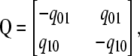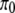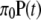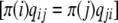Abstract
The insect chemoreceptor superfamily comprises the olfactory receptor (Or) and gustatory receptor (Gr) multigene families. These families give insects the ability to smell and taste chemicals in the environment and are thus rich resources for linking molecular evolutionary and ecological processes. Although dramatic differences in family size among distant species and high divergence among paralogs have led to the belief that the two families evolve rapidly, a lack of evolutionary data over short time scales has frustrated efforts to identify the major forces shaping this evolution. Here, we investigate patterns of gene loss/gain, divergence, and polymorphism in the entire repertoire of ∼130 chemoreceptor genes from five closely related species of Drosophila that share a common ancestor within the past 12 million years. We demonstrate that the overall evolution of the Or and Gr families is nonneutral. We also show that selection regimes differ both between the two families as wholes and within each family among groups of genes with varying functions, patterns of expression, and phylogenetic histories. Finally, we find that the independent evolution of host specialization in Drosophila sechellia and D. erecta is associated with a fivefold acceleration of gene loss and increased rates of amino acid evolution at receptors that remain intact. Gene loss appears to primarily affect Grs that respond to bitter compounds while elevated Ka/Ks is most pronounced in the subset of Ors that are expressed in larvae. Our results provide strong evidence that the observed phenomena result from the invasion of a novel ecological niche and present a unique synthesis of molecular evolutionary analyses with ecological data.
DROSOPHILA has emerged as one of the most valuable models for understanding chemoreception. Its value stems from a relatively simple anatomical structure, the vast genetic tools available, and the recent identification of what are believed to be the complete olfactory receptor (Or) and gustatory receptor (Gr) repertoires (Clyne et al. 1999, 2000; Gao and Chess 1999; Vosshall et al. 1999; Robertson et al. 2003; Hallem et al. 2004). In Drosophila melanogaster, the Or and Gr families comprise 60 genes each, encoding 62 and 68 proteins, respectively (Robertson et al. 2003). These genes are peripheral components of the chemosensory system. They are predicted to encode 7 transmembrane proteins that bind environmental chemicals and trigger nerve signals to higher processing centers in the brain. It has been demonstrated that in most cases, only one Or gene is expressed in any given olfactory receptor neuron and that this Or determines not only the odors to which the neuron is sensitive, but also the neuron's response dynamics (Hallem et al. 2006). Less is known about Grs, but it is clear that multiple Gr genes are often expressed in a single gustatory receptor neuron (Amrein and Thorne 2005). Although the two families are common to diverse insects, they share little sequence similarity with each other and do not appear to be homologous with functionally similar OR and GR genes found in vertebrates (Hallem et al. 2006).
Comparisons of the Or and Gr families from distantly related insects (Drosophila, mosquito, and honeybee) have uncovered dramatic changes in gene family size and content (Hill et al. 2002; Robertson et al. 2003; Robertson and Wanner 2006) and fueled the widely held suspicion that these genes evolve rapidly. It is not clear, however, how chemoreceptors evolve over short time scales, as comparisons of closely related species have been limited by a lack of whole-genome sequences. This void was recently filled by the release of 11 new Drosophila genomes, 4 of which belong to species in the melanogaster subgroup that, together with D. melanogaster itself, share a common ancestor within the past 12 million years (Figure 1). Taking advantage of all 11 new genomes, two recent studies documented overall stasis in the size of the Or family across the genus as a whole (Guo and Kim 2007; Nozawa and Nei 2007). But there has been no investigation of changes in the size of the Gr family and no comprehensive investigation of sequence evolution among closely related species for either family (but see Tunstall et al. 2007 for a study of 11 genes).
Figure 1.—

Species tree depicting the members of the melanogaster subgroup examined in this study. Numbers at nodes are estimates of the divergence times in millions of years (estimated by the mutational distance method using whole-genome data, S. Kumar, unpublished data). The two shaded triangles highlight independent host specialization events. D. sechellia has evolved to specialize exclusively on Morinda citrifolia, a coastal shrub that is toxic to all the other species. D. erecta has evolved to specialize on Pandanus candelabrum, a tree that grows in dense stands in the swampy areas of West Africa (Lachaise and Tsacas 1974). During the ∼3 months of every year when P. candelabrum is fruiting, D. erecta uses it exclusively (Rio et al. 1983). It is not clear, however, what D. erecta does for the rest of the year. It may suffer a drastic reduction in population size and opportunistically exploit alternative hosts (two individuals were once reared from non-Pandanus fruit; Rio et al. 1983), or it may enter some sort of diapause. D. melanogaster and D. simulans are cosmopolitan species while the other three are African endemics. The P. candelabrum illustration is reprinted with permission from Watson and Dallwitz (1992); the M. citrifolia illustration is modified from that found on a public domain website (http://www.fs.fed.us/global/iitf/pdf/shrubs/Morinda%20citrifolia.pdf).
