

Abstract
Epistasis seems to play a significant role in the manifestation of heterosis. However, the power of detecting epistatic interactions among quantitative trait loci (QTL) in segregating populations is low. We studied heterosis in Arabidopsis thaliana hybrid C24 × Col-0 by testing near-isogenic lines (NILs) and their triple testcross (TTC) progenies. Our objectives were to (i) provide the theoretical basis for estimating different types of genetic effects with this experimental design, (ii) determine the extent of heterosis for seven growth-related traits, (iii) map the underlying QTL, and (iv) determine their gene action. Two substitution libraries, each consisting of 28 NILs and covering ∼61 and 39% of the Arabidopsis genome, were assayed by 110 single-nucleotide polymorphism (SNP) markers. With our novel generation means approach 38 QTL were detected, many of which confirmed heterotic QTL detected previously in the same cross with TTC progenies of recombinant inbred lines. Furthermore, many of the QTL were common for different traits and in common with the 58 QTL detected by a method that compares triplets consisting of a NIL, its recurrent parent, and their F1 cross. While the latter approach revealed mostly (75%) overdominant QTL, the former approach allowed separation of dominance and epistasis by analyzing all materials simultaneously and yielded substantial positive additive × additive effects besides directional dominance. Positive epistatic effects reduced heterosis for growth-related traits in our materials.
HETEROSIS is a fundamental issue in plant breeding and designates the improved vigor of F1 hybrids in comparison with their parental homozygous lines. Ever since its discovery in the early 20th century (East 1908; Shull 1908), heterosis has been exploited systematically in breeding of crops and is considered a major asset to meeting world food needs (Duvick 1999). However, its genetic and molecular bases are still unknown. The most prevailing genetic theories include dominance (Davenport 1908; Bruce 1910; Jones 1917), overdominance (Hull 1945; Crow 1948), and epistasis (Powers 1944; Williams 1959).
Classical approaches of quantitative genetics to elucidate the genetic basis of heterosis include (i) generation means or diallel analyses and (ii) estimates of variance components reflecting different types of genetic effects (cf. Hallauer and Miranda 1981). Regarding the latter approach, design III proposed by Comstock and Robinson (1952) has been most widely used to estimate the average degree of dominance, but it ignores the effects of epistasis in estimates of additive and dominance variance components. The triple testcross (TTC) design (Kearsey and Jinks 1968) extends design III and provides a test for contribution of epistasis to heterosis. Nevertheless, none of these approaches yields detailed information about the different types of genetic effects involved in heterosis, because only genomewide estimates are obtained and/or different types of genetic effects are confounded.
Mapping of quantitative trait loci (QTL) with the approach devised by Lander and Botstein (1989) represented a major step forward to characterize the contribution of individual genomic regions to heterosis. While distinction between dominance and overdominance is straightforward unless closely linked loci mimic pseudo-overdominance, the power for investigating the role of epistasis by testing interactions between marker pairs in two-way analyses of variance (ANOVA) is low. Identification of individual QTL by a one-dimensional genome scan already implies testing a large number of possible positions for statistical significance (Kearsey 2002), but the problem of multiple tests increases in a quadratic manner when all pairs of marker loci are studied. Consequently, extremely high critical thresholds must be applied for each individual test to warrant a given genomewide type I error rate, which reduces the power of detecting epistatic interactions between QTL. Furthermore, when populations segregating for the entire genetic background are used, these complex interactions often mask the effects of individual loci (Semel et al. 2006).
Difficulties in defining specific heterotic phenotypes and individual loci that control them result predominantly from epistatic interactions among many segregating loci throughout the genome when F2, backcross, or recombinant inbred line (RIL) populations are used (Li et al. 2001; Luo et al. 2001). Genetic resolution of QTL mapping is increased when near-isogenic lines (NILs) (Eshed and Zamir 1995; Zamir 2001) are employed, because they segregate only for a single chromosomal region. Libraries of chromosome substitution lines have proven useful in fine mapping and cloning of QTL (Frary et al. 2000; Monforte and Tanksley 2000; Fridman et al. 2004) and have also been suggested to solve the problem of validating the presence of QTL identified in previous studies with segregating populations (Kearsey 2002). Recently, Semel et al. (2006) studied heterosis in a population of introgression lines of tomato, which carried single, marker-defined segments from a wild relative. On the basis of the comparison of each introgression line with the elite parent and their F1 cross, they concluded that heterosis for traits related to yield and reproductive fitness was mainly attributable to QTL with overdominant gene action.
Arabidopsis thaliana L. has emerged as the leading model species in plant genetics and functional genomics. It also holds great promise for investigating the genetic and molecular causes of heterosis due to the availability of well-developed genomics tools and the ease with which appropriate large mapping populations can be generated and manipulated. Recent studies demonstrated a substantial amount of heterosis for biomass yield and growth-related traits with a wide variation among individual hybrids (Barth et al. 2003; Meyer et al. 2004). First, QTL analyses on heterosis in cross Col × Ler were performed with testcross progenies of RILs using a TTC design (Kearsey et al. 2003) and design III (Syed and Chen 2005). A detailed analysis of heterosis for biomass-related traits in the cross C24 × Col-0 was conducted in a previous study (Kusterer et al. 2007a) and in an accompanying study (Kusterer et al. 2007b, this issue) with TTC progenies of RILs. Generation means analyses and estimates of variance components provided strong evidence for directional dominance and indicated an important role of digenic and/or higher-order epistatic effects for all biomass-related traits (Kusterer et al. 2007a). QTL mapping with these materials further indicated a polygenic basis of heterosis, with overdominance and/or additive × additive epistasis being the main types of gene action (Kusterer et al. 2007b).
The goals of this study were to investigate the genetic basis of heterosis for growth-related traits in Arabidopsis hybrid C24 × Col-0 by examining TTC progenies from a library of NILs. In detail, our objectives were to (i) provide the theoretical basis for estimating different types of genetic effects with this experimental design, (ii) determine the extent of heterosis for growth-related traits in NILs, (iii) map the underlying QTL, and (iv) estimate their gene action.
THEORY
Under the F2-metric (Cockerham 1954; Yang 2004) and a genetic model including additive × additive epistasis, the genotypic value of a genotype V = (v1, … , vq) can be expressed as
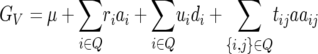 |
(1) |
(Melchinger et al. 2007, accompanying article, this issue), where μ is the mean of the F2 generation in linkage equilibrium produced from the cross of parents P1 and P2; ai is the additive effect of locus i (which is positive or negative depending on whether parent P2 or P1, respectively, carries the favorable allele at this locus); di is the dominance effect of locus i; aaij is the additive × additive effect between loci i and j; vi = 0, 1, or 2 if the genotype at QTL i is homozygogous P1, heterozygous, or homozygous P2, respectively; ri = vi − 1, ui = 2vi − vi2 − 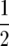 , and tij = (vi − 1)(vj − 1); and Q is the set of all QTL segregating for the trait in the cross P1 × P2.
, and tij = (vi − 1)(vj − 1); and Q is the set of all QTL segregating for the trait in the cross P1 × P2.
