

Abstract
Many genes duplicated by whole-genome duplications (WGDs) are more similar to one another than expected. We investigated whether concerted evolution through conversion and crossing over, well-known to affect tandem gene clusters, also affects dispersed paralogs. Genome sequences for two Oryza subspecies reveal appreciable gene conversion in the ∼0.4 MY since their divergence, with a gradual progression toward independent evolution of older paralogs. Since divergence from subspecies indica, ∼8% of japonica paralogs produced 5–7 MYA on chromosomes 11 and 12 have been affected by gene conversion and several reciprocal exchanges of chromosomal segments, while ∼70-MY-old “paleologs” resulting from a genome duplication (GD) show much less conversion. Sequence similarity analysis in proximal gene clusters also suggests more conversion between younger paralogs. About 8% of paleologs may have been converted since rice–sorghum divergence ∼41 MYA. Domain-encoding sequences are more frequently converted than nondomain sequences, suggesting a sort of circularity—that sequences conserved by selection may be further conserved by relatively frequent conversion. The higher level of concerted evolution in the 5–7 MY-old segmental duplication may reflect the behavior of many genomes within the first few million years after duplication or polyploidization.
GENE duplication (GD) is widespread in eukaryotic genomes. Individual genes may duplicate and spread in a genome, and duplication of a whole genome is a source of large numbers of duplicated genes with relatively long half-lives (Lynch and Conery 2000). Gene duplication is proposed to be a primary source of genetic material available for evolution of genes with new functions (Stephens 1951; Ohno 1970; Taylor and Raes 2004); one member of a duplicated gene pair may mutate and acquire unique functionality (Lynch et al. 2001; Tocchini-Valentini et al. 2005), with the fitness of the organism insulated by the homeolog. A duplicated gene may be lost, be inactivated, or develop a new function (Kellis et al. 2004); or a pair of duplicated genes may subdivide their ancestor's function (Lynch and Conery 2000). Mutation in regulatory elements of duplicated genes could contribute to novel gene expression patterns and alter morphological development (Ohta 2003).
A growing body of empirical data raises perplexing questions about the classical “functional divergence” model for duplicate gene evolution (Stephens 1951; Ohno 1970; Taylor and Raes 2004), specifically that duplicated genes are expected to accelerate in evolutionary rate due to a decrease in selective constraint, facilitating the acquisition of new functions by one member of a duplicated gene pair. Analysis of seventeen nonallelic duplicates in Xenopus laevis shows evidence of purifying selection, perhaps for regulatory divergence, on each duplicated gene (Hughes and Hughes 1993). For three recently duplicated (0.25–1.2 MYA) Arabidopsis genes, both progenitor and derived copies show significantly reduced specieswide polymorphism (Moore and Purugganan 2003), attributed by the authors to positive selection at the very early stage of these duplicate genes followed by fixation of favorable new alleles. While models such as positive selection and regulatory divergence (Hughes and Hughes 1993; Moore and Purugganan 2003) may be responsible for unexpectedly low divergence among some duplicated genes, recent data suggest that concerted evolution of duplicated genes is very widespread (Chapman et al. 2006).
The mere presence of two copies of a DNA sequence in the same genome raises a possibility that might contribute to genomewide concerted evolution of duplicated genes. Specifically, gene conversion, often accompanied by crossing over, can homogenize genetic variation to render similar DNA sequences identical (Galtier 2003). One model for recombination suggests that gene conversion may be explained by repair of unmatched bases during the formation of heteroduplex DNA (Holliday 1966). Gene conversion is often involved in homogenization of small tracts of paralogous DNA sequences, usually between several and several hundred base pairs (Petes et al. 1991), whereas the homogenization of larger tracts of DNA is generally believed to involve crossing over (Szostak and Wu 1980). Traditionally, gene conversion was used to describe the evolution of rRNA (Brown et al. 1972) and histone genes (Ohta 1984), both occurring in tandem clusters having tens of copies in an organism. Gene conversion has also been proposed to affect the evolution of various multigene families (Sawyer 1989; White and Crother 2000; Mondragon-Palomino and Gaut 2005).
The impact of gene conversion may have been neglected in prior research, partly for lack of a batch of duplicate genes having clear age estimation. Genomewide analyses of duplicated genes of eukaryotes have been performed (Lynch and Conery 2000; Kondrashov et al. 2002), inferring the ages of duplicates on the basis of synonymous substitution rate (Ks). However, this may be misleading, for duplicate genes with similar sequences can in some cases be quite old (Gao and Innan 2004).
Whole-genome duplication (WGD) produces large numbers of simultaneously duplicated genes, suitable for surveying levels and patterns of conversion events. Recently, on the basis of phylogenetic dating of duplicated genes, gene conversion has been suggested as a mechanism to explain low divergence rates between paralogs produced by ancient large-scale duplication events in yeast (Gao and Innan 2004). A comprehensive analysis of Caenorhabditis elegans duplicated genes inferred gene conversion to have occurred in 2% of them (Semple and Wolfe 1999). Phylogenetic patterns of homeologs duplicated prior to the mouse–rat divergence (Ezawa et al. 2006) indicated that at least 18% have been affected by gene conversion. A pairwise search method proposed by Sawyer (1989) in Arabidopsis paralogs produced by whole-genome duplication found no evidence of conversion (Zhang et al. 2002). However, Chapman et al. (2006) suggested gene conversion as a possible mechanism by which to explain sequence and functional conservation among paleologs (ancient, duplicated genes at homeologous locations) in the Arabidopsis and rice genomes after large-scale duplication events.
The availability of largely complete sequences for two divergent Oryza subspecies, indica and japonica, provides the means to explore the frequency and extent of recent conversion among paralogs of a range of ages, formed by whole-genome duplication ∼70 MYA and before the divergence of major cereals (Paterson et al. 2004; Wang et al. 2005), segmental chromosomal duplication (SD) ∼5–7 MYA, and before the divergence of two rice subspecies, japonica and indica (Wang et al. 2005; Rice Chromosomes 11 and 12 Sequencing Consortia 2005), and widespread single-gene duplication. These mechanisms result in duplicated genes with different distribution patterns: groups of paralogs in synteny or colinearity with one another, produced by large-scale duplication events and paralogs in proximal (“tandem”) clusters, produced by single-gene duplication events. We adopted several approaches to detect possible conversion events in these various types of paralogs. To reveal conversions after indica–japonica divergence, we inspected homologous gene quartets comprising each member of a duplicated gene pair in each of the two subspecies, for both genes duplicated by segmental/genomewide events, and proximally duplicated genes. Recent conversions would result in a phylogenetic topology change from that expected (Figure 1). To infer possible conversions prior to the split of the subspecies, we detected high-local-similarity segments between paralogs, on the basis of the consideration that conversion may render paralogous sequences identical. To explore possible conversion during the 41–47 MY since rice–sorghum divergence, we compared the rice paralogs and their homologous sorghum sequences. Our results indicate extensive concerted evolution in both rice subspecies, including both conversion and crossing over, among genes duplicated by segmental/genomewide events, and proximally duplicated genes. Our results shed light on the road to independent evolution of duplicated genes, with several lines of evidence suggesting that the road is longer for sequences conserved by selection in that such sequences appear to be further conserved by relatively frequent conversion.