A molecular evolutionary analysis of this kind is of broad interest for several reasons. For one, the chemoreceptor superfamily has many attributes that facilitate general inferences regarding molecular evolution. Its large size provides unusual statistical power. Its decomposition into two approximately equal-sized families (Ors and Grs) allows for compelling parallel studies and contrasts. And the fact that Or and Gr genes are distributed throughout the genome and do not show strong codon bias (a sign of selection on silent sites; Akashi 1994) makes them ideal subjects for classic tests of neutrality.
A second feature of the chemoreceptor superfamily that renders a study of its evolutionary behavior interesting is the fact that the functions and expression patterns of its constituent genes are rapidly being characterized. The past five years have witnessed the publication of nearly 100 articles on Drosophila Or and Gr genes. This information can guide biologically meaningful analyses of variation in evolutionary behavior within the superfamily. That variation, in turn, may provide insight into as yet undescribed functions, guiding further molecular genetic studies.
Last, because the sequenced Drosophila species within the melanogaster subgroup are ecologically diverse, an analysis of lineage-specific evolution may provide insight into the role of chemoreceptors in ecological adaptation. For example, while D. melanogaster, D. simulans, and D. yakuba are generalist flies exploiting a broad array of rotting fruit, D. sechellia and D. erecta have independently specialized on novel host fruit, Morinda citrifolia and Pandanus candelabrum, respectively (Figure 1; Tsacas and Bachli 1981; Rio et al. 1983; Louis and David 1986). The acquisition of these novel hosts was likely associated with dramatic changes in the microhabitats to which the two species are regularly exposed, including not only food plants, but also resting and mating sites, microclimates, and natural enemies. It therefore makes sense that Or and Gr genes, which represent the interface between the fly and its chemical environment, would experience novel evolutionary forces and display unusual patterns of evolution along these lineages. Indeed, a previous study demonstrated accelerated gene loss and amino acid substitution in D. sechellia Or and Gr genes compared to D. simulans (McBride 2007). With three additional species (one specialist and two generalists), forming a tree with six additional lineages (one specialist and five generalists; Figure 1), we can examine the generality of these phenomena and characterize them in greater detail.
Here we present a detailed molecular evolutionary analysis of the chemoreceptor superfamily among five closely related species of Drosophila from the melanogaster subgroup. We focus on (1) fundamental questions regarding the molecular evolution of the superfamily as a whole and contrasts between its two constituent Or and Gr families, (2) variation in the evolutionary behavior of discrete functional/expression/phylogenetic groups within each family, and (3) lineage-specific evolution associated with host specialization.
MATERIALS AND METHODS
Annotations:
We annotated Or and Gr genes in the comparative analysis freeze 1 (CAF1; Drosophila 12 Genomes Consortium 2007) genome assemblies of D. simulans, D. sechellia, D. yakuba (unreconciled version), D. erecta, and D. ananassae using two semiredundant automated pipelines followed by manual revision. The pipelines both involved searching the new genomes for sequences similar to the D. melanogaster Or and Gr proteins from Robertson et al. (2003) using TBLASTN (Altschul et al. 1998) and then predicting preliminary gene structures in the regions surrounding the resulting hits using GeneWise (Birney et al. 2004). One pipeline fed GeneWise a 40-kb region surrounding each TBLASTN hit with an E-value < 0.1. The other pipeline used chaining software (Kent et al. 2003) to establish the coordinates of the genomic slices read in by GeneWise, with the addition of 1 kb of flanking sequence. We manually curated the resulting predictions to ensure reasonable starts, stops, intron/exon structure, and splice sites. We filled all assembly gaps present within receptor gene coding regions (with the exception of a single 5′ gap in DanaGr23aA) and confirmed putative nonsense mutations by direct resequencing from the genome strains (plus an additional outbred strain for D. sechellia and D. erecta). Orthologs were defined as unique reciprocal best hits that shared at least one adjacent upstream or downstream neighbor (i.e., were microsyntenic).