If we define the parameters 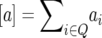 ,
, 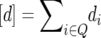 ,
, 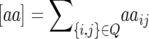 ,
,  and denote the cytoplasmic effect attributable to seed parent P1 vs. seed parent P2 by c, then we can express the generation means of (1) parents P1 and P2 and their F1 cross, (2) near-isogenic lines NIL1-i of P1 harboring exclusively the genomic region of locus i from parent P2 in the genetic background of parent P1, (3) near-isogenic lines NIL2-i of P2 harboring exclusively the genomic region of locus i from parent P1 in the genetic background of parent P2, and (4) triple testcross progenies (crosses with P1, P2, and the F1) of each NIL1-i or NIL2-i as
and denote the cytoplasmic effect attributable to seed parent P1 vs. seed parent P2 by c, then we can express the generation means of (1) parents P1 and P2 and their F1 cross, (2) near-isogenic lines NIL1-i of P1 harboring exclusively the genomic region of locus i from parent P2 in the genetic background of parent P1, (3) near-isogenic lines NIL2-i of P2 harboring exclusively the genomic region of locus i from parent P1 in the genetic background of parent P2, and (4) triple testcross progenies (crosses with P1, P2, and the F1) of each NIL1-i or NIL2-i as
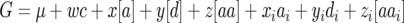 |
(2) |
with coefficients w, x, y, z, xi, yi, and zi as given in Table 1. Note that coefficients x, y, and z correspond exactly to the coefficients rj, uj, and tjk, respectively, for j ≠ i and k ≠ i, whereas coefficients xi, yi, and zi are obtained by subtracting values of x, y, and z from values of ri, ui, and tij, respectively.
TABLE 1.
Genetic expectations for the generation means of generations P1, P2, and F1 in both reciprocal forms (a, P1 × P2; b, P2 × P1), and NILs as well as their triple testcross progenies with testers P1, P2, and F1
| Genetic parametera
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Generationa | μ | c | [a] | [d] | [aa] | ai | di | [aai] |
| P1 | 1 | 1 | −1 | −0.5 | 1 | 0 | 0 | 0 |
| P2 | 1 | 0 | 1 | −0.5 | 1 | 0 | 0 | 0 |
| F1-a (P1 × P2) | 1 | 1 | 0 | 0.5 | 0 | 0 | 0 | 0 |
| F1-b (P2 × P1) | 1 | 0 | 0 | 0.5 | 0 | 0 | 0 | 0 |
| NIL1-i | 1 | 1 | −1 | −0.5 | 1 | 2 | 0 | −2 |
| P1 × NIL1-i | 1 | 1 | −1 | −0.5 | 1 | 1 | 1 | −1 |
| P2 × NIL1-i | 1 | 0 | 0 | 0.5 | 0 | 1 | −1 | 0 |
| F1-a × NIL1-i | 1 | 1 | −0.5 | 0 | 0.25 | 1 | 0 | −0.5 |
| NIL2-i | 1 | 0 | 1 | −0.5 | 1 | −2 | 0 | −2 |
| P1 × NIL2-i | 1 | 1 | 0 | 0.5 | 0 | −1 | −1 | 0 |
| P2 × NIL2-i | 1 | 0 | 1 | −0.5 | 1 | −1 | 1 | −1 |
| F1-b × NIL2-i | 1 | 0 | 0.5 | 0 | 0.25 | −1 | 0 | −0.5 |
| General coefficient | 1 | w | x | y | z | xi | yi | zi |
For a detailed explanation of the terminology and definition of genetic effects, see materials and methods.
MATERIALS AND METHODS
Plant materials:
Seeds from the Arabidopsis accessions C24 (provided by J. P. Hernalsteens, Vrije Universiteit Brussels, Belgium) and Col-0 (provided by G. Rédei, University of Missouri, Columbia, MO) were used to establish the plant materials employed in this study. The F2 generation was used to produce two substitution libraries of NILs (supplemental Table 1 at http://www.genetics.org/supplemental/), subsequently denoted as NIL1 and NIL2, by three to five generations of backcrossing, as described in detail by O. Törjék, R. C. Meyer, M. Zehnsdorf, M. Teltow, G. Strompen, H. Witucka-Wall, A. Blacha and A. Altman (unpublished results). Each NIL1-s (s = 1, 2, … , 28) derived from the cross C24 × Col-0 harbors exactly one chromosome segment from parent Col-0 in the genetic background of parent C24. Conversely, each NIL2-s (s = 1, 2, … , 28) harbors exactly one chromosome segment from parent C24 in the genetic background of parent Col-0. The presence and chromosomal position of the substituted segment in each NIL as well as absence of other chromosomal segments from the donor parent were examined by single-nucleotide polymorphism (SNP) analyses according to Törjék et al. (2003). A set of 110 SNPs chosen according to their physical distance in the Arabidopsis genome was used to achieve a uniform coverage of the entire Arabidopsis genome. The approximate length of each substituted segment was estimated by (1) determining the map distance between the two most distant markers carrying the marker alleles of the donor parent of the substituted segment and (2) adding half the distance of these markers to their adjacent markers carrying the marker genotype of the recurrent parent. The NILs were propagated via single-seed descent. To facilitate production of testcross seed, we also established male-sterile NILs of C24 and Col-0, subsequently referred to as P1 and P2, respectively, by crossing C24 and Col-0 with the male-sterile line N75 and recurrent backcrossing to parent C24 or Col-0 for six generations, accompanied by marker-assisted selection with SNPs for a rapid and efficient recovery of the recurrent parent genome. Subsequently, selfing and sibbing were performed for seed increase of the male-sterile NILs. The F1 generation was produced in two reciprocal forms: P1 × P2 (F1-a) and P2 × P1 (F1-b). In addition, testcross progenies were produced according to a TTC design (Kearsey and Jinks 1968) by mating each NIL1-s as pollen parent with P1, P2, and F1-a and each NIL2-s as pollen parent with P1, P2, and F1-b.
Experimental design:
The entire set of 56 NILs plus 16 NILs that harbored more than one chromosomal segment from the donor parent together with checks (included 12 times as entries on the main-plot level) were evaluated in a split-plot design with three replicates. Main plots were arranged in a 12 × 7 α-design (Patterson and Williams 1976). Each main plot comprised four entries: one NIL and its three testcross progenies produced by the TTC design. Likewise, each main plot of the checks also comprised four entries: parents P1 and P2, as well as the F1 generation in both reciprocal forms. In all instances, the entries within each main plot were randomly assigned to the subplots. Each subplot consisted of 10 plants per entry.
Plant cultivation:
Seeds were sown under sterile conditions on Murashige–Skoog medium. At the two-leaf stage, ∼10 days after sowing (DAS), each seedling was transferred to sterilized soil (Euflor GmbH: 90% peat, 7% perlite, and 3 vol% sand at pH 5–6; salt content <1.5 g/liter; nitrogen availability <300 mg/liter N; phosphate availability <300 mg/liter P2O5; and calcium oxide availability <400 mg/liter K2O). Plants in soil were irrigated with tap water. Standard light and temperature regime under greenhouse conditions was 16 hr light (20,000 lux) at 21° and 8 hr dark at 18°.
Traits measured:
Rosette diameter (RD) (in millimeters) was recorded on individual plants 22 days (RD22) and 29 days (RD29) after sowing. Leaf area (LA) was determined also on individual plants from digital images taken at 22 days (LA22) and 29 days (LA29) after sowing by using the software BARABI (L. Mauch and S. Mauch, unpublished data). The absolute growth rate per day (GR) (in millimeters per day) was determined as (RD29 − RD22)/7 to allow a direct comparison with results from our previous study with a generation means analysis of the same cross (Kusterer et al. 2007a). All plants of a subplot were harvested without the root system at 29 DAS and bulked into a plastic jar. Biomass yield (BY) (in milligrams) was recorded after drying in an oven to practically 0% moisture content. Dry matter content (DMC) (in percentage) was calculated as the ratio between dry and fresh biomass, multiplied by 100.
