

Abstract
An analysis of litter size and average piglet weight at birth in Landrace and Yorkshire using a standard two-trait mixed model (SMM) and a recursive mixed model (RMM) is presented. The RMM establishes a one-way link from litter size to average piglet weight. It is shown that there is a one-to-one correspondence between the parameters of SMM and RMM and that they generate equivalent likelihoods. As parameterized in this work, the RMM tests for the presence of a recursive relationship between additive genetic values, permanent environmental effects, and specific environmental effects of litter size, on average piglet weight. The equivalent standard mixed model tests whether or not the covariance matrices of the random effects have a diagonal structure. In Landrace, posterior predictive model checking supports a model without any form of recursion or, alternatively, a SMM with diagonal covariance matrices of the three random effects. In Yorkshire, the same criterion favors a model with recursion at the level of specific environmental effects only, or, in terms of the SMM, the association between traits is shown to be exclusively due to an environmental (negative) correlation. It is argued that the choice between a SMM or a RMM should be guided by the availability of software, by ease of interpretation, or by the need to test a particular theory or hypothesis that may best be formulated under one parameterization and not the other.
MIXED linear models (Henderson 1984) are broadly used to predict breeding values and to estimate variance components for traits of interest in livestock and plant breeding and play an important role in evolutionary and theoretical quantitative genetics (Lande 1979; Cheverud 1984; Walsh 2003). In genetic improvement programs, the objective of selection includes typically several correlated traits. The classical approach for a multiple-trait analysis is to use models posing that the nature of the correlation between response variables (phenotypes) is due to linear associations between unobservables, such as additive genetic values or nongenetic sources, like permanent or temporary environmental effects.
Structural equation models represent an extension of the standard linear model to account for links (feedback and/or recursiveness) involving either the phenotypes directly or latent variables; they are well established in econometrics and sociology (Goldberger 1972; Jöreskog 1973; Duncan 1975). These models were discussed in the early genetics literature by Wright (1921) but this work has not received much attention in quantitative genetics. Recently, Xiong et al. (2004) proposed the use of structural equation models for modeling and identifying genetic networks. In a quantitative genetics context, Gianola and Sorensen (2004) studied the consequences of the existence of simultaneous and recursive relationships between phenotypes on genetic parameters and presented statistical methods for inference. A recent application to study the relationship between somatic cell score and milk yield in goats is in de los Campos et al. (2006). Here we are concerned with an illustration of the implementation of structural equation models for the analysis of litter size and average litter weight in two breeds of Danish pigs.
Litter size is an important trait in pig genetic improvement programs (Rothschild and Bidanel 1998) and there is now convincing evidence that it has responded successfully to selection (i.e., Sorensen et al. 2000; Noguera et al. 2002). Several studies have also reported negative associations between litter size and individual birth weight (Kerr and Cameron 1995; Roehe 1999; Sorensen et al. 2000). Further, Sorensen et al. (2000) report an increase in the proportion of piglets born dead at higher litter size values.
Litter size is basically determined by ovulation rate and embryo mortality (Blasco et al. 1995); these processes take place mainly at the early stages of gestation. Piglet weight at birth is mostly determined by growth in late gestation. One could then postulate a one-way causal path establishing an effect of litter size on piglet weight at birth. This specification defines a recursive two-trait system. On the other hand, simultaneity occurs when trait 1 affects trait 2 and vice versa.
The objective of this study is, first, to show that recursive models can be interpreted as alternative parameterizations of standard linear models. We discuss identifiability of dispersion parameters, a topic that is intimately connected to the possibility of drawing inferences from the various parametric forms of a given model. Second, we address the statistical problems involved in deciding whether the association between traits is mediated by additive genetic and/or environmental covariances or via recursion only. The results are illustrated using data on litter size and average litter weight in pigs.
MATERIALS AND METHODS
Data:
Data from two breeds were analyzed: Landrace and Yorkshire. The traits analyzed were total number born per litter and average litter weight at birth (referred to as litter size and average piglet weight, hereinafter). The Landrace data set included 5178 litter size records and a pedigree file of 8800 individuals. The raw means for litter size and average piglet weight were 14.23 piglets and 1.36 kg., respectively, with standard deviations 3.62 piglets and 0.35 kg. The Yorkshire data set consisted of 3938 litter size records and a pedigree file of 7143 individuals. The raw means for litter size and average piglet weight were 13.01 piglets and 1.30 kg., respectively, with standard deviations 3.40 piglets and 0.22 kg. The raw correlations between traits were 0.01 in Landrace and −0.43 in Yorkshire.
Piglet weight at birth is strongly genetically determined by maternal effects (Grandinson et al. 2002), and, as a consequence, average piglet weight (as well as litter size) was considered a trait of the sow.