A second set of D. simulans Or/Gr annotations was created using the six syntenic genome assemblies produced by the Drosophila Population Genomics Project (http://www.dpgp.org; Begun et al. 2007). Each of these assemblies constitutes low coverage shotgun sequence data from a single inbred strain assembled via alignment to the D. melanogaster genome. By extracting regions syntenic to D. melanogaster Or/Gr genes from each assembly, we were able to gather a sample of six D. simulans alleles for most loci (used for analyses of polymorphism within D. simulans). We also constructed a single representative “syntenic” allele for each D. simulans Or/Gr gene. This single allele was usually a full-length coding sequence chosen randomly from one of the six assemblies. When no single assembly contained a full-length coding sequence, however, we constructed the representative allele by piecing together segments from different syntenic assemblies. Since the syntenic assemblies seemed to contain fewer mistakes than the CAF1 D. simulans assembly (e.g., many putative nonsense mutations resulting from low-quality reads in the CAF1 assembly were masked by a stringent phred filter in the syntenic assemblies), these representative syntenic alleles were substituted for those derived from the CAF1 assembly for protein tree inference and analyses of divergence whenever possible. In particular, this substitution was made for almost all genes with orthologs in D. melanogaster, but not for genes absent in D. melanogaster (since the latter are not covered by the syntenic assemblies; see Final Allele column in supplemental Table 1B at http://www.genetics.org/supplemental/ for the source assembly of each simulans allele used in our analyses). We did not resequence putative nonsense mutations from the CAF1 D. simulans assembly unless they were supported by, or not covered by, the syntenic assemblies.
Protein tree inference:
We used Bayesian methods to infer the phylogenetic relationships among all annotated Or and Gr genes from all five D. melanogaster subgroup species plus D. ananassae. Pseudogenes were repaired (frame corrected) and included, as long as ≥20% of the full coding sequence was present. Treating Ors and Grs separately, we aligned translated coding sequences using ClustalW (Thompson et al. 1994) under two alignment parameter settings: a relaxed setting (−gapopen = 9 −gapext = 0.18) and the default setting. We then inferred a single protein tree for each family using the MrBayes MPI software package (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003; Altekar et al. 2004); mcmc nchains = 8 ngen = 1,000,000 samplefreq = 100 Temp = 0.03). Chain conversion was assessed as recommended in the user manual: (1) the potential scale reduction factors were all very close to 1, (2) the average standard deviation of split frequencies were all <0.05, (3) plots of the iteration vs. the log probability of the data (using the sump command) showed no trends, and (4) the likelihoods for separate runs on each data set were very close. Using TreeJuxtaposer (Munzner et al. 2003), the trees resulting from the two alternative alignment settings were compared and found to have only minor differences, excluding the following two cases. First, the relaxed Or tree has the Or67d and Or83c orthologs grouped with the Or56a, Or43a, Or49b, Or30a clade with a posterior of 0.9; the default tree has the Or67d and Or83c orthologs more closely related to the Or65c, Or65b, Or65a, Or47b clade. Second, the default Gr tree provides a posterior of 0.95 for a node that places the Gr22a orthologs as an outgroup to Gr22f, Gr22c, Gr22b, Gr22d, Gr22e, and thus changes some of the relationships within this clade; the relaxed tree provided a posterior <0.75. Trees based on the relaxed alignment settings are reported on here. In Figures 2 and 3, we collapsed all nodes with <75% posterior support and pruned all D. ananassae genes and all but one representative branch per D. melanogaster subgroup ortholog set. No nodes were collapsed in, nor genes/orthologs pruned from, the trees in supplemental Figures 1 and 2 at http://www.genetics.org/supplemental/. Tree manipulation was performed using TreeDyn (Chevenet et al. 2006).
Figure 2.—

Bayesian protein tree for the Or family illustrated with an arbitrary root. All nodes with posterior support (red numbers) <75% have been collapsed and terminal branches have been pruned to display only a single representative branch per ortholog set (see supplemental Figure 1 at http://www.genetics.org/supplemental/ for unaltered version). Dots to the right of the tree indicate the number of corresponding orthologs found in each of the melanogaster subgroup species: blue, melanogaster; orange, simulans; green, sechellia; yellow, yakuba; black, erecta. Red slashes through dots indicate pseudogenes. Partially deleted fragments that lack >80% of their coding regions are treated as absent (no dot).
Figure 3.—

Bayesian protein tree for the Gr family illustrated with an arbitrary root. All nodes with posterior support (red numbers) <75% have been collapsed and terminal branches have been pruned to display only a single representative branch per ortholog set (see supplemental Figure 2 at http://www.genetics.org/supplemental/ for unaltered version). Dots to the right of the tree indicate the number of corresponding orthologs found in each of the melanogaster subgroup species: blue, melanogaster; orange, simulans; green, sechellia; yellow, yakuba; black, erecta. Red slashes through dots indicate pseudogenes. Partially deleted fragments that lack >80% of their coding regions are treated as absent (no dot).