Statistical analyses:
Means and standard errors of all generations were calculated as best linear unbiased estimates in a mixed-model analysis of the split-plot experiment with SAS PROC MIXED (SAS Institute 2004). Midparent heterosis was calculated as MPH = 100 × 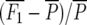 , where
, where 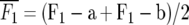 and
and 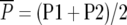 , and corresponding standard errors were determined according to Mood et al. (1974).
, and corresponding standard errors were determined according to Mood et al. (1974).
All further analyses were based on the subset of main-plot entries of the 56 NILs, which harbored one chromosome segment from the donor parent. The error variances for the main and subplots were calculated as combined estimates over all types of progenies. Heritability (h2) on an entry-mean basis was estimated as 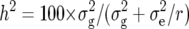 , where
, where 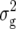 corresponds to the genetic variance pooled across all types of progenies (NIL1, NIL2, and each series of TTC progenies),
corresponds to the genetic variance pooled across all types of progenies (NIL1, NIL2, and each series of TTC progenies), 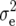 = main-plot error + subplot error, and r corresponds to the number of replicates. In addition, we calculated the coefficient of variation (CV) by
= main-plot error + subplot error, and r corresponds to the number of replicates. In addition, we calculated the coefficient of variation (CV) by 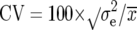 , where
, where 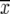 is the overall mean of all entries. Phenotypic (rp) and genotypic (rg) correlations among traits were calculated from estimates of variances and covariances pooled over all types of progenies. Computations were performed with adjusted entry means, using the software PLABSTAT (Utz 2000).
is the overall mean of all entries. Phenotypic (rp) and genotypic (rg) correlations among traits were calculated from estimates of variances and covariances pooled over all types of progenies. Computations were performed with adjusted entry means, using the software PLABSTAT (Utz 2000).
Generation means analysis:
Observations of generations P1, P2, F1-a, F1-b, NIL1-s, NIL2-s, P1 × NIL1-s, P1 × NIL2-s, P2 × NIL1-s, P2 × NIL2-s, F1-a × NIL1-s, and F1-b × NIL2-s were used to estimate the genetic parameters in a mixed model with replicates, main plots, and subplots as random effects and all other factors as fixed effects chosen according to the genetic model given in Equation 2. Model 1 included cytoplasmic (c), additive ([a]), and dominance ([d]) effects, as well as the additive (as) and dominance (ds.) effects of each NIL: G = μ + wc + x[a] + y[d] + xsas + ysds. Model 2 additionally included epistatic effects ([aa], [aas]): G = μ + wc + x[a] + y[d] + z[aa] + xsas + ysds + zs[aas]. The lack-of-fit of each model was tested with a 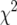 -test statistic, which corresponds approximately to a Wald F-test in the context of a mixed linear model, with denominator degrees of freedom approximated by the method of Kenward and Roger (1997). The genetic parameters in each model were estimated as a solution of the normal equations
-test statistic, which corresponds approximately to a Wald F-test in the context of a mixed linear model, with denominator degrees of freedom approximated by the method of Kenward and Roger (1997). The genetic parameters in each model were estimated as a solution of the normal equations 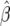 = (X′VX)−1 X′VY, where
= (X′VX)−1 X′VY, where 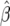 is the column vector of estimated genetic parameters, X is the design matrix with elements determined by the genetic model, V is the variance–covariance matrix of Y, and Y is the vector of original phenotypic observations. The variance–covariance matrix V was replaced by its restricted maximum-likelihood estimate. Standard errors of the genetic parameter estimates were calculated as the square root of the diagonal elements of matrix (X′VX)−1. Significance tests of the genetic effects were performed with a Wald t-test; furthermore, we used a sequential Bonferroni correction of P-values according to Holm (1979). Significance tests for means of as, ds, and [aas] across all NILs were also performed with a Wald t-test, but without a Bonferroni correction.
is the column vector of estimated genetic parameters, X is the design matrix with elements determined by the genetic model, V is the variance–covariance matrix of Y, and Y is the vector of original phenotypic observations. The variance–covariance matrix V was replaced by its restricted maximum-likelihood estimate. Standard errors of the genetic parameter estimates were calculated as the square root of the diagonal elements of matrix (X′VX)−1. Significance tests of the genetic effects were performed with a Wald t-test; furthermore, we used a sequential Bonferroni correction of P-values according to Holm (1979). Significance tests for means of as, ds, and [aas] across all NILs were also performed with a Wald t-test, but without a Bonferroni correction.
Qualitative mode-of-inheritance classification of QTL:
In addition to the generation means analysis of NILs described in the previous section, we performed QTL mapping and a classification of the mode-of-inheritance according to Semel et al. (2006). The method is described here for NIL1 with the same principles applying analogously to NIL2. Each NIL1-s and P1 × NIL1-s was compared by a t-test to parent P1 as well as to each other. If either of them was significantly different from P1, the corresponding NIL was considered as harboring a QTL. Because the number of replications for the NIL1-s and the P1 × NIL1-s was identical but the number of replications of P1 was 12 times higher, the comparison of NIL1-s and P1 × NIL1-s with P1 was tested at a significance level of P < 0.01, whereas the comparison of NIL1-s with P1 × NIL1-s was tested at P < 0.05, adopting the procedure of Semel et al. (2006).
Classification of the qualitative mode-of-inheritance of a QTL was based on its phenotypic effect, which was considered to be the effect of the significant genotype (NIL1-s or P1 × NIL1-s) relative to P1 (positive values for increasing QTL, in which that of the substitution line was higher than that of P1, and negative values for decreasing ones). If both the NIL1-s and the P1 × NIL1-s had a significant effect in the same direction, the higher value was considered the QTL phenotypic effect. If both the NIL1-s and the P1 × NIL1-s were significant but in opposite directions relative to P1, the substitution line was considered as harboring two QTL: one is increasing, and the other is decreasing. The mode-of-inheritance of a QTL was determined according to the decision tree given by Semel et al. (2006, Figure 4). If NIL1-s was significantly different from P1 and the P1 × NIL1-s phenotype was in between NIL1-s and P1, we distinguished three cases: (i) if P1 × NIL1-s was significantly different from NIL1-s but not from P1, the introgressed QTL allele was considered recessive; (ii) if P1 × NIL1-s differed from both parents or did not differ from either of them, the QTL effect was considered additive; and (iii) if P1 × NIL1-s differed from P1 but not from NIL-s, the introgressed QTL allele was classified as dominant. In the fourth case, if P1 × NIL1-s was significantly higher or lower in value than both its parents, the QTL was classified as overdominant.
RESULTS
Generation means:
Means of parents P1 and P2 were not significantly (P < 0.05) different from each other for all growth-related traits except DMC and BY, for which P2 slightly surpassed P1 (Table 2). Significant (P < 0.05) reciprocal differences were observed in the F1 crosses, with P1 × P2 exceeding P2 × P1 by 6–29% in all traits but DMC. MPH was lowest (15%) for RD22, intermediate (26%) for BY, and highest (33%) for LA29. An exception was DMC, which showed only marginal negative MPH.
TABLE 2.