Models and likelihoods:
A description is provided of a standard mixed model (SMM) and a recursive mixed model (RMM). The SMM postulates the following linear structures for yLij (subscript L represents litter size) and yWij (subscript W represents average piglet weight) of the jth pair of records from female i,
 |
(1a) |
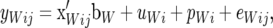 |
(1b) |
where  (k = L, W) is the appropriate row of a known incidence matrix, bk is a vector containing effects of herd years, seasons, and parity number, uki is an additive genetic effect of individual i, pki is a permanent environmental effect of individual i, and ekij is a residual effect (the lengths of the vectors of additive genetic effects and data are different, but to simplify notation, it is assumed throughout that after an appropriate relabeling, a common subindex i can be used for y, u, and p).
(k = L, W) is the appropriate row of a known incidence matrix, bk is a vector containing effects of herd years, seasons, and parity number, uki is an additive genetic effect of individual i, pki is a permanent environmental effect of individual i, and ekij is a residual effect (the lengths of the vectors of additive genetic effects and data are different, but to simplify notation, it is assumed throughout that after an appropriate relabeling, a common subindex i can be used for y, u, and p).
The following distributions were assigned to the location parameters:
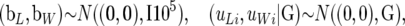 |
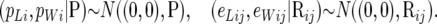 |
(2) |
Above, I is the identity matrix (of appropriate order),
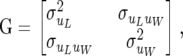 |
(3) |
and
 |
(4) |
The terms 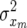 and
and 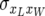 (x = u, p; m = L, W) in (3) and (4) are variance and covariance components associated with the distribution of additive genetic effects (x = u) and permanent environmental effects (x = p) for litter size and for individual piglet weight.
(x = u, p; m = L, W) in (3) and (4) are variance and covariance components associated with the distribution of additive genetic effects (x = u) and permanent environmental effects (x = p) for litter size and for individual piglet weight.
A possible approach to modeling the residual term Rij is as follows. Assume that the residual terms for individual piglet weight at birth that contribute to a given average piglet weight are conditionally normally and independently distributed, given litter size, with residual variance  , where
, where  is the residual component of variance of individual piglet weight at birth and
is the residual component of variance of individual piglet weight at birth and 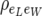 is the residual correlation between litter size and individual piglet weight at birth. Also assume that the residual terms for litter size are normally distributed with variance
is the residual correlation between litter size and individual piglet weight at birth. Also assume that the residual terms for litter size are normally distributed with variance 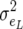 . Then the marginal (with respect to litter size) residual covariance between two individual piglet weight at birth records is
. Then the marginal (with respect to litter size) residual covariance between two individual piglet weight at birth records is 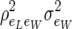 and the residual covariance matrix is equal to
and the residual covariance matrix is equal to
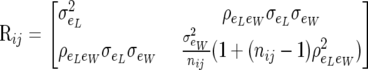 |
(5) |
In (5), the off-diagonal term 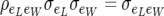 , and nij is the known number of records contributing to the average piglet weight of female i in parity j. There are three identifiable parameters in (5). This residual dispersion matrix can also be written as
, and nij is the known number of records contributing to the average piglet weight of female i in parity j. There are three identifiable parameters in (5). This residual dispersion matrix can also be written as
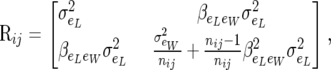 |
(6) |
where 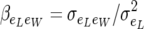 is the residual regression of individual piglet weight at birth on litter size. Matrix Rij is positive definite since
is the residual regression of individual piglet weight at birth on litter size. Matrix Rij is positive definite since  . The residual covariance matrix (5) for nij = 1 is denoted by R.
. The residual covariance matrix (5) for nij = 1 is denoted by R.
The heritabilities for the two traits are
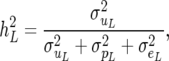 |
 |
(7) |
and the coefficients of correlation are
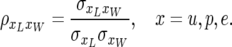 |
(8) |
Writing yij = (yLij, yWij)′, Equations 1 can be expressed as
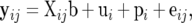 |
(9) |
where
 |
It follows that the sampling model for yij is the Gaussian process
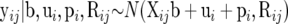 |
(10) |
and the contribution to the likelihood by yij is
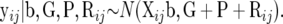 |
(11) |
The RMM assumes the following linear relationships between the jth pair of records from individual i and location parameters,
 |
(12a) |
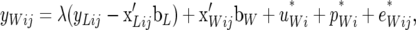 |
(12b) |
where λ is the recursive parameter. The first term in the right-hand side of (12b) indicates that, according to the model, average piglet weight is linearly related to the deviation of litter size from its group mean, and the strength of this relationship is measured by λ. On the other hand, Gianola and Sorensen (2004) postulate recursiveness or simultaneity between traits involving the observed phenotypes, rather than the unobserved deviations. We return to this point in the discussion.