Figure 1.—

Phylogenetic topology changes indicating gene conversion in a quartet of homologs from taxa A and B. Squares symbolize a duplication event in the common ancestral genome of A and B, and circles symbolize the divergence of the two taxa. The expected phylogenetic relationship of the homologous quartets is displayed (a) if no conversion has occurred, (b) if gene A2 is converted by A1 (donor), (c) if gene B1 is converted by B2, and (d) if both the above conversions occurred.
MATERIALS AND METHODS
Sequence data:
The Oryza sativa ssp. japonica genome sequences (ver. 4) were downloaded from TIGR (http://www.tigr.org/) and indica sequences from RISE (http://rise.genomics.org.cn/). We downloaded the japonica gene model sequences, 62,827 in total, from TIGR, and used them to determine genes in the indica genome by running the public software Blat under default parameters. Transposable element (TE)-related genes and alternatively spliced transcripts were removed, resulting in a data set containing 42,652 genes. We further restricted our search to require that the matched length exceeds 90% of the japonica query sequence, and that the query and subject sequences were known to be on the same chromosome in both subspecies. In sum, for 56.7% of the japonica gene models (35,641), we found homologs on the corresponding indica chromosome. In most cases there is a one-to-one correspondence, and physical location confirms that the genes are likely to be true orthologs (see below). This stringent selection of the gene set for study assures the credibility of the analysis and, actually, a most up-to-date estimation of rice gene number is only ∼32,000 (Itoh et al. 2007). We did not involve the predicted indica genes in our analysis, for if we used genes predicted independently in each of the two subspecies, possible mistakes due to annotation errors would be doubled.
Detection of duplicated segments and colinear quartets:
To detect possible interchromatid gene conversion, we inferred large homologous chromosomal segments by considering gene colinearity. japonica duplicated genes were defined by running BLAST (Altschul et al. 1990) at an E-value <10−5 in both subspecies. Colinear paralogs in the japonica genome sequence were revealed by running ColinearScan (Wang et al. 2006) with a minimal paralogous segment length of 4 colinear genes and probability of occurrence <0.01, after purging repetitive genes (>10 copies in a chromosome).
To confirm orthology, we checked whether the japonica genes and the indica homologs shared the same neighboring genes. If they shared the left or right or both neighboring genes, we supposed that they were physically orthologs (supplemental file 1 at http://www.genetics.org/supplemental/). These resulted in a data set of 676 quartets of colinear indica–japonica genes. All but one of the indica paralogous pairs were in perfect colinearity and the one exceptional pair was removed from further analysis.
Detection of tandem genes and homologous quartets:
To reveal possible gene conversion among tandem genes, BLASTCLUST (Altschul et al. 1990) was run to group the japonica proteins to define gene families on the basis of the following parameters: minimal matched sequence similarity, 0.8; minimal matched sequence length, 50 amino acids. On the basis of the detected gene families, we defined proximal/tandem gene clusters as having at least five paralogs within <200 kb. We found 125 such proximal clusters. By checking the neighboring genes, as described to confirm the colinear orthologs above, we identified indica orthologs for the proximally duplicated genes. Two japonica tandem genes and their two indica orthologs comprised a quartet of tandem genes. If two japonica tandem genes have the same indica ortholog, they were excluded from further analysis, since in such cases we could not distinguish gene conversion and gene duplication. This resulted in a data set of 1290 tandem quartets (supplemental file 2 at http://www.genetics.org/supplemental/).
Detection of whole-gene conversion:
The colinear and tandem quartets were aligned with Clustalw (Thompson et al. 1994), and alignments for which gaps account for >20% of the alignment length were discarded. To detect possible whole-gene conversion, we used phylogenetic analysis to identify possible topology change in the quartets of indica–japonica homologs. Since paralogs in each subspecies were formed by duplication prior to divergence of the subspecies, we expected paralogs in each subspecies to be more distant from one another than from their respective orthologs in the other subspecies. If the paralogs were more similar to one another than to their cross-subspecies orthologs, we inferred that gene conversion occurred after indica–japonica divergence. This was referred to as a putative whole-gene conversion event (Figure 1). A bootstrap test was performed to evaluate the significance of putative gene conversions. Columns of the aligned sequences were randomly selected with replacement to form random multiple alignments, and the similarity between random sequences was checked for whether the paralogs are nearer to one another. This process was repeated 1000 times to produce a bootstrap frequency indicating the confidence level of the supposed conversion.
Detection of partial-gene conversion:
We adopted two lines of exploration for gene conversion affecting only portions of a gene. First, we searched for possible conversion after indica–japonica divergence in local regions of aligned sequences, using the homologous quartets obtained above. An approach integrating dynamic programming and phylogenetic analysis similar to that proposed by Lin et al. (2006) was adopted. Arrays were defined to reflect the difference or distance between the homologs. At nucleotide position i, we recorded the difference between two aligned bases in paralogs of each subspecies (Dij = 0 or 1; j = 1, 2) and the difference between the orthologous pairs (Bik = 0 or 1, k = 1, 2). We averaged the orthologous distance arrays to get Bi = (Bi1 + Bi2)/2. If no partial sequence conversion was involved, the paralogs in each species should be more distant, i.e., Dij − Bi ≥ 0. Dynamic programming was used to reveal high-scoring segmental sequence, extending from nucleotide position m to n, that maximized 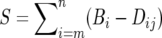 in each of the two paralogous pairs. If S > 0 and m − n ≥ 10 bases, we performed a bootstrap test to check the possibility that occurrence of such a segmental sequence could be explained by chance. Bootstrap samples were produced by selecting the aligned columns with replacement and the above programming search was performed for each sampled alignment. This procedure was repeated 1000 times. We counted the high-scoring segments that were shorter in length and with smaller score values to obtain a bootstrap frequency. We discovered shorter high-scoring segments by a recursive procedure after masking the larger ones. This helped to reveal the genes affected by multiple conversion events. Gaps were removed from the alignment in the above analysis. The original procedure by Lin et al. (2006) was not directly duplicated here since the japonica–indica divergence was quite recent in time (Zhu and Ge 2005), making their test methods too stringent.
in each of the two paralogous pairs. If S > 0 and m − n ≥ 10 bases, we performed a bootstrap test to check the possibility that occurrence of such a segmental sequence could be explained by chance. Bootstrap samples were produced by selecting the aligned columns with replacement and the above programming search was performed for each sampled alignment. This procedure was repeated 1000 times. We counted the high-scoring segments that were shorter in length and with smaller score values to obtain a bootstrap frequency. We discovered shorter high-scoring segments by a recursive procedure after masking the larger ones. This helped to reveal the genes affected by multiple conversion events. Gaps were removed from the alignment in the above analysis. The original procedure by Lin et al. (2006) was not directly duplicated here since the japonica–indica divergence was quite recent in time (Zhu and Ge 2005), making their test methods too stringent.