Means and associated standard errors in generations P1, P2, F1 and NILs and their triple testcross progenies with testers P1, P2, and F1 as well as midparent heterosis (MPH), heritability (h2), and coefficient of variation (CV) for growth-related traits in Arabidopsis hybrid C24 × Col-0
| Generations | Na | RD22 (mm) | RD29 (mm) | GR (mm/day) | LA22 (mm2) | LA29 (mm2) | DMC (%) | BY (mg) |
|---|---|---|---|---|---|---|---|---|
| Checks | ||||||||
| P1 | 12 | 38.18 (a) | 79.38 (a, b) | 5.89 (b, c) | 507.2 (a, b) | 2405 (a, b) | 7.02 (a, b) | 54.53 (a, b) |
| P2 | 12 | 39.34 (a, b) | 81.89 (b, c) | 6.08 (c) | 497.4 (a, b) | 2393 (a, b) | 7.48 (d, e) | 59.54 (c, d, e) |
| P1 × P2 | 12 | 46.94 (e) | 99.60 (e) | 7.52 (f) | 701.6 (f) | 3435 (e) | 6.94 (a, b) | 80.70 (g) |
| P2 × P1 | 12 | 42.02 (c, d) | 91.81 (d) | 7.11 (e) | 551.8 (c, d) | 2925 (c) | 6.91 (a) | 62.46 (c, d, e) |
| SE | 0.81 | 1.77 | 0.19 | 19.0 | 98 | 0.08 | 2.51 | |
| MPH (%)b | 14.8 ± 0.6 | 18.7 ± 0.4 | 22.3 ± 1.9 | 24.8 ± 0.2 | 32.5 ± 0.1 | −4.4 ± 1.2 | 25.5 ± 0.7 | |
| Entries | ||||||||
| NIL1 | 28 | 39.00 (a, b) | 76.23 (a) | 5.32 (a) | 517.4 (a, c) | 2301 (a) | 6.90 (a) | 53.42 (a) |
| P1 × NIL1 | 28 | 40.37 (b, c) | 80.37 (b) | 5.72 (b) | 556.3 (d) | 2492 (b) | 7.10 (b) | 59.18 (b, c) |
| P2 × NIL1 | 28 | 46.41 (e) | 96.59 (e) | 7.17 (f, e) | 673.9 (f) | 3207 (d) | 6.91 (a) | 72.32 (f) |
| F1 × NIL1 | 28 | 42.91 (d) | 88.89 (d) | 6.57 (d) | 599.7 (e) | 2849 (c) | 6.90 (a) | 63.94 (d, e) |
| NIL2 | 28 | 39.54 (a, b) | 82.24 (b, c) | 6.10 (c) | 490.3 (a) | 2487 (b) | 7.34 (c, d) | 59.34 (b, d) |
| P1 × NIL2 | 28 | 51.39 (f) | 103.48 (f) | 7.44 (f, e) | 830.9 (g) | 3792 (f) | 6.94 (a, b) | 91.37 (h) |
| P2 × NIL2 | 28 | 41.48 (c, d) | 84.36 (c) | 6.13 (c) | 540.6 (b, c, d) | 2506 (b) | 7.47 (e) | 64.75 (e) |
| F1 × NIL2 | 28 | 46.22 (e) | 89.89 (d) | 6.24 (c) | 675.5 (f) | 2924 (c) | 7.28 (c) | 75.84 (f, g) |
| SE | 0.72 | 1.50 | 0.15 | 17.7 | 84 | 0.06 | 2.17 | |
| h2 (%)c | 0.83 ± 0.04 | 0.78 ± 0.05 | 0.68 ± 0.07 | 0.83 ± 0.03 | 0.78 ± 0.05 | 0.66 ± 0.07 | 0.75 ± 0.05 | |
| CV (%) | 9.4 | 10.0 | 14.1 | 16.1 | 17.4 | 5.9 | 18.5 | |
For a description of growth-related traits see materials and methods. Means followed by the same letter are not significantly different at P < 0.05 based on pairwise comparisons (t-test) using the insert-and-absorb algorithm with sweeping.
The number of entries.
MPH (%) = 100 × 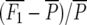 , where
, where 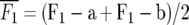 and
and 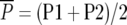 .
.
h2 (%) refers to the heritability on an NIL-entry-mean basis by using pooled estimates of variance components across the NILs and their triple testcross progenies.
The means of NIL1 and NIL2 did not differ significantly (P < 0.05) from those of their respective recurrent parent P1 or P2 with a single exception (Table 2). The mean of crosses P1 × NIL1 significantly (P < 0.05) exceeded the mean of NIL1 in all traits except RD22 but surpassed the mean of parent P1 only in RD22 and LA22. Similarly, the mean of crosses P2 × NIL2 outyielded the mean of NIL2 in most traits, surpassing parent P2 only in RD22. Interestingly, the mean of P2 × NIL1 exceeded significantly (P < 0.05) that of P2 × P1 by 5–20% in all traits except GR and DMC. Likewise, the mean of P1 × NIL2 surpassed that of P1 × P2 by ∼10% in all traits except GR and DMC. As expected in the absence of epistasis, the mean of F1-a × NIL1 did not differ significantly from the overall mean of P1 × NIL1 and P2 × NIL1. In contrast, the mean of F1-b × NIL2 was significantly smaller (P < 0.05) than the overall mean of P1 × NIL2 and P2 × NIL2 in RD29 and GR. Means of individual NILs for all growth-related traits are given in supplemental Table 2 (http://www.genetics.org/supplemental/).
Heritabilities, CVs, and trait correlations:
Heritability (h2) ranged between 66% for GR and 83% for RD22 and LA22 (Table 2). The CV was highest (18%) for BY and lowest (6%) for DMC. Phenotypic correlations (rp) among RD22, RD29, LA22, LA29, and BY were positive and high, ranging from 0.77 to 0.97 (supplemental Table 3 at http://www.genetics.org/supplemental/). Genotypic correlations (rg) were slightly greater than corresponding estimates of phenotypic correlations.
Generation means analysis:
Model 1 (without epistasis) yielded a poorer fit to the observed data than model 2 (with epistasis) based on 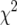 -tests (Table 3). Hence, presentation of the results in Table 3 is restricted to model 2. Nevertheless, significant (P < 0.01) deviations from model 2 were found for all traits except GR (Table 3).
-tests (Table 3). Hence, presentation of the results in Table 3 is restricted to model 2. Nevertheless, significant (P < 0.01) deviations from model 2 were found for all traits except GR (Table 3).
TABLE 3.