The system defined by (12) can be retrieved subtracting the mean on both sides of (9) and multiplying by Λ, to get
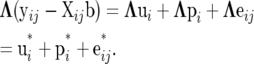 |
(13) |
The reduced form of (13) is
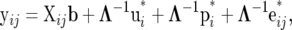 |
(14) |
which is the same as (9), where
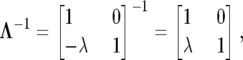 |
and
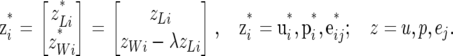 |
It follows from the Gaussian form of the distributions (2) that
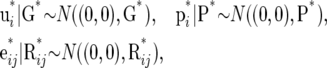 |
(15) |
where
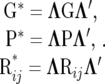 |
(16) |
Therefore the sampling model for yij under the RMM is the Gaussian process
 |
(17) |
and the contribution to the likelihood by yij is
 |
(18) |
If λ were known this is the same likelihood as (11) due to the one-to-one relationship
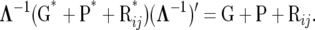 |
(19) |
However, with unknown λ, the left-hand side of (19) contains 10 parameters and the right-hand side 9. There are thus an infinite number of matrices involving the left-hand side of (19) that satisfy the equality, for any given G + P + Rij. In other words, disregarding identifiability at the level of the mean for both models, the RMM as defined above generates an unidentifiable likelihood.
Likelihood identification under the SMM and the RMM:
The subject of identifiability of the SMM and the RMM at the level of the mean is well known (e.g., Searle 1971) and is not discussed. In likelihood (11) of the SMM there are nine identifiable dispersion parameters associated with G, P, and Rij. This model with nondiagonal covariance matrices for u, p, and e is labeled SMMupe.
The RMM has an extra parameter, and a constraint needs to be introduced to achieve identification. One possible constraint is to assume that the phenotypic covariance on the recursive scale is zero. That is, denoting the mean of yL by μL,
 |
(20) |
This places the following interpretation on λ,
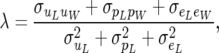 |
(21) |
the phenotypic regression of average litter weight on litter size. Expanding (19) it is easy to show that the constraint (20) guarantees a one-to-one relationship between the dispersion parameters of the RMM and those of the SMMupe and the likelihoods become equivalent. In this setting the RMM subject to the chosen constraint and the unconstraint SMMupe are two different identifiable parameterizations of the same likelihood model.
From the point of view of a likelihood analysis, inferences on the recursive scale can be obtained by fitting the SMMupe and transforming the estimated parameters appropriately, and vice versa. However, it is not statistically meaningful to ask whether the data have been generated by the SMMupe or by the recursive process described by the RMM subject to constraint (20), since both specifications lead to the same likelihood.
Generating an identifiable likelihood model to address the nature of the relationship between traits:
Here we present a statistically meaningful way to address the question whether the data have been generated by a recursive mechanism.
The starting point is the SMM defined in (3), (4), (5), and (9) but with a diagonal matrix for all the dispersion structures; that is,
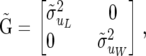 |
(22) |
 |
(23) |
and
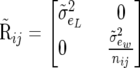 |
(24) |
The contribution to the likelihood by the pair of records yij is
 |
(25) |
There are six dispersion parameters associated with this model (the covariance matrices of u, p, and e have 0 off-diagonal elements), which is labeled SMM0.
The RMM that is developed here postulates that the relationship between data and location parameters is now
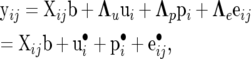 |
(26) |
where ui, pi, and eij are the same stochastic variables as in the SMM0 with covariance matrices (22), (23), and (24), and with
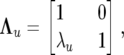 |
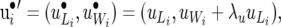 |
(27) |
and similarly for Λp, Λe,  , and
, and  . Note that the Λ's in (26) have the same structure as the Λ−1 in (14). Contrary to the generation of recursion in (13), the recursive model defined by (26) is not obtained by a linear transformation of the SMM and the two models lead to different marginal (with respect to random effects) distributions of the data. The linear structure specified by (26) and (27) has an interesting property: the components of average litter weight
. Note that the Λ's in (26) have the same structure as the Λ−1 in (14). Contrary to the generation of recursion in (13), the recursive model defined by (26) is not obtained by a linear transformation of the SMM and the two models lead to different marginal (with respect to random effects) distributions of the data. The linear structure specified by (26) and (27) has an interesting property: the components of average litter weight 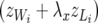 , z = u, p, e, have a term
, z = u, p, e, have a term 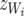 independent of litter size and a component
independent of litter size and a component 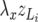 dependent on litter size.
dependent on litter size.