Nomenclature:
After detailed discussion with several other research groups interested in Drosophila chemosensory genes (including the authors of Drosophila Odorant Receptor Nomenclature Committee 2000; Robertson et al. 2003; Guo and Kim 2007; Vieira et al. 2007), we agreed upon the following scheme for naming Or and Gr genes in the new Drosophila genomes. First, according to the community standard, all genes identified in a new genome assembly were given a four-letter species-specific prefix (e.g., genes identified in the D. yakuba assembly always begin with Dyak). Second, a gene with a one-to-one ortholog in D. melanogaster was named after the D. melanogaster copy (e.g., the D. yakuba ortholog of DmelOr83b was named DyakOr83b). Third, a gene with multiple orthologs in D. melanogaster (resulting from a duplication along the D. melanogaster lineage) was named after the D. melanogaster ortholog with the lowest number or letter (e.g., the D. yakuba copy of DmelOr19a and DmelOr19b was named DyakOr19a). Fourth, a gene that duplicated along the lineage of a new species creating multiple orthologs for a single D. melanogaster gene, was named after the single D. melanogaster ortholog with the addition of a hyphen and a unique numeral (e.g., the two D. yakuba duplicates of the gene that is named DmelOr67a in D. melanogaster were named DyakOr67a-1 and DyakOr67a-2). Fifth, a gene without an ortholog in D. melanogaster (due to a deletion along the D. melanogaster lineage) was named by adding an “L” (for “like”) and a number to the end of the name of the D. melanogaster gene to which it was most closely related (e.g., a D. yakuba gene that has no ortholog in D. melanogaster, but is closely related to DmelOr98a, was named DyakOr98aL1). Finally, new isoforms of known D. melanogaster genes were given a unique upper case letter suffix (e.g., a new D. yakuba isoform of Or69a, which already has two isoforms in D. melanogaster named DmelOr69aA and DmelOr69aB, was named DyakOr69aC). Note that although we did not come across this problem, situations may arise where a gene in a non-melanogaster species is not closely related to any genes found in the D. melanogaster genome. For example, Guo and Kim (2007) annotated two sets of Or genes in D. grimshawi, D. willistoni, D. virilis, and D. mojavensis that are <20% similar at the amino acid level to the nearest D. melanogaster gene. They named these genes by adding an “N” (for “new”) and a unique numeral to the appropriate species prefix (e.g., DgriOrN1 and DgriOrN2). Finally, in supplemental Figures 1 and 2 at http://www.genetics.org/supplemental/ we have appended a “_P” to the end of the name of verified pseudogenes. Supplemental Table 1 at http://www.genetics.org/supplemental/ includes special columns that show how our names for genes in the Or family correspond to those from Guo and Kim (2007) and Nozawa and Nei (2007).
Divergence analyses 1—Ka/Ks:
We inferred the ratio of replacement to silent substitution (Ka/Ks) for each chemoreceptor gene present in the D. melanogaster subgroup by maximum likelihood as implemented in PAML (Yang 1997). Our inference for each gene was based on a manually curated ClustalW alignment of the D. melanogaster, D. simulans, D. sechellia, D. yakuba, and D. erecta orthologs (pseudogenes excluded) plus the nearest outgroup sequence (usually the D. ananassae ortholog, but sometimes a closely related paralog). Using a branch model, we assigned one Ka/Ks ratio to the outgroup branch, and a second independent ratio to all other branches (model = 2, NSsites = 0). The outgroup ratio was then discarded leaving a single Ka/Ks ratio characteristic of the divergence of the given gene within the D. melanogaster subgroup. For this analysis and for all analyses described below, we assumed the topology illustrated in Figure 1, placing D. yakuba and D. erecta as sister species. We compared the median and mean Ka/Ks ratios of interesting subsets of chemoreceptor genes using nonparametric two-sample Wilcoxon rank-sum tests or parametric t-tests/ANOVAs. Although log-transformed data were substituted for raw data for parametric analyses of variation within the Or family, the Gr data did not require such a transformation. Our analyses excluded genes that have been lost (or duplicated) along any lineage, except when explicitly comparing “lost” genes to those that remain intact in all species.