Genetic effects for growth-related traits in Arabidopsis thaliana hybrid C24 × Col-0 estimated by the generation-means analysis described in the theory section
| Parametera | RD22 (mm) | RD29 (mm) | GR (mm/day) | LA22 (mm2) | LA29 (mm2) | DMC (%) | BY (mg) | |
|---|---|---|---|---|---|---|---|---|
| μ | 40.04 ± 0.90** | 81.66 ± 1.92** | 5.95 ± 0.20** | 511.1 ± 21.7** | 2395 ± 107** | 7.11 ± 0.09** | 56.59 ± 2.70** | |
| c | 1.77 ± 0.50 | 5.75 ± 0.93** | 0.57 ± 0.09** | 55.4 ± 12.5** | 336 ± 55** | −0.10 ± 0.05 | 7.39 ± 1.33** | |
| [a] | 1.47 ± 0.47 | 4.13 ± 0.88** | 0.38 ± 0.09** | 22.8 ± 11.8 | 162 ± 51 | 0.18 ± 0.05* | 6.20 ± 1.25** | |
| [d] | 7.11 ± 1.41** | 22.33 ± 2.60** | 2.17 ± 0.26** | 175.8 ± 35.1** | 1233 ± 153** | −0.26 ± 0.13 | 22.60 ± 3.73** | |
| [aa] | 1.39 ± 1.29 | 7.26 ± 2.38 | 0.84 ± 0.24 | 51.4 ± 32.1 | 452 ± 140 | 0.06 ± 0.12 | 8.05 ± 3.41 | |
| as | Mean | −2.40 ± 0.49** | −0.87 ± 1.04 | 0.22 ± 0.11* | −72.2 ± 11.9** | −128 ± 58* | −0.05 ± 0.05 | −8.23 ± 1.49** |
| ds | Mean | 1.91 ± 0.42** | 2.43 ± 0.87** | 0.07 ± 0.09 | 45.4 ± 10.3** | 103 ± 49* | 0.10 ± 0.04* | 5.26 ± 1.25** |
| [aas] | Mean | 6.07 ± 0.76**,c | 7.46 ± 1.60** | 0.20 ± 0.17 | 170.3 ± 18.3** | 425 ± 89** | 0.17 ± 0.08* | 15.86 ± 2.30** |
| ds/|as|b | Mean | 0.49 | — | — | 0.41 | 0.55 | 0.82 | 0.38 |
| [aas]/|as|c | Mean | 0.99 | 1.11 | — | 1.01 | 1.05 | 1.31 | 1.01 |
| Total no. QTLd | 10 | 1 | 0 | 12 | 5 | 3 | 7 | |
| as | No. pos.e | 1 | 1 | 1 | 1 | |||
| as | No. neg. | 7 | 10 | 2 | 1 | 5 | ||
| ds | No. pos. | 3 | 2 | 1 | 3 | 1 | ||
| [aas] | No. pos. | 8 | 1 | 12 | 4 | 3 | 7 | |
| Model with [aa], [aas] | ||||||||
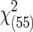 |
130.36** | 88.86** | 67.28 | 142.78** | 99.61** | 87.57** | 153.96** | |
| Model without [aa], [aas] | ||||||||
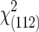 |
288.87** | 182.71** | 160.87** | 373.12** | 229.93** | 167.64** | 349.87** | |
For a description of growth-related traits see materials and methods. *P < 0.05, **P < 0.01.
For a detailed explanation of the terminology and definition of genetic effects, see materials and methods.
Mean of the ratio ds/|as| for NIL1-s and NIL2-s with a significant genetic effect (as and/or ds).
Mean of the ratio [aas]/|as| for NIL1-s and NIL2-s with a significant genetic effect (as and/or [aas]).
Total number of NILs with a significant genetic effect (as and/or ds and/or [aas]).
Number of NILs with positive or negative effects for as, ds, or [aas].
Cytoplasmic effects (c) were significant (P < 0.01) for all traits except RD22 and DMC and exceeded 10% of the mean μ for LA22, LA29, and BY (Table 3). Estimates of the additive effect [a] were significantly (P < 0.01) greater than zero only for RD29, GR, and BY. As expected, estimates of the dominance effect [d] were significantly (P < 0.01) positive for all traits but DMC and ranged between 18% (RD22) and 51% (LA29) of μ. Estimates of epistatic effects [aa] were always positive but not significantly (P < 0.05) different from zero for any trait.
The mean of as effects across NIL1 and NIL2 was negative for all traits but GR and highly significant (P < 0.01) only for RD22, LA22, and BY (Table 3). In contrast, the corresponding means of ds and [aas] were consistently positive and significant (P < 0.05) for all traits but GR. Means of [aas] were about three to four times larger than those of ds except for DMC. Likewise, the ratio of [aas]/|as| was on average much larger than the ratio of ds/|as| for NILs with a significant QTL.
The number of NILs with a significant QTL effect (as and/or ds and/or [aas] significant according to a t-test with P < 0.05 and sequential Bonferroni correction) was largest (12 and 10) for LA22 and RD22, intermediate (7 and 5) for BY and LA29, and smallest (≤3) for DMC, RD29, and GR (Table 3). The majority (74%) of significant QTL was observed for NIL2. Significant estimates of as predominantly had a negative sign, whereas significant estimates of ds and [aas] were consistently positive for all traits. The largest number of significant estimates across all traits was observed for [aas] and as, with many of the NILs with significant QTL showing both a positive estimate of [aas] and a negative estimate of as.
Qualitative mode-of-inheritance classification of QTL:
The number of QTL detected according to the method of Semel et al. (2006) ranged from 2 for GR to 15 for LA22 (Figure 1). Most (71%) of the detected QTL had a positive effect. Three-quarters of the 56 detected QTL showed overdominant gene action and 20% showed recessive gene action. Many of the detected QTL were in common among traits and in common with QTL detected by the generation-means approach (supplemental Table 4 at http://www.genetics.org/supplemental/).
Figure 1.—

Distribution of QTL mode-of-inheritance for growth-related traits in Arabidopsis thaliana hybrid C24 × Col-0. Each vertical bar represents the number of QTL for a specific trait, labeled according to the mode-of-inheritance categories (Semel et al. 2006): R, recessive; A, additive; D, dominant; and O, overdominant. The bars above the 0 line represent the number of increasing QTL, whereas the negative bars represent the number of decreasing QTL relative to P1 for NIL1-s or P2 for NIL2-s.
DISCUSSION
Better-parent heterosis vs. midparent heterosis:
In many autogamous crops such as wheat, rice, or tomato, better-parent heterosis (BPH) is of interest from an agronomic and a breeding point of view, because its amount relative to the performance of the better parent is crucial for the decision of whether or not to embark on a hybrid breeding program. In most allogamous crops such as maize, this question is obsolete because hybrids outperform lines by far. If one is interested in the genetic causes of heterosis, following Shull (1908) it is most plausible to compare the hybrid with the average performance of its parental inbreds (and not only the better parent), because the F1 inherited half of its nuclear genome from each parent. On the basis of quantitative genetic theory (Melchinger et al. 2007), MPH can be expressed as the sum of augmented dominance effects di* across the set of QTL Q, whereas BPH corresponds to the sum of (di* − ai*) across Q, where ai* denotes the augmented additive effect at QTL i. Consequently, the genetic causes of BPH are more complex than those of MPH and include the latter. For these reasons, we focused our analyses on MPH in contrast to Semel et al. (2006), who considered BPH.
Midparent heterosis and genomewide genetic effects:
In the present study, the level of MPH was consistently smaller and that for BY only half the size of that reported in our previous article using the same cross C24 × Col-0 of Arabidopsis (Kusterer et al. 2007a). A possible explanation for this discrepancy is that environmental conditions for plant growth, particularly during the early stages of plant development, were more favorable in the present study, as reflected by larger values for RD22 and RD29 of the common checks. For crops such as barley (Einfeldt et al. 2005) and sorghum (Haussmann et al. 1998), it is well known that MPH is higher under stress conditions than under favorable environmental conditions for plant growth. Interestingly, MPH was larger for LA22 and LA29 than for RD22 and RD29. This supports the hypothesis that complex traits generally display higher MPH than their component traits (Williams 1959).
The cytoplasmic effect c, which in our study reflects mainly the difference between reciprocal F1 crosses, was highly significant (P < 0.01) for most traits and in favor of the cytoplasm from parent P1. By comparison, significant reciprocal differences in the same direction as reported for RD22, RD29, and GR were found for the F1 cross in our previous study (Kusterer et al. 2007a). However, when other generations were also included, no significant cytoplasmic effects were observed. This may suggest an interaction between the cytoplasm and nuclear loci, depending on the level of heterozygosity of the nuclear genotype. Nevertheless, maternal effects (e.g., genetic imprinting) influencing seed quality in the seed parent could be alternative explanations (Stokes et al. 2007).