The sampling model for yij is
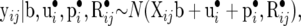 |
(28) |
and the contribution to the likelihood from yij is
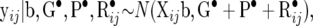 |
(29) |
where  ,
, 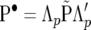 , and
, and 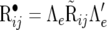 . This form of recursive model is labeled RMMupe. There are nine identifiable parameters in the dispersion matrix of this likelihood and when λu = λp = λe = 0 (or when Λu = Λp = Λe = I), likelihood (29) is equal to (25). A comparison between RMMupe and RMM with λu = λp = λe = 0, which is labeled RMM0, is to jointly test whether or not there is recursion at the level of the unobservable additive genetic values, permanent environmental and environmental effects. Alternatively, since likelihoods (29) and (11) are equivalent [it is easy to see that, for example,
. This form of recursive model is labeled RMMupe. There are nine identifiable parameters in the dispersion matrix of this likelihood and when λu = λp = λe = 0 (or when Λu = Λp = Λe = I), likelihood (29) is equal to (25). A comparison between RMMupe and RMM with λu = λp = λe = 0, which is labeled RMM0, is to jointly test whether or not there is recursion at the level of the unobservable additive genetic values, permanent environmental and environmental effects. Alternatively, since likelihoods (29) and (11) are equivalent [it is easy to see that, for example, 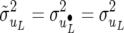 and
and  ] the comparison can be interpreted as testing whether or not the covariance matrices of the random effects of the SMMupe have a diagonal structure. Indeed, note that
] the comparison can be interpreted as testing whether or not the covariance matrices of the random effects of the SMMupe have a diagonal structure. Indeed, note that
 |
(30) |
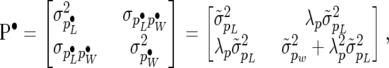 |
(31) |
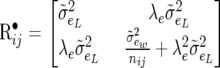 |
(32) |
The bottom diagonal element in (32) is very similar to the corresponding element in (6). However, when the trait is not average (that is, when nij = 1), the second term in the bottom diagonal element of (6) vanishes. Since, for example,  , by inspection of (30), (31), and (32) with (3), (4), and (6) it is obvious that the β's under the SMM are identical to the
, by inspection of (30), (31), and (32) with (3), (4), and (6) it is obvious that the β's under the SMM are identical to the 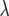 's in the RMM.
's in the RMM.
We also need
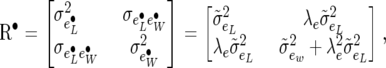 |
(33) |
which is matrix  for nij = 1. When λu = λp = λe = 0, the above covariance matrices become equal to (22), (23), and (24).
for nij = 1. When λu = λp = λe = 0, the above covariance matrices become equal to (22), (23), and (24).
Under the RMMupe, the heritability of average litter weight for nij = nT for all i, j is defined as
 |
(34) |
Prior and posterior distributions:
For the RMMupe, the joint prior distribution of all parameters is assumed to admit the factorization
 |
(35) |
where u• is the vector that contains the pairs  for all individuals in the pedigree, and p• is the vector that contains all permanent environmental effects
for all individuals in the pedigree, and p• is the vector that contains all permanent environmental effects  of females with records. The vector b is allocated an improper uniform distribution and vectors u• and p• are assumed to be normally distributed
of females with records. The vector b is allocated an improper uniform distribution and vectors u• and p• are assumed to be normally distributed
 |
where A is the known additive genetic relationship matrix, and
 |
The 2 × 2 matrices G•, P•, and R• follow inverse Wishart distributions
 |
 |
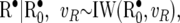 |
where the hyperpriors 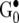 ,
, 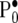 , and
, and  are known matrices of dimension 2 × 2 and the v's are known degrees of freedom. The conditional density for the total data
are known matrices of dimension 2 × 2 and the v's are known degrees of freedom. The conditional density for the total data 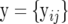 is equal to
is equal to
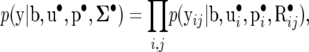 |
(36) |
where Σ• is a block diagonal with blocks 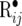 associated with each pair of records yij.
associated with each pair of records yij.
The posterior distribution of the RMMupe, up to a proportionality constant, is obtained by multiplication of the joint prior (35) by (36), giving
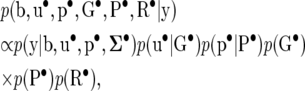 |
(37) |
which is also the posterior distribution of SMMupe, the standard two-trait mixed model with nondiagonal covariance matrices associated with all the random effects. Inferences based on RMMupe can be drawn from the posterior distribution (37) and the recursive parameters can easily be constructed from (30), (31), and (33),
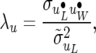 |
(38) |
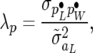 |
(39) |
and
 |
(40) |
A variety of submodels can be generated either by assuming some or all of the λ's equal or by setting some of them equal to zero.
