J. A. Blake, D. Botstein, H. Butler et al., 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun, D. J., 2001. The frequency distribution of nucleotide variation in Drosophila simulans. Mol. Biol. Evol. 18: 1343–1352. [DOI] [PubMed] [Google Scholar]
- Betancourt, A. J., and D. C. Presgraves, 2002. Linkage limits the power of natural selection in Drosophila. Proc. Natl. Acad. Sci. USA 99: 13616–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne, N., and A. Eyre-Walker, 2003. The problem of counting sites in the estimation of the synonymous and nonsynonymous substitution rates: implications for the correlation between the synonymous substitution rate and codon usage bias. Genetics 165: 1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne, N., and A. Eyre-Walker, 2006. Variation in synonymous codon use and DNA polymorphism within the Drosophila genome. J. Evol. Biol. 19: 1–11. [DOI] [PubMed] [Google Scholar]
- Birney, E., D. Andrews, M. Caccamo, Y. Chen, L. Clarke et al., 2006. Ensembl 2006. Nucleic Acids Res. 34: D556–D561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer, M., 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone, A., A. Zinovyev and F. Kepes, 2003. Codon adaptation index as a measure of dominating codon bias. Bioinformatics 19: 2005–2015. [DOI] [PubMed] [Google Scholar]
- Diniz-Filho, J. A. F., C. E. R. De Sant'Ana and L. M. Bini, 1998. An eigenvector method for estimating phylogenies. Evolution 52: 1247–1262. [DOI] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium, 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218. [DOI] [PubMed] [Google Scholar]
- Dumont, V. B., J. C. Fay, P. P. Calabrese and C. F. Aquadro, 2004. DNA variability and divergence at the notch locus in Drosophila melanogaster and D. simulans: a case of accelerated synonymous site divergence. Genetics 167: 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, K. A., J. P. Bielawski and Z. Yang, 2001. Substitution rates in Drosophila nuclear genes: implications for translational selection. Genetics 157: 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret, L., 2002. Evolution of synonymous codon usage in metazoans. Curr. Opin. Genet. Dev. 12: 640–649. [DOI] [PubMed] [Google Scholar]
- Duret, L., and D. Mouchiroud, 1999. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 96: 4482–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., 1985. Phylogenies and the comparative method. Am. Nat. 125: 1–15. [Google Scholar]
- Felsenstein, J., 1989. PHYLIP - Phylogeny inference package (version 3.2). Cladistics 5: 164–166. [Google Scholar]
- Goldman, N., and Z. Yang, 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11: 725–736. [DOI] [PubMed] [Google Scholar]
- Goodstadt, L., and C. P. Ponting, 2006. Phylogenetic reconstruction of orthology, paralogy, and conserved synteny for dog and human. PLoS Comput. Biol. 2: e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumbling, G., and V. Strelets, 2006. FlyBase: anatomical data, images and queries. Nucleic Acids Res. 34: D484–D488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey, P. H., and A. Purvis, 1991. Comparative methods for explaining adaptations. Nature 351: 619–624. [DOI] [PubMed] [Google Scholar]
- Heger, A., and C. P. Ponting, 2007. Evolutionary rate analyses of orthologues and paralogues from twelve Drosophila genomes. Genome Res. (in press). [DOI] [PMC free article] [PubMed]
- Hey, J., and R. M. Kliman, 2002. Interactions between natural selection, recombination, and gene density in the genes of Drosophila. Genetics 160: 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh, A. E., H. B. Fraser and D. P. Wall, 2005. Adjusting for selection on synonymous sites in estimates of evolutionary distance. Mol. Biol. Evol. 22: 174–177. [DOI] [PubMed] [Google Scholar]
- Inagaki, Y., and A. J. Roger, 2006. Phylogenetic estimation under codon models can be biased by codon usage heterogeneity. Mol. Phylogenet. Evol. 40: 428–434. [DOI] [PubMed] [Google Scholar]
- Jia, M., and Y. Li, 2005. The relationship among gene expression, folding free energy and codon usage bias in Escherichia coli. FEBS Lett. 579: 5333–5337. [DOI] [PubMed] [Google Scholar]
- Kliman, R. M., and A. Eyre-Walker, 1998. Patterns of base composition within the genes of Drosophila melanogaster. J. Mol. Evol. 46: 534–541. [DOI] [PubMed] [Google Scholar]