Our estimates of genomewide genetic effects ([a], [d], [aa]) were in close agreement with those from generation means analysis in our previous study (Kusterer et al. 2007a), where seven generations with various degrees of inbreeding were evaluated. Estimates of [a] from both studies agreed in sign but partly differed in size. In both studies, estimates of [d] showed by far the largest values and were significantly positive except for DMC. Likewise, [aa] effects were mostly positive but nonsignificant with a single exception. This is in agreement with results of Kearsey et al. (2003) from the cross Col × Ler, where significant [aa] effects were observed for only one of 22 traits. Nevertheless, it is not possible to draw conclusions about the predominant type of gene action at QTL on the basis of these estimates, because they reflect only net effects over the entire genome and QTL effects with positive and negative sign may cancel in the sum. Thus, QTL mapping is necessary for drawing inferences about the type of gene action prevailing in individual genomic regions.
Characterization of the NIL libraries:
We used two chromosome segment substitution libraries, NIL1 and NIL2, each consisting of 28 NILs. The length of the substitution chromosome segments ranged between 1 and 33.5 cM (supplemental Table 1 at http://www.genetics.org/supplemental/). In total, the substituted chromosome segments covered 261 cM in NIL1 and 165 cM in NIL2. Thus, only ∼61 and 39% of the Arabidopsis genome was covered by the substituted segments. Large gaps in each library existed on chromosomes 3, 4, and 5. Since some of the substituted segments overlapped, an estimated proportion of 18% in NIL1 and 19% in NIL2 of the Arabidopsis genome was represented by more than one NIL in each library. These gaps and overlapping regions must be kept in mind when interpreting the QTL results from our study.
Consistency of QTL across both NIL libraries and traits:
Of the 9 QTL detected for the seven traits in library NIL1 and 29 QTL detected in library NIL2, six regions overlapped (supplemental Table 1 at http://www.genetics.org/supplemental/), suggesting that we presumably mapped the same QTL twice. The comparison of QTL for different traits revealed that all 10 QTL detected for RD22 were also found for LA22 (supplemental Table 4 at http://www.genetics.org/supplemental/), as expected from the high correlation among these traits (supplemental Table 3 at http://www.genetics.org/supplemental/). However, only 1 of the 5 QTL detected for LA29 was also found for RD29, suggesting that these nonconsensus QTL hardly influenced leaf length but rather leaf width and/or leaf number. A high proportion of common QTL regions was also observed between LA22, LA29, and BY.
QTL mapping with NILs and RILs:
It is appealing to compare our results from QTL mapping with NILs with those from our companion study (Kusterer et al. 2007b), where RILs were used for QTL mapping to investigate the causes of heterosis. While both NILs and RILs represent immortalized homozygous genotypes, they do not provide exactly the same information on QTL positions and effects but are in many aspects complementary, as demonstrated in a recent study by Keurentjes et al. (2007). With NILs, inferences about the presence of QTL refer to the entire substituted chromosome segment of the corresponding NIL and not to exact positions. Only if two or more NILs have partly overlapping chromosome segments, is it possible to further narrow down the region and make more refined statements (Kearsey 2002; Keurentjes et al. 2007). Furthermore, no inferences about the number of QTL present in the substituted chromosome segment can be made. If a substituted chromosome segment harbors more than one QTL, chances of QTL detection are increased if QTL are in coupling phase, but reduced with QTL in repulsion phase. In the latter case, the corresponding NIL may show no significant difference from its recurrent parent and, hence, these QTL remain undetected.
By comparison, in composite interval mapping (CIM) with RILs one-dimensional scans are performed across the entire genome. Thus, in principle it is possible to make more refined statements about (i) the number of QTL present in a certain genomic region, provided the map resolution is sufficiently high, (ii) the position of the QTL, and (iii) their genetic effects. However, confidence intervals for point estimates of QTL positions (Manichaikul et al. 2006) and QTL effects (Utz et al. 2000) are typically rather wide unless the sample size is extremely large. In the present study, the average length of the substituted chromosome segments in libraries NIL1 and NIL2 amounted to 10.4 cM. This was about half the length of confidence intervals of QTL detected by CIM with RILs (Kusterer et al. 2007b; Kearsey et al. 2003), even though the sample size of RILs (N = 234, Kusterer et al. 2007b; N = 85, Kearsey et al. 2003) was much higher than the number of NILs per library (N = 28).
For all traits except RD29 and DMC, the number of QTL detected with NILs in our study surpassed substantially the number of QTL detected with RILs in our companion article (Kusterer et al. 2007b) and also in cross Col × Ler (Kearsey et al. 2003). This was unexpected because experimental expenditures with NILs (N = 168 progenies) were considerably smaller than those with RILs (N = 260, Kearsey et al. 2003; N = 702, Kusterer et al. 2007b). One reason was presumably the higher precision of the greenhouse experiment in the present study, as reflected by smaller CVs for all traits, especially RD22 and BY (data not shown). Another factor contributing to the higher power of QTL detection with NILs was the smaller number of multiple tests. With RILs, a total of 105 tests were performed with CIM, corresponding to the marker intervals between the 110 SNP markers scanned for the presence of QTL. The multiple-test problem with NILs relates to the number of lines evaluated in each NIL library, which was only 28. Moreover, for traits with truly polygenic inheritance, clusters of QTL with smaller effects in one region would most likely remain undetected in QTL mapping with CIM, because the LOD score of each QTL is below the critical threshold level. However, if these QTL happen to have identical sign and reside in the same substituted chromosome segment, chances are fairly high that the corresponding NIL will be significantly different from its recurrent parent. Furthermore, RIL populations have a lower power than NIL populations to detect small-effect QTL due to segregation of multiple QTL in the genetic background (Keurentjes et al. 2007). Altogether, we conclude that the potential of NILs in QTL mapping has been largely overlooked up to now and goes far beyond the validation of QTL detected by QTL analyses in segregating populations, as pointed out by Kearsey (2002) and Keurentjes et al. (2007).
Comparison of gene effects estimated by TTC progenies of NILs and RILs:
With the incomplete coverage of the Arabidopsis genome by our NIL libraries, only about one-half of the 20 genomic regions detected in our companion study (Kusterer et al. 2007b) by QTL mapping with the RILs were covered by NILs. For RD22, we could confirm the presence of QTL with additive effects reported in our study with RILs for the 2 QTL residing on chromosomes 3 and 5 and covered by NILs (supplemental Table 1 at http://www.genetics.org/supplemental/). For RD29 and GR, none of the QTL regions detected by RILs and covered by NILs could be confirmed. For DMC, we could confirm with the NILs 1 QTL on chromosome 2, showing an additive effect, and 1 QTL on chromosome 4, showing epistatic effects. For BY, two of the three QTL regions detected with RILs were covered by NILs and these confirmed the presence of 2 QTL with significant additive effects. In addition to the 20 main-effect QTL, Kusterer et al. (2007b) also reported nine digenic epistatic interactions among QTL across all five traits. Six of these QTL × QTL interactions were recovered in the present study by NILs covering at least one of the QTL regions and displaying a significant [aas] effect. No comparison could be made for LA22 and LA29, because these traits were not recorded in our companion study. In conclusion, about half of the QTL regions detected with RILs and covered by NILs could be confirmed by our novel generation-means analysis. This proportion was unexpectedly high considering (i) the power of QTL detection and the sizeable confidence intervals of QTL positions expected with the sample size (N = 234) in our companion study and (ii) the conceptual differences in the QTL-mapping approaches based on RILs and NILs discussed in the previous section.