Implementation:
If the number of piglets born was the same for all litters, nT, say, then  , where
, where  denotes the residual covariance matrix (32) with nij replaced by nT. In this case, the structure of
denotes the residual covariance matrix (32) with nij replaced by nT. In this case, the structure of 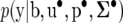 in (37) simplifies considerably. To take advantage of this simplification in the computations one can augment the piglet weight data with the so-called missing single records
in (37) simplifies considerably. To take advantage of this simplification in the computations one can augment the piglet weight data with the so-called missing single records 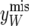 , so that nij = nT for all ij, where nT is the largest number of records contributing to average piglet weight in the data set. This technique is known as data augmentation (Tanner and Wong 1987) and the general idea is as follows. Given observed data y and a model indexed by parameters θ, the posterior distribution
, so that nij = nT for all ij, where nT is the largest number of records contributing to average piglet weight in the data set. This technique is known as data augmentation (Tanner and Wong 1987) and the general idea is as follows. Given observed data y and a model indexed by parameters θ, the posterior distribution 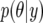 is proportional to
is proportional to 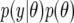 . When the model is fitted using MCMC, drawing samples from this posterior distribution may be computationally demanding. However, it may be easy to draw samples from
. When the model is fitted using MCMC, drawing samples from this posterior distribution may be computationally demanding. However, it may be easy to draw samples from
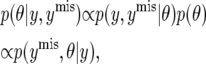 |
where ymis stands for the missing data. The strategy requires generating ymis from 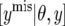 . In the present case,
. In the present case, 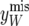 is generated from
is generated from
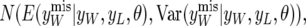 |
where θ is the vector of all parameters indexing the model.
After a little experimentation, a length of the Gibbs chain equal to 1 million was chosen. In Tables 1 and 2 we report Monte Carlo standard errors of estimates of various posterior means to give an idea of the accuracy of the Monte Carlo computations.
TABLE 1.
Monte Carlo estimates of posterior means of chosen parameters (posterior standard deviations in parentheses) based on SMMupe
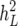 |
 |
ρu | ρp | ρe | |
|---|---|---|---|---|---|
| Landrace | 0.08 (0.02) | 0.24 (0.06) | 0.16 (0.19) | 0.07 (0.32) | 0.01 (0.06) |
| Yorkshire | 0.07 (0.02) | 0.29 (0.04) | −0.21 (0.16) | −0.24 (0.44) | −0.73 (0.03) |
| MSE | 0.63 | 1.3 | 5.0 | 7.8 | 1.0 |
h2, heritability with subscripts L (litter size) and W (average piglet weight) for nT = 25 individuals; ρ, correlations with subscripts u, p, and e involving additive genetic, permanent, and environmental effects; MSE, Monte Carlo standard error ×104.
TABLE 2.
Monte Carlo estimates of posterior means of chosen parameters (posterior standard deviations in parentheses) based on RMMupe
 |
 |
λu | λp | λe | |
|---|---|---|---|---|---|
| Landrace | 0.08 (0.02) | 0.24 (0.06) | 0.022 (0.026) | 0.050 (0.189) | 0.000 (0.003) |
| Yorkshire | 0.07 (0.02) | 0.22 (0.03) | −0.028 (0.022) | −0.028 (0.067) | −0.034 (0.003) |
| MSE | 0.60 | 1.3 | 5.6 | 9.5 | 1.7 |
h2, heritability with subscripts L (litter size) and W• (average piglet weight) for nT = 25 individuals; λ with subscripts u, p, and e involves additive genetic, permanent, and environmental effects; MSE, Monte Carlo standard error ×104.
Model testing:
Checking for systematic differences between a given model and the observed data discloses the quality of fit of the posed model. An attractive way to study the fit of a model is to use posterior predictive model checking (Gelman et al. 1996, 2004). The approach is simple to implement, is flexible, and provides a graphical exploration of residual-type diagnostics. The key feature is the construction of the so-called discrepancy measures that describe particular putative features of the data that the model may fail to account for. To be more specific, consider testing for the presence of recursion at the level of permanent environmental effects. Absence or presence of recursion at the level of additive genetic effects or residuals is studied in a similar way. Let (yLij, yWij), i = 1, 2, … , denote observed data and for parity j = 1, define the discrepancy measure
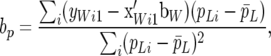 |
(41) |
the change of average piglet weight per unit change of permanent environmental effect associated with litter size. In (41), the sum is over all females with first parity records, and 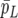 is the average pLi across females. If the observed data had been generated under RMM0 one would expect a value of bp in the vicinity of zero. If parameters were known, one could compare the observed value of bp to its sampling distribution, with a significant difference indicating model failure with respect to the discrepancy measure. This is equivalent to simulating data
is the average pLi across females. If the observed data had been generated under RMM0 one would expect a value of bp in the vicinity of zero. If parameters were known, one could compare the observed value of bp to its sampling distribution, with a significant difference indicating model failure with respect to the discrepancy measure. This is equivalent to simulating data 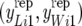 , i = 1, 2, … , under the RMM0, if parameters were known, computing
, i = 1, 2, … , under the RMM0, if parameters were known, computing 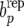 in each replicate, and deciding whether the observed value of bp is an atypical value in the distribution of
in each replicate, and deciding whether the observed value of bp is an atypical value in the distribution of 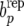 . Specifically and in the current context, one is testing whether the null model RMM0 is failing to account for a recursive mechanism present in the observed data.
. Specifically and in the current context, one is testing whether the null model RMM0 is failing to account for a recursive mechanism present in the observed data.