- Kliman, R. M., and J. Hey, 1994. The effects of mutation and natural selection on codon bias in the genes of Drosophila. Genetics 137: 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland, C. G., 1991. Codon bias and gene expression. FEBS Lett. 285: 165–169. [DOI] [PubMed] [Google Scholar]
- Marais, G., and G. Piganeau, 2002. Hill-Robertson interference is a minor determinant of variations in codon bias across Drosophila melanogaster and Caenorhabditis elegans genomes. Mol. Biol. Evol. 19: 1399–1406. [DOI] [PubMed] [Google Scholar]
- Maside, X., A. W. Lee and B. Charlesworth, 2004. Selection on codon usage in Drosophila americana. Curr. Biol. 14: 150–154. [DOI] [PubMed] [Google Scholar]
- McVean, G. A., and J. Vieira, 1999. The evolution of codon preferences in Drosophila: a maximum-likelihood approach to parameter estimation and hypothesis testing. J. Mol. Evol. 49: 63–75. [DOI] [PubMed] [Google Scholar]
- McVean, G. A., and J. Vieira, 2001. Inferring parameters of mutation, selection, and demography from patterns of synonymous site evolution in Drosophila. Genetics 157: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama, E. N., and D. L. Hartl, 1993. Codon usage bias and base composition of nuclear genes in Drosophila. Genetics 134: 847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama, E. N., and J. R. Powell, 1997. Codon usage bias and tRNA abundance in Drosophila. J. Mol. Evol. 45: 514–523. [DOI] [PubMed] [Google Scholar]
- Petrov, D. A., and D. L. Hartl, 1999. Patterns of nucleotide substitution in Drosophila and mammalian genomes. Proc. Natl. Acad. Sci. USA 96: 1475–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, J. R., and E. N. Moriyama, 1997. Evolution of codon usage bias in Drosophila. Proc. Natl. Acad. Sci. USA 94: 7784–7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, J. R., E. Sezzi, E. N. Moriyama, J. M. Gleason and A. Caccone, 2003. Analysis of a shift in codon usage in Drosophila. J. Mol. Evol. 57(Suppl. 1): S214–S225. [DOI] [PubMed] [Google Scholar]
- Richards, S., Y. Liu, B. R. Bettencourt, P. Hradecky, S. Letovsky et al., 2005. Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 15: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Trelles, F., R. Tarrio and F. J. Ayala, 2000. Evidence for a high ancestral GC content in Drosophila. Mol. Biol. Evol. 17: 1710–1717. [DOI] [PubMed] [Google Scholar]
- Sharp, P. M., and W. H. Li, 1986. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 24: 28–38. [DOI] [PubMed] [Google Scholar]
- Sharp, P. M., and W. H. Li, 1987. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15: 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, P. M., and W. H. Li, 1989. On the rate of DNA sequence evolution in Drosophila. J. Mol. Evol. 28: 398–402. [DOI] [PubMed] [Google Scholar]
- Sharp, P. M., T. M. Tuohy and K. R. Mosurski, 1986. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 14: 5125–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields, D. C., P. M. Sharp, D. G. Higgins and F. Wright, 1988. “Silent” sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol. Biol. Evol. 5: 704–716. [DOI] [PubMed] [Google Scholar]
- Singh, N. D., P. F. Arndt and D. A. Petrov, 2005. Genomic heterogeneity of background substitutional patterns in Drosophila melanogaster. Genetics 169: 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater, G. S. C., and E. Birney, 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenico, M., A. T. Lloyd and P. M. Sharp, 1994. Codon usage in Caenorhabditis elegans: delineation of translational selection and mutational biases. Nucleic Acids Res. 22: 2437–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K., S. Subramanian and S. Kumar, 2004. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol. Biol. Evol. 21: 36–44. [DOI] [PubMed] [Google Scholar]
- Wan, X., D. Xu and J. Zhou, 2003. A new informatics method for measuring synonymous codon usage bias, pp. 1101–1018 in Intelligent Engineering Systems Through Artificial Neural Networks, Vol. 13, edited by C. H. Dagli, J. Gosh, A. L. Buczak, O. Ersoy and M. J. Embrechts. ASME Press, New York.
- Wright, F., 1990. The ‘effective number of codons’ used in a gene. Gene 87: 23–29. [DOI] [PubMed] [Google Scholar]
- Zeeberg, B., 2002. Shannon information theoretic computation of synonymous codon usage biases in coding regions of human and mouse genomes. Genome Res. 12: 944–955. [DOI] [PMC free article] [PubMed] [Google Scholar]