Inferences about the mode of gene action involved in heterosis:
In the present study, we used two approaches to arrive at inferences about the prevailing mode of gene action contributing to heterosis. The approach recently devised by Semel et al. (2006) relies on the comparison of triplets consisting of a NIL, its recurrent parent, and the cross between these two genotypes. The majority of QTL detected in our study with this approach showed overdominance, especially for RD22, LA22, and BY (Figure 1). This is in agreement with the results of Semel et al. (2006), who found a prevalence of QTL with overdominance for traits related to reproductive fitness in introgression lines of tomato carrying chromosome segments of a wild relative. However, this approach does not allow investigation of epistatic interactions between the substituted segment and the remainder genome that occur even with a uniform, homozygous genetic background of all three genotypes included in the comparison for each triplet.
With our approach to evaluate NILs and their TTC progenies in addition to the parents and their reciprocal F1 crosses, it was possible to separate dominance effects from epistatic effects by the generation-means analysis described in the theory section. In model 2, epistasis was restricted to additive × additive effects, because according to quantitative genetics theory (e.g., Cockerham 1984) they are expected to be of greatest importance in autogamous species such as Arabidopsis. However, the presence of other types of digenic and/or higher-order epistasis cannot be ruled out and may explain the significant lack-of-fit for model 2 in all traits but GR (Table 3).
Our results revealed that many of the QTL that were considered overdominant according to the qualitative mode-of-inheritance classification devised by Semel et al. (2006) showed a significant ds effect in the range of partial to complete dominance (ds/|as| ≤ 1) and a positive additive × additive effect [aas] with the genetic background. Since Arabidopsis as a weed was exposed to diverse selection pressures during its evolution, one may expect that each parent harbors complexes of genes at different loci displaying positive main effects and positive interactions for growth-related traits as a major determinant of competition and reproductive fitness. This hypothesis is in agreement with the findings of Syed and Chen (2005), who concluded that heterozygosity in individual chromosomes or segments combined with epistasis may be the reason for the observed heterosis in the related cross Col × Ler.
In comparison with dominance effects ds, epistatic effects [aas] were of considerably greater importance in our study, as reflected by the number of NILs displaying such effects and their magnitude (Table 3). This is in excellent agreement with the significant estimates of epistatic variances 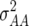 and
and 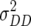 in our previous study with RILs (Kusterer et al. 2007a), the sum of which surpassed the estimates of
in our previous study with RILs (Kusterer et al. 2007a), the sum of which surpassed the estimates of 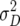 for all traits but GR and DMC. In rice, too, epistatic interactions were an important component of heterosis (Yu et al. 1997; Hua et al. 2003). Heterosis is reduced by positive additive × additive effects, because they increase parental performance but do not affect the F1 performance (Melchinger et al. 2007).
for all traits but GR and DMC. In rice, too, epistatic interactions were an important component of heterosis (Yu et al. 1997; Hua et al. 2003). Heterosis is reduced by positive additive × additive effects, because they increase parental performance but do not affect the F1 performance (Melchinger et al. 2007).
On the basis of these results, we hypothesize that the moderate level of heterosis for growth-related traits in cross C24 × Col-0 is partly attributable to positive additive × additive epistasis. It is known that the Arabidopsis genome contains a substantial proportion of duplicated genes (Arabidopsis Genome Initiative 2000; Moore and Purugganan 2003; Maere et al. 2005). If these genes interact favorably with each other through buffering of crucial functions (Chapman et al. 2006), subfunctionalization (Force et al. 1999; Causier et al. 2005), or functional diversification (Blanc and Wolfe 2004), this would result in “fixed heterosis” in the parents, adopting a term originally coined by MacKey (1976). Recently, Abel (2006) demonstrated by an elegant design that “fixed heterosis” plays an important role in growth-related traits in Brassica napus L., a polyploid relative of Arabidopsis. Since the complete genome sequence of Col-0 is known (Arabidopsis Genome Initiative 2000) and very detailed knowledge of C24 vs. Col-0 polymorphisms will soon become available from resequencing by hybridization (Clark et al. 2007), it seems promising to study the hypothesis of fixed heterosis with our materials using refined sets of NILs constructed according to the proposal of Koumproglou et al. (2002). Furthermore, sets of NILs with smaller substituted chromosome segments could be used for cloning and detailed investigations of individual genes underlying the heterotic QTL detected in this study by zooming in interesting regions following the approach of Fridman et al. (2004).
Acknowledgments
Initial plant material for the NILs was generously provided by R. C. Meyer. We gratefully acknowledge the expert technical assistance of B. Devezi-Savula, N. Friedl, E. Kokai-Kota, M. Teltow, and M. Zehnsdorf. We are also indebted to two anonymous reviewers for helpful suggestions on an earlier version of the manuscript. This project was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation) under priority research program “Heterosis in Plants” (research grants ME931/4-1 and ME931/4-2, PI 377/7-1 and PI 377/7-2, AL387/6-1 and AL387/6-2).
References
- Abel, S., 2006. Raps als modell zur untersuchung der fixierten heterosis bei allopolyploiden pflanzen. Vortr. Pflanzenzüchtg. 69: 177–180. [Google Scholar]
- Arabidopsis Genome Initiative, 2000. Analysis of genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Barth, S., A. K. Busimi, H. F. Utz and A. E. Melchinger, 2003. Heterosis for biomass yield and related traits in five hybrids of Arabidopsis thaliana L. Heynh. Heredity 91: 36–42. [DOI] [PubMed] [Google Scholar]
- Blanc, G., and K. H. Wolfe, 2004. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16: 1679–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce, A. B., 1910. The Mendelian theory of heredity and the augmentation of vigor. Science 32: 627–628. [DOI] [PubMed] [Google Scholar]
- Causier, B., R. Castello, J. Zhou, R. Ingram, Y. Xue et al., 2005. Evolution in action: following function in duplicated floral homeotic genes. Curr. Biol. 15: 1508–1512. [DOI] [PubMed] [Google Scholar]
- Chapman, B. A., J. E. Bowers, F. A. Feltus and A. H. Paterson, 2006. Buffering of crucial functions by paleologous duplicated genes may contribute cyclicality to angiosperm genome duplication. Proc. Natl. Acad. Sci. USA 103: 2730–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, R., G. Schweikert, C. Toomajian, S. Ossowski, G. Zeller et al., 2007. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science 317: 338–342. [DOI] [PubMed] [Google Scholar]
- Cockerham, C. C., 1954. An extension of the concept of partitioning hereditary variance for analysis of covariances among relatives when epistasis is present. Genetics 39: 859–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham, C. C., 1984. Additive by additive variance with inbreeding and linkage. Genetics 108: 487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock, R. E., and H. F. Robinson, 1952. Estimation of average dominance of genes, pp. 494–516 in Heterosis, edited by J. W. Growen. Iowa State College Press, Ames, IA.
- Crow, J. F., 1948. Alternative hypotheses of hybrid vigor. Genetics 33: 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport, C. B., 1908. Degeneration, albinism and inbreeding. Science 28: 454–455. [DOI] [PubMed] [Google Scholar]
- Duvick, D. N., 1999. Heterosis: feeding people and protecting natural resources, pp. 19–29 in Genetics and Exploitation of Heterosis, edited by J. G. Coors and S. Pandey. American Society of Agronomy, Madison, WI.
- East, E. M., 1908. Inbreeding in corn. Report of the Connecticut Agricultural Experiment Station, pp. 419–428.