Divergence analyses 2—rate heterogeneity:
To investigate whether replacement and silent divergence within the Or and Gr families is clocklike, we followed a maximum-likelihood procedure provided by Langley and Fitch (1974). The aim was to investigate whether the observed data deviate from what would be expected given a constant Poisson clock. Briefly (Langley and Fitch 1974, p. 162), the likelihood of the observed number of substitutions in the mth protein along the ith branch (χm,i) is
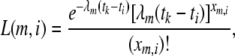 |
where λm is the proportionate substitution rate of the mth protein and tk and ti represent time points at the beginning and the end of a branch. Assuming independence across proteins and along branches, the likelihood of the entire data set is
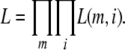 |
Our “observations” were inferred via maximum likelihood using PAML's codeml package (Yang 1997) by parsing rst files from the runs described under Divergence analyses 1—Ka/Ks and are provided in supplemental Table 2 at http://www.genetics.org/supplemental/. With one exception (Or19a), we considered genes that have only a single intact ortholog in all five subgroup species. λm was computed for a particular protein by taking the sum of all substitutions among its five D. melanogaster subgroup orthologs and dividing it by the sum of all substitutions in all proteins. Because the denominator in the likelihood cannot equal zero, ortholog sets in which one or more branches had zero observed substitutions were excluded.
To estimate the maximum likelihood, MCMC sampling with uniform proposal distributions was used to distribute mutations along branches. Three different chains were run, each with very different starting values. The computationally intensive portion of the routine was written in C and data were outputted to R, where statistical analyses and convergence diagnostics were carried out using the CODA package (Plummer et al. 2006). Convergence was considered successful if all three chains showed good mixing, converged to the same likelihood, and if the Geweke's convergence diagnostic (Geweke 1992), which compares the mean values within windows at the start and end of the chain following the burnin, supported stationarity. A likelihood-ratio test was used to test the constant-rate assumption.
To compare our results for Or/Gr genes to a random gene sample of similar size, we repeated the above procedure on a group of 50 protein-coding genes randomly chosen from the D. melanogaster group guide-tree alignments provided by the Drosophila 12 Genomes Consortium (2007).
Divergence analyses 3—index of dispersion:
To complement the rate-heterogeneity tests described above, we carried out a second test of the Poisson molecular clock at the same loci by estimating the index of dispersion [R(t), variance-to-mean ratio] for silent and replacement substitutions. We subsampled the orthologs for each gene in two different ways: (1) excluding D. sechellia [(Dere, Dyak), Dsim, Dmel] and (2) excluding D. simulans [(Dere, Dyak), Dsec, Dmel]. The rationale for this is that speciation between D. simulans and D. sechellia has occurred very recently (≤2 MY; Hey and Kliman 1993; Kliman et al. 2000; S. Kumar, unpublished data), and the stochasticity of coalescent events occurring in species trees with short branches can inflate estimates of R(t) (Hudson 1983). Analyzing D. simulans and D. sechellia separately should avoid this bias. R(t) was calculated following the procedure of Gillespie (1994); lineage weights are provided in supplemental Table 3 at http://www.genetics.org/supplemental/.
Under strict neutrality, R(t) is expected to equal one (Gillespie 1989, 1994). To evaluate the significance of departures from 1, we generated simulated data sets using a procedure similar to those previously described (Gillespie 1989; Zeng et al. 1998). First, ancestral sequences for each ortholog group were inferred using maximum likelihood as implemented in PAML's codeml (rateancestral = 1). These ancestral sequences were “evolved” according to the four-species topologies, with silent and replacement mutations along the sequence being Poisson distributed with means equal to those estimated from the actual data, but after the lineage weights had been applied. The Ors and Grs had their own transition and transversion probabilities that were estimated from their respective full data sets. The procedure was repeated 5000 times for each ortholog group, and R(t) was estimated for each resulting data set as described above.