Since parameters are not known, we use the idea of posterior predictive model checking (Gelman et al. 1996, 2004) and consider the posterior predictive distribution of 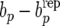 . This distribution reflects uncertainty about the parameters that enter in the discrepancy measure (41) as well as sampling variation. Note that the parameters are inferred from the “null model” RMM0 that assumes absence of recursion. The presence of recursion, not accounted for by model RMM0 would result in a distribution of bp −
. This distribution reflects uncertainty about the parameters that enter in the discrepancy measure (41) as well as sampling variation. Note that the parameters are inferred from the “null model” RMM0 that assumes absence of recursion. The presence of recursion, not accounted for by model RMM0 would result in a distribution of bp − 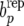 shifted from zero. This can also be construed as a test for a nonzero covariance between permanent environmental effects affecting litter size and those affecting average piglet weight. The exploration of recursion at the level of additive genetic effects and of residuals involves constructing bu −
shifted from zero. This can also be construed as a test for a nonzero covariance between permanent environmental effects affecting litter size and those affecting average piglet weight. The exploration of recursion at the level of additive genetic effects and of residuals involves constructing bu − 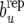 and be −
and be − 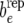 along the same lines.
along the same lines.
Often the diagnostic results of posterior predictive model checking are apparent visually, as is the case in this work. Other times it can be useful to compute a posterior predictive P-value to see whether the results could have arisen by chance under the null model (Gelman et al. 1996, 2004). These can be very easily computed from the MCMC output.
RESULTS
The familiar parameterization in a two-trait mixed-model analysis is based on model SMMupe. We therefore show in Table 1 Monte Carlo estimates of posterior means and standard deviations for chosen parameters based on the SMMupe for Landrace and Yorkshire. Due to the symmetry of all the posterior distributions referred to below, standard deviations rather than posterior intervals are reported. The numbers in the table indicate that there is a striking difference between the breeds, especially for the size and the sign of the correlation coefficients. For Landrace, a value in the vicinity of zero for all three correlation coefficients is in an area of high probability mass. For Yorkshire, only for the environmental correlation is the value of zero excluded in the 95% posterior interval.
Table 2 shows Monte Carlo estimates of posterior means and standard deviations for chosen parameters based on the RMMupe parameterization for Landrace and Yorkshire. There is a one-to-one relation between the parameters of the RMMupe and those of the SMMupe. The conclusions based on the recursive parameters are the same as those based on the correlation coefficients from Table 1.
Figures 1 and 2 show the posterior predictive distribution of discrepancies bu − 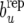 , bp −
, bp − 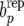 , and be −
, and be −  for Landrace and Yorkshire generated under RMM0. For Landrace, the Monte Carlo estimates of the posterior means (posterior standard deviations) for the three discrepancy measures are 0.067 (0.280), 0.019 (0.020), and −0.000 (0.003), reflecting lack of recursion at all levels. There is therefore lack of evidence suggesting that there is conflict between the data and the null model RMM0, with respect to the feature described by the discrepancy measure. For Yorkshire, the corresponding numbers are −0.057 (0.052), −0.030 (0.019), and −0.034 (0.002), supporting recursion at the level of the residual term only, a feature of the data that the null model fails to account for.
for Landrace and Yorkshire generated under RMM0. For Landrace, the Monte Carlo estimates of the posterior means (posterior standard deviations) for the three discrepancy measures are 0.067 (0.280), 0.019 (0.020), and −0.000 (0.003), reflecting lack of recursion at all levels. There is therefore lack of evidence suggesting that there is conflict between the data and the null model RMM0, with respect to the feature described by the discrepancy measure. For Yorkshire, the corresponding numbers are −0.057 (0.052), −0.030 (0.019), and −0.034 (0.002), supporting recursion at the level of the residual term only, a feature of the data that the null model fails to account for.
Figure 1.—



(Landrace) Estimates of posterior distributions (under RMM0) of discrepancies 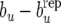 (left),
(left), 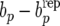 (center), and
(center), and 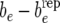 (right).
(right).
Figure 2.—



(Yorkshire) Estimates of posterior distributions (under RMM0) of discrepancies 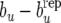 (left),
(left), 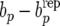 (center), and
(center), and 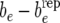 (right).
(right).
DISCUSSION
In a recent article, Gianola and Sorensen (2004) discussed the use of simultaneous equation models to analyze and interpret systems of traits that may be subject to feedback and recursive relationships. Here we report an application of a recursive mixed model for the analysis of litter size and average piglet weight in two breeds of Danish pigs. The recursive relationship defined by model (12) establishes that average piglet weight is linearly related to the deviation of litter size from its group mean. The traditional specification, like that in Gianola and Sorensen (2004), postulates that average piglet weight is linearly related to litter size, rather than to its deviation from the mean. The system defined by (12) is free of some identifiability problems at the level of parameters entering the mean that are common to both traits. It seems also appealing that deviations from a midvalue, rather then absolute values, exert an influence on average piglet weight. Ultimately, these are two different models and a way of discerning between them is by computing their posterior probabilities, in the light of the data. This was not studied in the present work.