Second, assuming uniform mutation rates in the third nucleotides of the coding sequences, if a local region of two paralogous genes was affected by conversion, sequence similarity between the paralogs should be higher than for the rest of the gene. This would help to reveal genes affected by older conversion events. Therefore, first we scanned for local high similarity along the coding sequences of the japonica paralogs with a sliding window (30 codons in size and 10 codons as sliding step) relative to the overall average of the pair of paralogs. A chi-square test was adopted to compare the numbers of matched and mismatched sites in the local region to those in the full length of the paralogous genes. If the paralogs are significantly similar in several neighboring windows, we merged the windows into one. The search was performed on the basis of the third codon sites, which were supposed to be largely neutral. We searched for such high-local-similarity (HLS) segments, both between colinear gene pairs and between proximal/tandem gene pairs. The HLS segments were further required to be more similar at the first and second codon sites than in the full length of the paralogs (supplemental files 3 and 4 at http://www.genetics.org/supplemental/). The HLS segments were not directly assumed to be affected by conversion, and where possible we also evaluated phylogenetic incongruity of indica–japonica colinear quartets to identify events likely to have occurred since subspecies divergence. However, the level of HLS segments implies the possibility of many more ancient conversions, perhaps occurring during the much longer period between duplication and indica–japonica divergence.
Conversion after rice–sorghum divergence:
To infer possible gene conversion after rice–sorghum divergence, we identified sorghum genes or coding sequence segments putatively homologous to the rice japonica colinear paralogous sequences in the GD blocks, by searching assembled Sorghum bicolor methyl-filtered sequences and a sorghum EST database (containing 39,654 nonredundant items) from TIGR Plant Transcript Assemblies (http://plantta.tigr.org/). For the HLS rice homologous segments, by performing a BLASTX search their sorghum homologous sequences were determined at thresholds of an E-value <1.0 × 10−10, DNA length ≥51 nucleotides. If two rice HLS paralogous segments are more similar to one another than they are to the sorghum homologs, resulting in an aberrant tree topology, we inferred a possible gene conversion after rice–sorghum divergence (Figure 1). Bootstrap tests with 1000 recursive samples with replacement were performed, similar to those done to test candidate whole-gene conversions (above).
Factors related to gene conversion:
The HLS segments in japonica genes were linked to protein domain families by performing searches against the Pfam database (version 19, 8183 models) at an E-value <10−4. To assess possible correlation of gene conversion with tandem cluster size and gene transcriptional orientation, HLS segment numbers per gene pair and per gene were calculated.
RESULTS AND DISCUSSION
Duplicated blocks in the rice genome:
We revealed 319 duplicated blocks in the japonica genome and focused our interchromatid conversion search on the 21 largest duplicated pairs, each containing ≥27 colinear genes. Among these, 20 blocks including 1449 colinear gene pairs were produced by the GD 70 MYA (Paterson et al. 2004), and one including 278 gene pairs colinear between chromosome 11 and chromosome 12 was produced by a large SD 5–7 MYA (Paterson et al. 2004; Rice Chromosomes 11 and 12 Sequencing Consortia 2005; Wang et al. 2005).
The fact that these duplication events predated the divergence of the japonica and indica subspecies of Oryza sativa ∼0.4 MYA (Zhu and Ge 2005), provided a means to infer the occurrence of gene conversion on the basis of phylogenetic incongruity. GD and SD paralogs in each subspecies should normally be much more diverged than orthologs. Gene conversion between paralogs after the indica–japonica speciation would result in an aberrant tree topology, with paralogs more similar to one another than to their orthologs in the other subspecies (Figure 1).
Concerted evolution is more abundant in a single recent segmental duplication than in ancient duplicates across the entire genome:
For the 675 indica and japonica quartets of colinear homologs that we found in GD and SD blocks (see materials and methods), we looked for gene tree topology change. Two japonica colinear genes (Os11g02630 and Os12g02550, Os11g04750 and Os12g04530) were affected by possible whole-gene conversion at bootstrap confidence level > 0.9 (Table 1). These two gene pairs are both located in the SD block on chromosomes 11 and 12 and represent 1.8% of the 109 quartets in this block for which we were able to identify their indica homologs. No indica colinear paralogs in the SD region were detected to be affected by whole-gene conversion.
TABLE 1.
Converted interchromatid paralogs revealed by analyzing homologous quartets in japonica and indica
| Paralog 1a | Paralog 2 | Functionb | Conversion descriptionc |
|---|---|---|---|
| Os01g57370 | Os05g42250 | Cation channel family protein, expressed | japonica partial conversion; 1736–1764; BC = 0.971; donor, japonica paralog 1 |
| Os08g37350 | Os09g28940 | Ubiquitin carboxyl-terminal hydrolase family protein, expressed | japonica partial conversion; 122–137; BC = 0.977; donor, japonica paralog 2 |
| Os11g01860 | Os12g01910 | Pentatricopeptide, putative, paralog 1 expressed | japonica partial conversion; 273–347; BC = 1; donor, japonica paralog 1 |
| Os11g02630 | Os12g02550 | Expressed protein | japonica whole conversion; BC = 0.949; donor, japonica paralog 1 |
| Os11g02730 | Os12g02700 | Harpin-induced protein 1 containing protein, expressed | indica partial conversion; 326–533; BC = 0.996; donor, indica paralog 2 |
| Os11g03230 | Os12g02980 | GDA1/CD39 family protein, expressed | japonica partial conversion; 869–944; BC = 0.964; donor, japonica paralog 2 |
| indica partial conversion; 565–625; BC = 0.992; donor, indica paralog 2 | |||
| Os11g03540 | Os12g03290 | AP2 domain containing protein, expressed | indica partial conversion; 814–947; BC = 0.916; donor, indica paralog 2 |
| Os11g04020 | Os12g03830 | Major facilitator superfamily antiporter, putative, paralog 1 expressed | indica partial conversion; 528–602; BC = 0.94; donor, indica paralog 2 |
| Os11g04230 | Os12g04060 | Transcription initiation factor IIE, putative | indica partial conversion; 394–559; BC = 0.996; donor, indica paralog 2 |
| Os11g04360 | Os12g04150 | Cell-death-associated protein, putative, paralog 2 expressed | japonica partial conversion; 745–761; BC = 0.977; donor, japonica paralog 2 |
| indica partial conversion; 340–363; BC = 0.994; donor, indica paralog 2 | |||
| Os11g04460 | Os12g04220 | Calcium-transporting ATPase 4, plasma-membrane type, putative, expressed | indica partial conversion; 1878–2173; BC = 0.972; donor, indica paralog 1 |
| Os11g04750 | Os12g04530 | Hypothetical protein | japonica whole conversion; BC = 0.911; donor, japonica paralog 1 |
| Os11g04760 | Os12g04540 | Hypothetical protein | indica partial conversion; 473–482; BC = 0.975; donor, indica paralog 1 |
| Os11g04840 | Os12g04700 | Phosphatidylinositol-4-phosphate 5-kinase 4, putative, paralog 2 expressed | japonica partial conversion; 23–67; BC = 0.982; donor, japonica paralog 2 |
The gene names are for japonica gene models, and indica orthologs were decided by sequence search.