Polymorphism analyses in D.simulans:
To investigate patterns of polymorphism at Or/Gr loci, we took advantage of the six syntenic D. simulans genome assemblies. We extracted the coding sequence of each Or/Gr gene from all six assemblies (see Annotations) and aligned them to the inferred sequence of the most recent common ancestor (MRCA) of D. simulans and D. melanogaster (ancestral sequence for each gene inferred via maximum likelihood during the PAML runs described in Divergence analyses 1—Ka/Ks). These alignments included many gaps because the coverage from any given assembly was relatively low; the average number of alleles available per site was 3.5. Genes with putative nonsense mutations in any of the six assemblies were not considered. We then wrote automated procedures in Python (http://www.python.org) that used the alignments to infer the number of silent and replacement substitutions/polymorphisms that had occurred at each locus along the D. simulans lineage. Inferences were parsimony based and minimized first the number of total changes and second the number of replacement changes required to explain the variation observed at any given codon site. To reduce the likelihood of including ancestral polymorphisms in our analysis, we ignored all polymorphisms for which one allele was shared with D. melanogaster and the other allele was shared with D. yakuba. Using the resulting substitution and polymorphism counts, we tested for recent positive/purifying selection by (1) conducting a McDonald–Kreitman (MK) test (McDonald and Kreitman 1991) on each individual gene and (2) examining the distribution of the neutrality index (NI, ratio of silent to replacement divergence divided by the ratio of silent to replacement polymorphism) (Rand and Kann 1996) for the Or and Gr families as wholes. These tests excluded genes with fewer than six polymorphisms, six fixations, six silent variants, or six replacement variants (i.e., with any row or column sum less than six). We also compared Ors to Grs by tallying the number of silent and replacement polymorphisms/substitutions across all genes within each family and then asking whether the resulting pooled MK tables were significantly different using a three-way contingency test.
To ask whether the pattern of polymorphism and divergence observed at Or and Gr loci was significantly different from that characterizing the rest of the genome, we repeated the above analyses on a set of 3222 genes scattered throughout the genome using alignments provided by Begun et al. (2007). Since the D. erecta, D. sechellia, and D. ananassae alleles were not included in these alignments, we inferred the sequence of the MRCA of D. melanogaster and D. simulans via parsimony using the D. simulans, D. melanogaster, and D. yakuba alleles only (rather than extracting these ancestral sequences from PAML runs on six species alignments). The Or/Gr data were reanalyzed in the same way for comparison.
Gene loss in specialists:
We examined potential variation in the rate of chemoreceptor gene loss among lineages using a maximum-likelihood framework implemented in a new extension of the program Brownie (O'Meara et al. 2006; see appendix). In this analysis, the status of each Or/Gr gene present in the MRCA of the D. melanogaster subgroup was traced across an ultrametric phylogeny including the five focal species (Figure 1). The likelihood of inferred gene loss events (considered irreversible) was then estimated under five alternative models. The simplest model assigned a single rate of loss to the entire subgroup. The remaining four models assigned an independent rate of loss to each of two groups of lineages defined a priori. We assessed the relative fit of alternative models using corrected Akaike information criteria (AICc) (Hurvich and Tsai 1989) and Akaike weights. Ors and Grs were examined separately.
We investigated the possibility that the Gr genes lost along specialist lineages were a nonrandom subsample of Grs in two different ways. First, we asked whether the lost genes were phylogenetically clustered. Using our unrooted Bayesian Gr tree (Figure 7), we computed the total length of the smallest subtree containing all lost genes; the smallest subtree was defined as that which included the fewest total genes. We then compared this length to a null distribution derived by repeating this procedure for 10,000 random samples of lost Gr genes (of the same size as the real set). The smallest subtree for each data set was identified by comparing a list of the lost genes to a bipartition table using automated Python scripts and the total lengths of these subtrees were computed in PAUP* (Swofford 2002). Second, we asked whether the same genes were lost independently along multiple lineages more often than expected by chance. We simulated losses along the D. melanogaster subgroup topology shown in Figure 4 by assigning loss events to randomly selected genes while holding the number of losses and the branches on which these losses occur constant. We then compared the number of “overlaps” (genes lost independently along two or more lineages) from the real data set to the distribution derived from the simulations. We considered three types of overlaps—those among specialist lineages, those among generalist lineages, and those between specialist and generalist lineages.
Figure 7.—

Bayesian phylogeny of all Gr genes present in the MRCA of the melanogaster subgroup (same as in Figure 3) shown as unrooted phylogram. All nodes have at least 75% posterior probability. Gene names in boldface type denote volatile CO2 (green), putative bitter (orange), putative sweet (blue), and pheromone (purple) receptors. Red branches (solid or dashed) subtend genes that have been lost along either or both of the two specialist lineages. Dashed branches (red or black) subtend genes that have been lost along any of the generalist lineages. The tree is broken up into 10 subfamilies labeled A–H. The thick dotted line in the middle divides the tree into two parts—the upper left of which is the smallest subtree that includes all specialist losses (all red branches; see section entitled Specialists are losing a nonrandom set of Grs).