The structure of the residual dispersion matrix (5) has three parameters and was arrived at assuming that the covariance between residuals for single weight measurements is  . This leads to conditionally independent residuals, given litter size. A more general model would assume that the above covariance is
. This leads to conditionally independent residuals, given litter size. A more general model would assume that the above covariance is  , where
, where 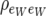 is the residual correlation between single weight measurements. Then residuals are no longer conditionally independent, given litter size. The resulting residual covariance matrix between litter size and average litter weight has four identifiable parameters and retrieves (5) when
is the residual correlation between single weight measurements. Then residuals are no longer conditionally independent, given litter size. The resulting residual covariance matrix between litter size and average litter weight has four identifiable parameters and retrieves (5) when 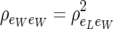 . This model has also its recursive parameterization counterpart.
. This model has also its recursive parameterization counterpart.
The saturated recursive model used in this work has nine identifiable dispersion parameters. A more parsimonious alternative with seven parameters postulates that the three recursive parameters λu, λp, and λe are equal. In general, the recursive parameterization can be an attractive approach to arrive at parsimonious models, especially in analyses involving many traits.
Special attention has been given here to identifiability at the level of the likelihood, despite the fact that inferences were based on posterior distributions. In principle, a Bayesian analysis with a nonidentifiable likelihood is possible if proper prior distributions are specified for all the parameters (Bernardo and Smith 1994). In fact, depending on the prior distributions, a Bayesian analysis with a nonidentifiable likelihood may result in Bayesian learning, in the sense that the posterior and prior distributions of the nonidentified parameters are different (see, for example, Sorensen and Gianola 2002, p. 543). However, an MCMC implementation of a Bayesian model with “barely” identified parameters can lead to poor inferences due to extremely slow convergence and very short effective chain lengths. Achieving identifiability of parameters at the level of the likelihood will always lead to Bayesian learning and in general to better behavior of the MCMC algorithm. However, there may be situations where the constraints needed for identifiability may restrict inferences, and an unconstrained model using a careful prior specification could be considered instead.
The analyses of Yorkshire and Landrace data lead to markedly different inferences; we are not disturbed by this result. The breeds are distinct in various behavioral, physiological, and anatomical traits, as well as in outward appearance. From a breeding point of view, in Landrace, changes in litter size should not lead to associated changes in average litter weight. In Yorkshire, a change in environmental deviation of litter size of 1 unit (for example, due to culling) should result in a temporary reduction of average piglet weight of 36 g. In neither breed should successful selection for litter size have a direct effect on average piglet weight.
There is a rich literature dealing with various transformations of the data or reparameterizations that can lead to computationally more tractable analyses of the multivariate linear model (for example, Meyer 1987; Jensen and Mao 1988; Quaas 1988; Ducrocq and Besbes 1993; Groeneveld 1994; Gelfand et al. 1995; Thompson et al. 1995; Ducrocq and Chapuis 1997). While the recursive model can be viewed in this framework, the focus of the present work is that a recursive model whose likelihood is identifiable is an alternative parameterization of a standard mixed model. The two models provide different interpretations of the results, but are statistically equivalent. There is a one-to-one relationship between the parameters entering the likelihood in both models. This applies also in principle to simultaneous equation models, which in general require a larger number of constraints to achieve identifiability. However, it may not always be easy to define the equivalent standard model, say, to a model involving complex simultaneous and recursive relationships among many traits. Ultimately, the choice of parameterization should be guided by the availability of software (in simple situations like in the present work), by ease of interpretation, or by the need to test a particular theory or hypothesis. The mathematical formulation of such a hypothesis may be more naturally accomplished using one parameterization and not the other.
Acknowledgments
We are grateful to Gustavo de los Campos and Daniel Gianola for discussions and comments on an earlier version of this manuscript.
References
- Bernardo, J. M., and A. F. M. Smith, 1994. Bayesian Theory. Wiley, New York.
- Blasco, A., J. P. Bidanel and C. Haley, 1995. Genetics and neonatal survival, pp. 17–38 in The Neonatal Pig. Development and Survival, edited by M. A. Varley. CAB International, Wallingford, Oxon, UK.
- Cheverud, J. M., 1984. Quantitative genetics and developmental constraints on evolution by selection. J. Theor. Biol. 110: 155–171. [DOI] [PubMed] [Google Scholar]
- de los Campos, G., D. Gianola, P. Boettcher and P. Moroni, 2006. A structural equation model for describing relationships between somatic cell score and milk yield in dairy goats. J. Anim. Sci. 84: 2934–2941. [DOI] [PubMed] [Google Scholar]
- Ducrocq, V., and B. Besbes, 1993. Solution of multiple trait animal models with missing data on some traits. J. Anim. Breed. Genet. 110: 81–92. [DOI] [PubMed] [Google Scholar]
- Ducrocq, V., and H. Chapuis, 1997. Generalising the use of the canonical transformation for the solution of multivariate mixed model equations. Genet. Sel. Evol. 29: 205–224. [Google Scholar]
- Duncan, O. D., 1975. Introduction to Structural Equation Models. Academic Press, San Diego.