The function description is based on japonica gene models. All these paralogs have the same function, and any possible difference is specifically indicated.
We showed conversion type, subspecies, partial conversion position after alignment, bootstrap confidence (BC) level, and donor gene (inferred by checking tree incongruity displayed in Figure 1). The partial conversion position is that of aligned sequences (supplemental file 5 at http://www.genetics.org/supplemental/).
In the GD blocks, which include the vast majority of duplicated genes, no cases of whole-gene conversion could be inferred since divergence of the two subspecies ∼0.4 MYA.
Partial-gene conversion is found in both recent and ancient paralogs:
Gene conversion typically involves between several and several hundred base pairs (Petes et al. 1991), so might affect only a portion of a paralogous gene pair, resulting in significantly high sequence identity in a small window. We scanned the coding sequences of the japonica paralogs for high local similarity in the third codon sites relative to the degree of similarity found across the full lengths of paralogous gene pairs containing the HLS (using the chi-square test to infer significance). At a significance level of 0.01, we found a total of 548 HLS segments involving 31.7% (459/1449) of paralogs in the 20 large GD blocks, and 79 HLS segments involving 22.7% (63/278) of paralogs in the SD block on chromosomes 11 and 12 (supplemental file 3 at http://www.genetics.org/supplemental/). The HLS segments have an average length of 65 codons. Although the percentage of segments affected by conversion is lower in the SD than the GD block, one needs to keep in mind that conversion in the GD blocks reflected by HLS segments could have occurred >70 MYA vs. 5–7 MYA for SD paralogs. If there had been a depressed mutation rate in a subregion of gene sequence, it may be taken as possible conversion (Sawyer 1989). However, we have focused on synonymous sites—biological mechanisms leading to depressed mutation rates at the synonymous sites in partial-gene sequence are not clear. Therefore, the level of HLS segments could be taken as an upper limit of conversions.
While many of the HLS segments are presumably a result of conversion during the long period since duplication, phylogenetic incongruity of indica–japonica colinear quartets showed that 12 of the partial-conversion events have occurred since subspecies divergence (Table 1). A total of 10 (80%) of the partial-gene conversions after indica–japonica divergence involved paralogs in the large SD on chromosomes 11 and 12 (Figure 2), with the remaining 2 involving genes in the ancient GD blocks. This equates to partial conversion rates of 9.1% among the 109 indica–japonica colinear quartets found on the large SD block vs. 0.36% of 566 quartets involving 70-MY-old duplicated genes, a difference that is highly significant (P < 2.2 × 10−16, the smallest P-value calculable using the “R” package). The conversion rate has been similar in the two subspecies (6.4% in japonica vs. 7.3% in indica), with 5 events occurring between japonica paralogs, 6 between indica paralogs, and 2 in both japonica and indica paralogs (Os11g03230, Os12g02980; Os11g04360, Os12g04150 in Table 1).
Figure 2.—

Recursive crossings over on the initial part of rice chromosomes 11 and 12. (a) On the basis of patterns of difference, the genes can be divided into four regions: A, B, C, and D. Each region differs from its neighbors in the degree of divergence between paralogs, and/or in colinear gene density (Table 2). The paralogs showing whole-gene (square) or segmental (triangle) evidence of concerted evolution are indicated in both indica (blue) and japonica (red), and the paralogs converted in both subspecies are in light green. (b) A parsimonious reconstruction of the crossings over recursively occurred with time. Colors display divergence between paralogous chromosomal segments: the more intense the shading, the more diverged over time. Crossings over render the corresponding segments identical, denoted in white.
Introns tended not to be involved in the partial-gene conversions that we inferred. Of the 627 HLS segments, most are in intron-containing paralogs, and many of these paralogs (28%) are diverged in intron number. There are 238 introns flanked by HLS segments on both sides. However, only 1 of these introns shows high similarity comparable to that of HLS segments. Crossing over would be expected to produce some paralogous introns with high similarity, such as those observed in the SD on chromosomes 11 and 12. The scarcity of this finding indicates either rapid evolution of introns, not only in nucleotide mutation rate but also in intron number, or conversion without crossing over, the latter of which is supported by the finding that gene conversion, rather than crossing over, homogenized the bacterial RNA genes (Liao 2000).
Phylogenetic analysis using sorghum methyl-filtered and EST sequences suggests that a considerable percentage of paralogs may have been subjected to partial-gene conversion after rice–sorghum divergence. The GD paralogs (paleologs) were produced before the divergence of the major cereal lineages (Paterson et al. 2004). By searching the assembled methyl-filtered sorghum sequences, we retrieved high-quality sorghum matches (>0.5 amino acid similarity and >20 amino acids long) for 149 of 548 japonica HLS segments in genes on the 20 longest blocks produced by the 70-MYA GD. According to phylogenetic analysis, 38 of these 149 HLS segments showed incongruence consistent with conversion after rice–sorghum divergence, and 12 of the 38 cases were supported by bootstrap confidence levels >0.9. This yields an estimated conversion frequency of 8.1% in the 41 MY since rice–sorghum divergence (a similar estimate is obtained on the basis of EST analysis, not shown). This implies that thousands of colinear genes may have been converted after rice–sorghum divergence. However, this estimate must be considered an upper bound of the conversion rate since rice–sorghum divergence, in that we surely failed in some cases to obtain the true orthologs from the available sequences (despite using stringent criteria to define the homology).
Crossing over appears to have restructured recently duplicated chromosomal segments:
The findings that all whole-gene conversions, and many partial-gene conversions, are in the SD block led us to perform more exploration in this recently duplicated region of 3.5 Mb on chromosomes 11 and 12, representing <1% of the genome. We found that this small region has undergone an exceptionally high rate of concerted evolution. Many of the japonica colinear genes show high sequence similarity, however the distribution of sequence similarity between the paralogs is not uniform across the region. According to direct observation we divided the SD segment into four regions: A, B, C, and D (Figure 2a). t-tests confirmed the reasonableness of the classification in that the neighboring regions differed in both sequence similarity and average gene identity, and/or in colinear gene density (Table 2), indicating a complex evolutionary history. No single duplication, conversion, or crossing-over event could produce the observed differences in sequence similarity among neighboring regions. Therefore, a series of events could be the answer. One can quickly rule out a series of single-gene duplications, which could not explain why only the same two chromosomes were always involved or why the paralogous regions were preserved in the same order on the two chromosomes. A series of conversions or a series of crossings over could each lead to identical DNA sequence. However, the sequence similarity is not confined to the coding sequences of colinear genes, e.g., for the 20 paralogs in region A, introns and intergenic regions are both highly similar. This indicates that the events involved large chromosomal segments. So, our preferred model to explain the observation is a series of interchromatid crossing-over events, by which chromosomal segments replaced their paralogous counterparts on the other chromosome. A parsimonious model of three crossing-over events can explain the observation (Figure 2b): the first crossing over might involve regions A, B, and C, which kept the paralogous segments in high similarity and consequently facilitated further possible crossing over; the second involved only region C; and the third only region A (see Figure 2). The first two crossing-over events could have preceded the indica–japonica divergence for the existence of corresponding similar structure in indica sequences (data not shown) and based on Ks values [average Ks value is 0.015 in region A, 0.131 in B, and 0.077 in C, estimated by the evolutionary pathway model (Nei and Gojobori 1986)]. The third was near the two subspecies' divergence, but at present cannot be precisely determined since the indica sequence corresponding to region A is not available due to possible assembly problems. Therefore, variation in sequence similarity between the paralogs in these regions may partly reflect the antiquity of crossing over, rather than directly reflecting the age of segmental duplication as previously reported (Rice Chromosomes 11 and 12 Sequencing Consortia 2005; Wang et al. 2005). This suggests that the age of the duplication event may be somewhat underestimated. Superimposed on these crossovers are the two more recent single-gene conversions, and partial conversions that could be inferred statistically.