Figure 4.—

Lineage-specific gene loss and gain in the Or and Gr families. Cartoon phylogenies of the melanogaster subgroup show the distribution of gene loss events (red slashes) and duplications (blue dots) across lineages for the Or (left) and Gr (right) families. Generalist lineages are solid black lines and specialist lineages are dotted lines. The timing of events was inferred via parsimony. Species names are abbreviated to their first three letters.
Divergence in specialists:
To identify variation in substitution rates among lineages, with particular focus on contrasts between the lineages of host generalists and specialists, we implemented two additional PAML branch models on the five species plus outgroup alignments described previously (see Divergence 1—Ka/Ks). Only genes that remained intact in all five subgroup species were considered. Both models assigned the outgroup branch its own independent Ka/Ks ratio. In the “species” model, each branch in the D. melanogaster subgroup was also assigned its own unique Ka/Ks ratio (model = 1, NSsites = 0). In the “ecological” model, the D. sechellia and D. erecta lineages (ecological specialists) were assigned one ratio while the rest of the D. melanogaster subgroup lineages (ecological generalists) were assigned a different ratio (model = 2, NSsites = 0). We then compared the Ka/Ks ratios inferred for various focal lineages using paired Wilcoxon rank-sum tests (each individual species vs. the subgroup as a whole, specialists vs. generalists). Since the D. sechellia and D. erecta lineages were characterized by unusually high Ka/Ks, we further examined substitution along these lineages via sister-species comparisons (sechellia vs. simulans and erecta vs. yakuba) of Ks and Ka individually.
To examine potential heterogeneity in the observed lineage effects across groups of genes with different functions or expression profiles, we conducted two nested ANOVAs on log transformed Ka/Ks data from the ecological model. The first ANOVA applied to Or genes only and tested for the main effects of host ecology (generalist lineages vs. specialist lineages), life stage (genes expressed in adults only vs. adults plus larvae vs. larvae only), the interaction between host ecology and life stage, and the nested effect of individual Or genes within life stages. The interaction effect was of primary interest—addressing the possibility that Ka/Ks along specialist lineages may be elevated for genes expressed during certain life stages but not for genes expressed during other life stages. The second model was similar to the first except that it applied to Gr genes only, and the effect of tuning modality (whether individual genes respond to bitter compounds, sweet compounds, or unknown compounds) was substituted for the effect of life stage. For visual comparison in Figure 9, the large set of Gr genes that respond to unknown compounds was split into “conserved” and “unconserved” subsets on the basis of inferred whole subgroup Ka/Ks ratios. The intact orthologs of genes that have been lost were included in the Gr model to maintain balance among categories; otherwise, there would be only three bitter receptors.
Figure 9.—

Mean Ka/Ks ratios for subsets of Or and Gr genes along generalist (Gen) and specialist (Spec) lineages. (A) Ors: genes expressed in larvae only (dotted line) and in both larvae and adults (dashed line) are more elevated in specialists than are genes expressed in adults only (solid line). (B) Grs: putative sweet receptors (dotted line) are more elevated in specialists than are putative bitter receptors (dashed line) or Gr genes with unknown functions (most and least conserved subsets represented by solid and dashed-dotted lines, respectively). Mean Ka/Ks of the whole-genome set is plotted in shaded lines for comparison in both A and B.
To test whether the lineage variation observed at Or/Gr loci was specific to these gene families or characteristic of the genomes as a whole, we reran the species and ecological PAML models and repeated the paired Wilcoxon contrasts on a whole genome set of alignments provided by the Drosophila 12 Genomes Consortium (2007). Genes with zero silent substitutions along any focal lineage or set of lineages were excluded. Unpaired, two-sample Wilcoxon tests were used to compare the distribution of pairwise differences in Ka/Ks between specific lineages for the whole genome set with the distribution of pairwise differences in Ka/Ks between the same lineages at Or/Gr loci.
RESULTS AND DISCUSSION
Annotations of the Or and Gr families in five newly sequenced species of Drosophila:
Using the known sequences of D. melanogaster Or and Gr proteins as queries within two partially automated annotation pipelines, we identified a total of 328 Or and 369 Gr gene copies in the new genome assemblies of D. simulans, D. sechellia, D. yakuba, D. erecta, and D. ananassae (hereafter referred to using species names only). The coding regions of 13 of these genes overlapped with assembly gaps that we filled by direct resequencing. We also used direct resequencing to confirm or correct the nonsense mutations present in 36 and 24 genes, respectively. The majority (15) of genes with spurious nonsense mutations came from the low coverage sechellia assembly. The comparative analysis freeze 1 (CAF1) simulans assembly also appeared to contain mistakes that were not supported by any of the six alternative syntenic simulans assemblies (see materials and methods). We therefore relied on the syntenic assemblies whenever possible and resequenced CAF1 simulans assembly nonsense mutations only in areas not covered by the syntenic assemblies. Table 1 lists the number of intact genes, pseudogenes, and gene fragments annotated for each species. Supplemental Table 1 at http://www.genetics.org/supplemental/ provides detailed information associated with each annotation, including a description of verified/corrected nonsense mutations. Supplemental Table 4 at http://www.genetics.org/supplemental/ contains the coding sequences themselves. After consultation with several other research groups interested in Drosophila chemosensory genes, we adopted a naming scheme based on orthologous and paralogous relationships with previously named genes in melanogaster (described in materials and methods).