- Gelfand, A. E., S. K. Sahu and B. P. Carlin, 1995. Efficient parameterization for normal linear mixed models. Biometrika 82: 479–488. [Google Scholar]
- Gelman, A., X. L. Meng and H. Stern, 1996. Posterior predictive assessment of model fitness via realized discrepancies (with discussion). Stat. Sin. 6: 733–807. [Google Scholar]
- Gelman, A., J. B. Carlin, H. S. Stern and D. B. Rubin, 2004. Bayesian Data Analysis. Chapman & Hall, London/New York.
- Gianola, D., and D. Sorensen, 2004. Quantitative genetic models describing simultaneous and recursive relationships between phenotypes. Genetics 167: 1407–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberger, A. S., 1972. Structural equation methods in the social sciences. Econometrica 40: 979–1001. [Google Scholar]
- Grandinson, K., M. S. Lund, L. Rydhmer and E. Strandberg, 2002. Genetic parameters for piglet mortality traits crushing, stillbirth and total mortality, and their relation to birth weight. Acta Agric. Scand. Ser. A. Anim. Sci. 52: 167–173. [Google Scholar]
- Groeneveld, E., 1994. A reparameterization to improve numerical optimization in multivariate REML (co)variance component estimation. Genet. Sel. Evol. 26: 537–545. [Google Scholar]
- Henderson, C. R., 1984. Applications of Linear Models in Animal Breeding. University of Guelph, Guelph, ON, Canada.
- Jensen, J., and I. L. Mao, 1988. Transformation algorithms in analysis of single trait and of multiple trait models with equal design matrices and one random factor per trait: a review. J. Anim. Sci. 26: 2750–2761. [Google Scholar]
- Jöreskog, K. G., 1973. A general method for estimating a linear structural equation system, pp. 85–112 in Structural Equation Models in the Social Sciences, edited by A. S. Goldberger and O. D. Duncan. Seminar, New York.
- Kerr, J. C., and N. D. Cameron, 1995. Reproductive performance of pigs selected for components of efficient lean growth. Anim. Sci. 60: 281–290. [Google Scholar]
- Lande, R., 1979. Quantitative genetic analysis of multivariate evolution, applied to brain:body allometry. Evolution 33: 402–416. [DOI] [PubMed] [Google Scholar]
- Meyer, K., 1987. A note on the use of an equivalent model to account for relationships between animals in estimating variance components. J. Anim. Breed. Genet. 104: 163–168. [Google Scholar]
- Noguera, J. L., L. Varona, D. Babot and J. Estany, 2002. Multivariate analysis of litter size for multiple parities with production traits in pigs: II. Response to selection for litter size and correlated responses to production traits. J. Anim. Sci. 80: 2548–2555. [DOI] [PubMed] [Google Scholar]
- Quaas, R. L., 1988. Transformed mixed model equations: a recursive algorithm to eliminate A1. J. Dairy Sci. 72: 1937–1941. [Google Scholar]
- Roehe, R., 1999. Genetic determination of individual birthweight and its association with sows' productivity traits using Bayesian analysis. J. Anim. Sci. 77: 330–343. [DOI] [PubMed] [Google Scholar]
- Rothschild, M. F., and J. P. Bidanel, 1998. Biology and genetics of reproduction, pp. 313–343 in The Genetics of the Pig, edited by M. F. Rothschild and A. Ruvinsky. CAB International, Wallingford, Oxon, UK.
- Searle, S. R., 1971. Linear Models. Wiley, New York.
- Sorensen, D., and D. Gianola, 2002. Likelihood, Bayesian, and MCMC Methods in Quantitative Genetics. Springer-Verlag, Berlin/Heidelberg, Germany/New York.
- Sorensen, D., A. Vernersen and S. Andersen, 2000. Bayesian analysis of response to selection: a case study using litter size in Danish Yorkshire pigs. Genetics 156: 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner, M. A., and W. Wong, 1987. The calculation of posterior distributions by data augmentation. J. Am. Stat. Assoc. 82: 528–550. [Google Scholar]
- Thompson, R., R. E. Crump, J. Juga and P. M. Visscher, 1995. Estimating variances and covariances for bivariate animal models using scaling and transformation. Genet. Sel. Evol. 27: 33–42. [Google Scholar]
- Walsh, B., 2003. Evolutionary quantitative genetics, pp. 380–442 in Handbook of Statistical Genetics, Vol. I, edited by D. J. Balding, M. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Wright, S., 1921. Correlation and causation. J. Agric. Res. 210: 557–585. [Google Scholar]
- Xiong, M., J. Li and X. Fang, 2004. Identification of genetic networks. Genetics 166: 1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]