TABLE 2.
The density and sequence identity of colinear paralogs on chromosomes 11 and 12
| Region | Colinear gene number | Average gene number | Colinear gene density | P-value | Sequence identity | P-value |
|---|---|---|---|---|---|---|
| A | 18 | 20.5 | 0.878 | 0.991 | ||
| B | 136 | 237.5 | 0.573 | 0.0135 | 0.914 | 1.64 × 10−10 |
| C | 63 | 97 | 0.649 | 0.239 | 0.950 | 0.028 |
| D | 61 | 234.5 | 0.260 | 6.13 × 10−11 | 0.812 | 1.89 × 10−10 |
The P-values are shown between neighboring regions defined in Figure 2.
These findings cannot be explained by a misassembly of the rice sequence in the region(s) because (1) the 1st and 11th genes on chromosome 11 and the 12th and 14th genes on chromosome 12 have no complete match in corresponding regions on the other chromosome; (2) the 8th gene on chromosome 11 has two segmental DNA losses relative to its paralog on chromosome 12; (3) Ks values between the first 20 paralogous genes on the two chromosomes vary from 0 to 0.072; and (4) although some genes have Ks = 0, Ka values are not 0. [This may be due to relaxed selection pressure or strong diversifying selection soon after duplication, conversion, or crossing over, favoring evolution of new alleles that quickly reached fixation Moore and Purugganan (2003).] These facts could not be explained by misassembly with a sequencing error rate ∼1/10,000, instead supporting the proposition of concerted evolution of the sequences at one end of the two chromosomes.
The recursive crossings over in the terminal portion of rice chromosomes 11 and 12 somewhat resemble the human sex chromosomes. For example, human X and Y chromosomes share a small homologous terminal region, where X–Y crossing over is normal and frequent during male meiosis (Skaletsky et al. 2003). Rice chromosomes 11 and 12 appear to be homologs produced by the WGD event, but evidence is weaker outside of this terminal region, perhaps due to extensive heterochromatin (Yu et al. 2005; Rice Chromosomes 11 and 12 Sequencing Consortia 2005). The relationship between frequent crossings over between these two chromosomes and their probable more ancient homology is an interesting subject for further exploration.
Partial-gene conversion between tandem paralogs:
A particularly high rate of gene conversion is found between proximally or tandemly duplicated genes. Among a total of 848 genes in 125 tandem clusters, 820 pairs of segmental sequences in 511 (60.3%) of the clustered genes contained HLS segments at a significance level of 0.01 (supplemental file 4 at http://www.genetics.org/supplemental/). The higher percentage of HLS-segment-containing genes in the proximal clusters implies a higher conversion rate than in GD- or SD-duplicated blocks. There are 59 cases of introns flanked by HLS segments, but no intron show high similarity as compared with that of the HLS segments. This observation in proximal paralogs is consistent with that in colinear paralogs in duplicated chromosomal segments.
Some conversions between the proximal genes occurred since indica–japonica divergence. We analyzed the 1290 quartets involving proximal genes that duplicated before subspecies divergence and found 20 affected by gene conversion at bootstrap confidence level >0.9 (Table 3). There are 6 whole-gene conversion events affecting 4 homolog quartets, i.e., for 2 quartets both the japonica and the indica paralogs were each fully converted (Os02g15169, Os02g15178 and Os06g35320, Os06g35370 in Table 3). The alternative hypothesis that independent gene duplications in japonica and indica following their divergence could result in proximal paralogs that are very similar to one another, seems unlikely to account for such new paralogs occurring in precisely colinear physical locations in each of the two subspecies, suggesting that conversion is the more likely explanation of our findings. A total of 14 partial-gene conversions occurred in 13 quartets, 6 in japonica and 8 in indica, i.e., in 1 quartet the japonica and indica paralogs were both affected (Os02g12770, Os02g12780 in Table 3). One japonica proximal paralogous pair (Os03g57680, Os03g57690) was affected twice by partial conversion. These findings, together with those from colinear quartets in which both japonica and indica pairs showed conversion, indicate possible high conversion rates for specific genes. The proximal conversion rate since the indica–japonica split is ∼1.5%, lower than that between the young SD paralogs, but higher than that between the old GD paralogs. It must be noted that the proximal paralogs may have formed over a range of times. Those that are substantially younger than the SD have had less time for conversion to occur; those that are substantially older may be less prone to conversion.
TABLE 3.
Converted proximal paralogs revealed by analyzing homologous quartets in japonica and indica
| Paralog 1 | Paralog 2 | Function | Conversion description |
|---|---|---|---|
| Os01g02300 | Os01g02430 | Paralog 1: receptor kinase ORK10, putative, expressed; paralog 2: AK14, putative, expressed | indica partial conversion; 1334–1346; BC = 0.971; donor, indica paralog 2 |
| Os01g24560 | Os01g24570 | Papain family cysteine protease-containing protein, expressed | indica partial conversion; 909–975; BC = 0.996; donor, indica paralog 1 |
| Os01g26876 | Os01g27050 | Flavin-containing monooxygenase family protein, putative, expressed | indica partial conversion; 73–461; BC = 0.995; donor, indica paralog 1 |
| Os02g05940 | Os02g05950 | Leucine-rich repeat family protein, paralog 2 expressed | japonica partial conversion; 28–95; BC = 1; donor, japonica paralog 2 |
| Os02g12770 | Os02g12780 | Cytokinin dehydrogenase 1 precursor, putative | japonica partial conversion; 205–284; BC = 0.956; donor, japonica paralog 2 |
| indica partial conversion; 1229–1272; BC = 1; donor, indica paralog 1 | |||
| Os02g15169 | Os02g15178 | Glutelin type B 1 precursor, putative, expressed | japonica whole conversion; BC = 1 |
| indica whole conversion; BC = 1 | |||
| Os02g55649 | Os02g55658 | Expressed protein | indica whole conversion; BC = 0.939 |
| Os03g57680 | Os03g57690 | Aldehyde oxidase 1, putative, expressed | japonica partial conversion; 3510–3875; BC = 0.999; donor, japonica paralog 1 |
| japonica partial conversion; 2396–2519; BC = 0.994; donor, japonica paralog 1 | |||
| Os06g35320 | Os06g35370 | Exopolygalacturonase precursor, putative, expressed | japonica whole conversion; BC = 0.963; donor, japonica paralog 1 |
| indica whole conversion; BC = 0.962; donor, indica paralog 1 | |||
| Os06g47700 | Os06g47740 | Leucine-rich repeat family protein, paralog 1 expressed | japonica partial conversion; 308–450; BC = 1; donor, japonica paralog 1 |
| Os06g48000 | Os06g48010 | Peroxidase 16 precursor, putative | indica partial conversion; 399–598; BC = 0.997; donor, indica paralog 2 |
| Os07g32830 | Os07g32839 | Hypothetical protein | japonica partial conversion; 269–315; BC = 0.961; donor, japonica paralog 1 |
| Os07g32830 | Os07g33040 | Hypothetical protein | japonica partial conversion; 425–492; BC = 0.985; donor, japonica paralog 2 |
| Os07g32839 | Os07g32960 | Hypothetical protein | indica partial conversion; 317–359; BC = 1; donor, indica paralog 2 |
| Os09g27750 | Os09g27820 | 1-aminocyclopropane-1-carboxylate oxidase 1, putative, expressed | indica partial conversion; 531–800; BC = 1; donor, indica paralog 1 |
| Os10g36090 | Os10g36100 | Nonspecific lipid-transfer protein 2, putative, paralog 2 expressed | indica partial conversion; 188–297; BC = 1; donor, indica paralog 2 |
| Os10g39260 | Os10g39300 | Eukaryotic aspartyl protease family protein, expressed | indica whole conversion; BC = 0.999; donor, indica paralog 2 |
For aligned sequences, see supplemental file 6 at http://www.genetics.org/supplemental/.