- Einfeldt, C. H. P., S. Ceccarelli, S. Grando, A. Gland-Zwerger and H. H. Geiger, 2005. Heterosis and mixing effects in barley under drought stress. Plant Breed. 124: 350–355. [Google Scholar]
- Eshed, Y., and D. Zamir, 1995. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141: 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force, A., M. Lynch, F. B. Pickett, A. Amores, Y. L. Yan et al., 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151: 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frary, A., T. C. Nesbitt, S. Grandillo, E. Knaap, B. Cong et al., 2000. Fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289: 85–88. [DOI] [PubMed] [Google Scholar]
- Fridman, E., F. Carrari, Y. S. Liu, A. R. Fernie and D. Zamir, 2004. Zooming in on a quantitative trait for tomato yield using interspecific introgressions. Science 305: 1786–1789. [DOI] [PubMed] [Google Scholar]
- Hallauer, A. R., and J. B. Miranda, 1981. Quantitative Genetics in Maize Breeding. Iowa State University Press, Ames, IA.
- Haussmann, B. I. G., A. B. Obilana, A. Blum, P. O. Ayiecho, W. Schipprack et al., 1998. Hybrid performance of sorghum and its relationship to morphological and physiological traits under variable drought stress in Kenya. Plant Breed. 117: 223–229. [Google Scholar]
- Holm, S., 1979. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6: 65–70. [Google Scholar]
- Hua, J., Y. Xing, W. Wu, C. Xu, X. Sun et al., 2003. Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 100: 2574–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull, F. H., 1945. Recurrent selection for specific combining ability in corn. J. Am. Soc. Agron. 37: 134–145. [Google Scholar]
- Jones, D. F., 1917. Dominance of linked factors as a means of accounting for heterosis. Genetics 2: 466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey, M. J., 2002. QTL analysis: problems and (possible) solutions, pp. 45–58 in Quantitative Genetics, Genomics and Plant Breeding, edited by M. S. Kang. CABI, Oxford.
- Kearsey, M. J., and J. L. Jinks, 1968. A general method of detecting additive, dominance and epistatic variation for metrical traits. I. Theory. Heredity 23: 403–409. [DOI] [PubMed] [Google Scholar]
- Kearsey, M. J., H. S. Pooni and N. H. Syed, 2003. Genetics of quantitative traits in Arabidopsis thaliana. Heredity 91: 456–464. [DOI] [PubMed] [Google Scholar]
- Kenward, M. G., and J. H. Roger, 1997. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 53: 983–997. [PubMed] [Google Scholar]
- Keurentjes, J. J. B., L. Bentsink, C. Alonso-Blanco, C. J. Hanhardt, H. Blankestijn-de Vries et al., 2007. Development of a near isogenic line population of Arabidopsis thaliana and comparison of mapping power with recombinant inbred line population. Genetics 175: 891–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumproglou, R., T. M. Wilkes, P. Townson, X. Y. Wang, J. Beynon et al., 2002. STAIRS: a new genetic resource for functional genomic studies of Arabidopsis. Plant J. 31: 355–364. [DOI] [PubMed] [Google Scholar]
- Kusterer, B., J. Muminovic, H. F. Utz, H.-P. Piepho, S. Barth et al., 2007. a Analysis of a triple testcross design with recombinant inbred lines reveals a significant role of epistasis in heterosis for biomass-related traits in Arabidopsis. Genetics 175: 2009–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusterer, B., H.-P. Piepho, H. F. Utz, C. C. Schön, J. Muminovic et al., 2007. b Heterosis for biomass-related traits in Arabidopsis investigated by quantitative trait analysis of the triple-testcross design with recombinant inbred lines. Genetics 177: 1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z.-K., L. J. Luo, H. W. Mei, D. L. Wang, Q. Y. Shu et al., 2001. Overdominant epistatic loci are the primary genetic basis of inbreeding depression and heterosis in rice. I. Biomass and grain yield. Genetics 158: 1737–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, L. J., Z. K. Li, H. W. Mei, Q. Y. Shu, R. Tabien et al., 2001. Overdominant epistatic loci are the primary genetic basis of inbreeding depression and heterosis in rice. II. Grain yield components. Genetics 158: 1755–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKey, J., 1976. Genetic and evolutionary principles of heterosis. Heterosis in Plant Breeding, Proceedings of the 7th Congress of EUCARPIA, Budapest, June 24–29, 1974, pp. 17–33.
- Maere, S., S. De Bodt, J. Raes, T. Casneuf, M. Van Montagu et al., 2005. Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. USA 102: 5454–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul, A., J. Dupuis, S. Sen and K. W. Broman, 2006. Poor performance of bootstrap confidence intervals for the location of quantitative trait locus. Genetics 174: 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchinger, A. E., H. F. Utz, H. P. Piepho, Z. B. Zeng and C. C. Schön, 2007. The role of epistasis in the manifestation of heterosis: a systems-oriented approach. Genetics 177: 1815–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, R. C., O. Törjék, M. Becher and T. Altmann, 2004. Heterosis of biomass production in Arabidopsis. Establishment during early development. Plant Physiol. 134: 1813–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monforte, A. J., and S. D. Tanksley, 2000. Development of a set of near isogenic and backcross recombinant inbred lines containing most of the Lycopersion hirsutum genome in a L. esculentum genetic background: a tool for gene mapping and gene discovery. Genome 43: 803–813. [PubMed] [Google Scholar]
- Mood, A. M., F. A. Graybill and D. C. Boes, 1974. Introduction to the Theory of Statistics, Ed. 3. McGraw-Hill, New York.
- Moore, R. C., and M. D. Purugganan, 2003. The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. USA 100: 15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, H. D., and E. R. Williams, 1976. A new class of resolvable incomplete block designs. Biometrika 63: 83–93. [Google Scholar]
- Powers, L., 1944. An expansion of Jones' theory for the explanation of heterosis. Am. Nat. 78: 275–280. [Google Scholar]
- SAS Institute, 2004. SAS Version 9.1. SAS Institute, Cary, NC.
- Semel, Y., J. Nissenbaum, N. Menda, M. Zinder, U. Krieger et al., 2006. Overdominant quantitative trait loci for yield and fitness in tomato. Proc. Natl. Acad. Sci. USA 130: 12981–12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull, G. H., 1908. The composition of a field of maize. Am. Breed. Assoc. Rep. 4: 296–301. [Google Scholar]
- Stokes, D., C. Morgan, C. O'Neill and I. Bancroft, 2007. Evaluating the utility of Arabidopsis thaliana as a model for understanding heterosis in hybrid crops. Euphytica 156: 157–171. [Google Scholar]
- Syed, N. H., and Z. J. Chen, 2005. Molecular marker genotypes, heterozygosity and genetic interactions explain heterosis in Arabidopsis thaliana. Heredity 94: 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törjék, O., D. Berger, R. C. Meyer, C. Müssig, K. J. Schmid et al., 2003. Establishment of a high-efficiency SNP-based framework marker set for Arabidopsis. Plant J. 36: 122–140. [DOI] [PubMed] [Google Scholar]
- Utz, H. F., 2000. PLABSTAT: A Computer Program for Statistical Analysis of Plant Breeding Experiments. Institute of Plant Breeding, Seed Science and Population Genetics, University of Hohenheim, Stuttgart, Germany.
- Utz, H. F., A. E. Melchinger and C. C. Schön, 2000. Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 154: 1839–1849. [PMC free article] [PubMed] [Google Scholar]
- Williams, W., 1959. Heterosis and the genetics of complex characters. Nature 184: 527–530. [DOI] [PubMed] [Google Scholar]
- Yang, R. C., 2004. Epistasis of quantitative trait loci under different gene action models. Genetics 167: 1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, S. B., J. X. Li, C. G. Xu, Y. F. Tan, Y. J. Gao et al., 1997. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 94: 9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir, D., 2001. Improving plant breeding with exotic genetic libraries. Nat. Rev. Genet. 2: 983–989. [DOI] [PubMed] [Google Scholar]