Conversion tends to be more frequent between genes in close proximity. The average distance between the HLS segment containing proximally duplicated genes was 43.6 kbp, much smaller than the average distance of 67.1 kbp between all tandem genes (P = 0.021). This observation is consistent with findings regarding three leucine-rich repeat gene families in Arabidopsis (Mondragon-Palomino and Gaut 2005) and with genomewide exploration in mouse and rat (Ezawa et al. 2006).
To explore the influence of gene cluster size on conversion, we checked the predicted numbers of HLS-segment-containing genes in clusters of different sizes (Table 4). Increased tandem cluster size tended to be associated with a smaller frequency of HLS-segment-containing genes. The average frequency of HLS-segment-containing genes varies, especially for large clusters. The genes in large clusters (containing >10 genes) are less likely to be involved in conversion (0.13 vs. 0.28, P < 2.2 × 10−16). We further checked HLS frequencies between the neighboring genes, and found that the neighboring genes in large and small clusters have a similar chance to be involved in conversion (0.34 vs. 0.31, P = 0.489), whereas the neighboring genes have a much larger chance to be converted than the nonneighboring genes (0.33 vs. 0.21, P = 3.35 × 10−11). While the HLS segment frequency reflects in essence the possibility of conversion between paralogous genes, these findings suggest that physical distance or proximity is an important factor relating to gene conversion, as previously reported by Ezawa et al. (2006).
TABLE 4.
The relationship between conversion and proximal cluster size
| Cluster size | Cluster | HLS | Total pair | HLS per gene | Neighbor HLS | Neighbor gene | HLS per neighbor |
|---|---|---|---|---|---|---|---|
| 5 | 65 | 202 | 650 | 0.311 | 84 | 260 | 0.323 |
| 6 | 19 | 86 | 285 | 0.302 | 34 | 95 | 0.358 |
| 7 | 12 | 57 | 252 | 0.226 | 21 | 72 | 0.292 |
| 8 | 7 | 48 | 196 | 0.245 | 18 | 49 | 0.367 |
| 9 | 5 | 47 | 180 | 0.261 | 16 | 40 | 0.400 |
| 10 | 7 | 87 | 315 | 0.276 | 24 | 63 | 0.381 |
| 11 | 2 | 5 | 110 | 0.045 | 3 | 20 | 0.150 |
| 12 | 1 | 20 | 66 | 0.303 | 3 | 11 | 0.273 |
| 14 | 2 | 53 | 182 | 0.291 | 13 | 26 | 0.500 |
| 15 | 1 | 1 | 105 | 0.010 | 1 | 14 | 0.071 |
| 17 | 1 | 0 | 136 | 0.000 | 0 | 16 | 0.000 |
| 18 | 1 | 20 | 153 | 0.131 | 8 | 17 | 0.471 |
| 20 | 1 | 1 | 190 | 0.005 | 1 | 19 | 0.053 |
| 22 | 1 | 63 | 231 | 0.273 | 15 | 21 | 0.714 |
| Total | 125 | 690 | 3051 | 0.226 | 241 | 723 | Average: 0.333 |
“Cluster size” is the number of genes in a proximal gene cluster. “HLS” denotes the numbers of HLS segments between all gene pairs in clusters of the same size. “Total pair” denotes the summed numbers of pairs in tandem clusters of the same size, calculated by the formula: cluster no. × cluster size × (cluster size − 1)/2. “Neighbor HLS” denotes the numbers of HLS segments between neighboring genes. Neighbor gene number is calculated by multiplying (cluster size − 1) by cluster number.
It was previously reported that tandem genes on the same DNA strand have the same transcriptional orientation, facilitating strand mispairing that may lead to conversions (Hulbert et al. 2001). In rice proximal clusters, we found that genes having the same transcriptional orientation are prone to being converted. In 125 gene clusters, we found 2341 gene pairs of the same orientation, and 700 different. Of the HLS-segment-containing genes, 631 gene pairs are in the same orientation, and 189 are in different orientations, indicating proximal genes in the same orientation have the same chance to be involved in conversion as those in opposite orientation (P = 0.98). In neighboring paralogs, we found the same result. This finding is consistent with a mouse–rat comparison (Ezawa et al. 2006) but not with Arabidopsis multigene families (Mondragon-Palomino and Gaut 2005), the latter of which may constitute an extraordinary case.
Protein functional domains are prone to conversion:
Sequences encoding known protein functional domains are more likely to be involved in conversion than those not encoding domains. We counted the accumulated sequence lengths corresponding to domain and nondomain regions in the duplicated genes that contain HLS segments, for paralogs in both GD and SD blocks as well as tandem clusters. In colinear genes, 18.4% of domain regions are involved in HLS vs. 6.5% of nondomain regions (P < 2.2 × 10−16). In tandem genes, 13.9% of the domain regions are in HLS vs. 7.1% of nondomain regions (P < 2.2 × 10−16).
A road to independence of duplicated genes:
Our findings suggest concerted evolution of recently duplicated homeologous and/or paralogous genes, with a long road to regaining independence. The large SD block in rice chromosomes 11 and 12 thought to have emerged only 5–7 MYA, provides us a good chance to observe the evolution of hundreds of young paralogs. In this SD block, we found a whole-gene conversion frequency of 1.8% and partial-gene conversion frequency of 6.4% (7/109). The discovery of several possible crossovers as well as these whole-gene and partial conversions, indicate a high level of concerted evolution in this particular region and may reflect the behavior of many genomes within the first few million years after duplication or polyploidization. Indeed, crossovers may remove the evidence of earlier conversions causing us to underestimate conversion events.
In contrast, the 70-MY-old rice paralogs have now largely escaped concerted evolution. Among GD genes in the longest 20 blocks, no gene has been wholly converted and the partial-gene conversion frequency is only <0.36% (2/566) in the ∼0.4 MY since the japonica–indica divergence. Nonetheless, there appears to have been appreciable concerted evolution of these paralogs earlier in their evolutionary history—an estimated 8.3% were converted since the sorghum–rice split ∼41 MYA.
Key to estimating the rates of concerted evolution of recently duplicated genes was access to the entire genome—studies reporting independent evolution of small numbers of homeologs (Cronn et al. 1999) may have scanned too little of the gene space to find gene conversion.
Concerted evolution and classical evolutionary models:
In the present analysis, we explore a phenomenon that is not expected from classical evolutionary views, i.e., that duplicated genes even at distant locations in the genome may evolve more slowly than singleton genes. While models like purifying selection and regulatory divergence may explain the slow evolution of some paralogs (Hughes and Hughes 1993; Moore and Purugganan 2003), we show that gene conversion has also contributed to concerted evolution between rice paralogs.
What is the relationship between concerted evolution and classical evolutionary theory about gene duplication (Stephens 1951; Ohno 1970; Taylor and Raes 2004)? The mechanics of recombination (e.g., Holliday 1966) could make conversion or crossing over an unavoidable interruption of classical evolutionary processes, after which the homogenized paralogs restart their evolutionary journey in response to a spectrum of selection pressures. For example, as justified above, colinear paralogous genes on chromosomes 11 and 12 have clearly been homogenized by both crossing over and conversion. Nonetheless, we found these genes to display a range of characteristics of positive selection [Ka > Ks between 27 paralogs (9.7%)], relaxed purifying selection [Ka = Ks between 5 paralogs (1.7%)], or purifying selection [Ka < Ks between 246 paralogs (88.6%)]. Thus, a degree of concerted evolution between duplicated genes is not necessarily incompatible with classical evolutionary models favoring their divergence.
A degree of concerted evolution would fit very well with the recent proposal that the functional divergence of duplicated genes favored by classical models (Stephens 1951; Ohno 1970) may only be one extreme in a continuum of duplicated gene fates, the other extreme being occupied by functional conservation (Paterson et al. 2006). We found two cases in which chromosome 11/12 paralogs and three additional cases in which proximally duplicated genes appear to have each undergone conversion independently in both subspecies japonica and indica (Table 1; Table 3). In view of the small percentages of genes converted, these independent parallel conversions of the same genes are extremely unlikely to have occurred by chance (P < 0.003 for chromosome 11/12 paralogs; P < 10−12 for proximally duplicated genes). There is also a case in which two proximal paralogs have been involved in two partial-conversion events (P < 10−4). One explanation may be that concerted evolution of some genes is actually favored by selection, perhaps contributing to the spectrum of fates that we suggest to exist (Paterson et al. 2006). The small number of affected genes (Tables 1 and 3) show no obvious function or pathway that might be preferentially affected, playing a wide range of roles in cell death, cytokinin dehydrogenation, aldehyde oxidation, seed storage protein (glutelin), and lectin reception.
Summary and implications:
The prevalence of concerted evolution among the colinear (homeologous) and tandem paralogs is surprising, but consistent with previous findings for genes involved in olfaction, immune response, HLA, MHC, sex or reproductive isolation, mating type, multiallelic systems, and tissue- or time-specific expression (Bettencourt and Feder 2002; Mondragon-Palomino and Gaut 2005), and involving a wide range of species including yeast, fly, plants, and mammals.
Higher conversion rates in recent than in ancient duplicates, in domain than in nondomain regions, and in exons than in introns are all fully consistent with the need for DNA similarity as the physical basis of conversion and suggest a sort of circularity—that DNA sequences conserved by selection may be prone to further conservation by relatively frequent conversion. Our focus on the third codon sites should mitigate the contention that conservation of domain (or other) regions alone may be sufficient to explain the results. However, we note that analysis of the first and second nucleotides in the codon also support our discovery of gene conversion in an appreciable number of rice paralogs (supplemental files 3 and 4 at http://www.genetics.org/supplemental/).
Since ectopic recombination in plants is dramatically reduced by even small variations in DNA sequence (Li et al. 2006), one can envision an exponential decline in conversion frequencies, with genome duplication followed by rapid restoration of independent evolution for nonconserved DNA and a longer road to independence for conserved sequences. This implies that the sequences of all duplicated genes may be “sheltered,” albeit inadvertently, by more frequent conversion during the period of instability immediately following genome duplication. Such sheltering may be especially important to avoiding the deleterious effects of Muller's ratchet (Muller 1932) under asexual reproductive systems, perhaps partly explaining why so many apomicts (Bayer and Stebbins 1987) and other clonally propagated angiosperms are polyploids. For domain-rich genes, which tend to be preferentially preserved in duplicate (Chapman et al. 2006), the exons might be subject to concerted evolution for much longer time periods. The formation of heteroduplexes that may be necessary to permit conversion of ancient duplicated genes (Holliday 1966) may also occasionally lead to nonhomologous chromosomal associations during mitosis in rice (Lawrence 1931) and perhaps other organisms.
Acknowledgments
We thank numerous members of the Paterson lab for assistance and appreciate financial support from the National Science Foundation (DBI-0115903 and MCB-0450260 to A.H.P. and J.E.B.).
References
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers and D. J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Bayer, R. J., and G. L. Stebbins, 1987. Chromosome numbers, patterns of distribution, and apomixis in Antennaria (Asteraceae: Inuleae). Syst. Bot. 12: 305–319. [Google Scholar]
- Bettencourt, B. R., and M. E. Feder, 2002. Rapid concerted evolution via gene conversion at the Drosophila hsp70 genes. J. Mol. Evol. 54: 569–586. [DOI] [PubMed] [Google Scholar]
- Brown, R. D., E. Mattoccia and G. P. Tocchini-Valentini, 1972. On the role of RNA in gene amplification. Acta Endocrinol. Suppl. 168: 307–318. [DOI] [PubMed] [Google Scholar]
- Chapman, B. A., J. E. Bowers, F. A. Feltus and A. H. Paterson, 2006. Buffering of crucial functions by paleologous duplicated genes may contribute cyclicality to angiosperm genome duplication. Proc. Natl. Acad. Sci. USA 103: 2730–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronn, R. C., R. L. Small and J. F. Wendel, 1999. Duplicated genes evolve independently after polyploid formation in cotton. Proc. Natl. Acad. Sci. USA 96: 14406–14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezawa, K., S. Oota and N. Saitou, 2006. Proceedings of the SMBE Tri-National Young Investigators' Workshop 2005. Genome-wide search of gene conversions in duplicated genes of mouse and rat. Mol. Biol. Evol. 23: 927–940. [DOI] [PubMed] [Google Scholar]
- Galtier, N., 2003. Gene conversion drives GC content evolution in mammalian histones. Trends Genet. 19: 65–68. [DOI] [PubMed] [Google Scholar]
- Gao, L. Z., and H. Innan, 2004. Very low gene duplication rate in the yeast genome. Science 306: 1367–1370. [DOI] [PubMed] [Google Scholar]
- Holliday, R., 1966. Studies on mitotic gene conversion in Ustilago. Genet. Res. 8: 323–337. [DOI] [PubMed] [Google Scholar]
- Hughes, M. K., and A. L. Hughes, 1993. Evolution of genes in a tetraploid animal, Xenopus laevis. Mol. Biol. Evol. 10: 1360–1369. [DOI] [PubMed] [Google Scholar]
- Hulbert, S. H., C. A. Webb, S. M. Smith and Q. Sun, 2001. Resistance gene complexes: evolution and utilization. Annu. Rev. Phytopathol. 39: 285–312. [DOI] [PubMed] [Google Scholar]
- Itoh, T., T. Tanaka, R. A. Barrero, C. Yamasaki, Y. Fujii et al., 2007. Curated genome annotation of Oryza sativa ssp. japonica and comparative genome analysis with Arabidopsis thaliana. Genome Res. 17: 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, M., B. W. Birren and E. S. Lander, 2004. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428: 617–624. [DOI] [PubMed] [Google Scholar]
- Kondrashov, F. A., I. B. Rogozin, Y. I. Wolf and E. V. Koonin, 2002. Selection in the evolution of gene duplications. Genome Biol. 3: RESEARCH0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, W. J. C., 1931. The secondary association of chromosomes. Cytologia 2: 352–384. [Google Scholar]
- Li, L., M. Jean and F. Belzile, 2006. The impact of sequence divergence and DNA mismatch repair on homeologous recombination in Arabidopsis. Plant J. 45: 908–916. [DOI] [PubMed] [Google Scholar]
- Liao, D., 2000. Gene conversion drives within genic sequences: concerted evolution of ribosomal RNA genes in bacteria and archaea. J. Mol. Evol. 51: 305–317. [DOI] [PubMed] [Google Scholar]
- Lin, Y. S., J. K. Byrnes, J. K. Hwang and W. H. Li, 2006. Codon-usage bias versus gene conversion in the evolution of yeast duplicate genes. Proc. Natl. Acad. Sci. USA 103: 14412–14416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and J. S. Conery, 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155. [DOI] [PubMed] [Google Scholar]
- Lynch, M., M. O'Hely, B. Walsh and A. Force, 2001. The probability of preservation of a newly arisen gene duplicate. Genetics 159: 1789–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragon-Palomino, M., and B. S. Gaut, 2005. Gene conversion and the evolution of three leucine-rich repeat gene families in Arabidopsis thaliana. Mol. Biol. Evol. 22: 2444–2456. [DOI] [PubMed] [Google Scholar]
- Moore, R. C., and M. D. Purugganan, 2003. The early stages of duplicate gene evolution. Proc. Natl. Acad. Sci. USA 100: 15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, H. J., 1932. Some genetic aspects of sex. Am. Nat. 66: 118–138. [Google Scholar]
- Nei, M., and T. Gojobori, 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3: 418–426. [DOI] [PubMed] [Google Scholar]
- Ohno, S., 1970. Evolution by Gene Duplication. Springer, Berlin.
- Ohta, T., 1984. Some models of gene conversion for treating the evolution of multigene families. Genetics 106: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., 2003. Evolution by gene duplication revisited: differentiation of regulatory elements versus proteins. Genetica 118: 209–216. [PubMed] [Google Scholar]
- Paterson, A. H., J. E. Bowers and B. A. Chapman, 2004. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. USA 101: 9903–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson, A. H., B. A. Chapman, J. C. Kissinger, J. E. Bowers, F. A. Feltus et al., 2006. Many gene and domain families have convergent fates following independent whole-genome duplication events in Arabidopsis, Oryza, Saccharomyces and Tetraodon. Trends Genet. 22: 597–602. [DOI] [PubMed] [Google Scholar]
- Petes, T. D., R. E. Malone and L. S. Symington, 1991. Recombination in yeast, pp. 407–421 in The Molecular and Cellular Biology of the Yeast Saccharomyces, edited by J. Broach, E. Jones and J. Pringle. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Rice Chromosomes 11 and 12 Sequencing Consortia, 2005. The sequence of rice chromosomes 11 and 12, rice in disease resistance genes and recent gene duplications. BMC Biology 3: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer, S., 1989. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 6: 526–538. [DOI] [PubMed] [Google Scholar]
- Semple, C., and K. H. Wolfe, 1999. Gene duplication and gene conversion in the Caenorhabditis elegans genome. J. Mol. Evol. 48: 555–564. [DOI] [PubMed] [Google Scholar]
- Skaletsky, H., T. Kuroda-Kawaguchi, P. J. Minx, H. S. Cordum, L. Hillier et al., 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423: 825–837. [DOI] [PubMed] [Google Scholar]
- Stephens, S., 1951. Possible significance of duplications in evolution. Adv. Genet. 4: 247–265. [DOI] [PubMed] [Google Scholar]
- Szostak, J. W., and R. Wu, 1980. Unequal crossing over in the ribosomal DNA of Saccharomyces cerevisiae. Nature 284: 426–430. [DOI] [PubMed] [Google Scholar]
- Taylor, J. S., and J. Raes, 2004. Duplication and divergence: the evolution of new genes and old ideas. Ann. Rev. Genet. 38: 615–643. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocchini-Valentini, G. D., P. Fruscoloni and G. P. Tocchini-Valentini, 2005. Structure, function, and evolution of the tRNA endonucleases of Archaea: an example of subfunctionalization. Proc. Natl. Acad. Sci. USA 102: 8933–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., X. Shi, B. Hao, S. Ge and J. Luo, 2005. Duplication and DNA segmental loss in the rice genome: implications for diploidization. New Phytol. 165: 937–946. [DOI] [PubMed] [Google Scholar]
- Wang, X., X. Shi, Z. Li, Q. Zhu, L. Kong et al., 2006. Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice. BMC Bioinformatics 7: 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, M. E., and B. I. Crother, 2000. Gene conversions may obscure actin gene family relationships. J. Mol. Evol. 50: 170–174. [DOI] [PubMed] [Google Scholar]
- Yu, J., J. Wang, W. Lin, S. Li, H. Li et al., 2005. The genomes of Oryza sativa: a history of duplications. PLoS Biol. 3: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L., T. J. Vision and B. S. Gaut, 2002. Patterns of nucleotide substitution among simultaneously duplicated gene pairs in Arabidopsis thaliana. Mol. Biol. Evol. 19(9): 1464–1473. [DOI] [PubMed] [Google Scholar]
- Zhu, Q., and S. Ge, 2005. Phylogenetic relationships among A-genome species of the genus Oryza revealed by intron sequences of four nuclear genes. New Phytol. 167: 249–265. [DOI] [PubMed] [Google Scholar]