TABLE 1.
Counts of Or and Gr genes annotated in each species
|
Ors |
Grs |
|||||
|---|---|---|---|---|---|---|
| Species | Intact | Pseudo | Fragment | Intact | Pseudo | Fragment |
| melanogaster | 61 | 2 | 1 | 68 | 0 | 5 |
| simulans | 64 | 0 | 1 | 71 | 2 | 0 |
| sechellia | 57 | 6 | 1 | 60 | 12 | 1 |
| yakuba | 64 | 0 | 1 | 71 | 0 | 3 |
| erecta | 62 | 0 | 0 | 60 | 6 | 2 |
|
ananassae |
67 |
4 |
1 |
73 |
8 |
0 |
Individual isoforms were counted as separate genes. The intact category includes all loci segregating at least one intact allele. The pseudo (pseudogene) category includes loci at which all sampled alleles exhibited nonsense mutations. Fragments are degraded loci missing ≥80% of their original coding sequences. Note that comparison with the newly sequenced species indicates that DmOr98b, previously annotated as an intact gene, is actually a pseudogene with a small frameshifting indel.
We compared our Or annotations to those of two recent studies (Guo and Kim 2007; Nozawa and Nei 2007). The three annotation sets are in agreement regarding number and location of genes with the following general exception: both other groups annotated several gene fragments that we ignored. Most of these fragments are nearly identical segments of full-length genes already annotated from a given species and are located on short and/or unassembled contigs. We did not annotate these fragments because we suspect that many reflect assembly error caused by residual heterozygosity in the sequenced inbred strains. The same applies to a putative tandem duplicate of DyakGr92a that we ignored because we were unable to amplify the region extending from the last exon of the first copy to the first exon of the second copy. The loci shared between all three annotation sets also differ somewhat in sequence. These differences appear to be attributable to (1) gaps and spurious nonsense mutations that we filled and corrected (2) minor differences in the melanogaster proteins used as queries, and (3) use of alternative start codons. Finally, our simulans annotations differ substantially in sequence from those of the other two groups because of our use of the six syntenic assemblies.
Using our annotated coding sequences, we calculated the effective number of codons (ENC) (Wright 1990) used in each gene from each melanogaster subgroup species using the codonw server (http://bioweb.pasteur.fr/seqanal/interfaces/codonw.html). Overall, there is little evidence of codon bias, with both Ors and Grs having a mean ENC of ∼52 (Gr range = 36.38–61, Or range = 32.66–61; ENC for each gene is an average across all intact orthologs). There was also little variation in ENC among orthologs of the same gene from different species (supplemental Table 5 at http://www.genetics.org/supplemental/).
Bayesian trees provide confident inferences of branching order deep within both families:
Using Bayesian methods, we inferred unrooted protein trees for both the Or and Gr families. Figures 2 and 3 show abridged versions of these trees; supplemental Figures 1 and 2 at http://www.genetics.org/supplemental/ show full versions. Though computationally intensive, Bayesian methods are more powerful than the neighbor-joining method used by previous studies (Robertson et al. 2003; Guo and Kim 2007; Nozawa and Nei 2007) and provide more confident inferences of branching order deep within the family. Almost all nodes in both trees are strongly supported (Figures 2 and 3). Comparison of our Or tree to that of Guo and Kim (2007) did not reveal any major discrepancies.
Evolution of the Or and Gr families as wholes
To characterize the molecular evolution of the Or and Gr families within the melanogaster subgroup, we adopted three complementary approaches. First, we examined changes in copy number. Second, using comparative sequence data from all five species, we examined rates of orthologous sequence divergence at silent and replacement sites (Ka/Ks, overall rate heterogeneity, and the index of dispersion). Third, using population genetic data from just one species, we contrasted divergence to polymorphism at silent and replacement sites (neutrality index).
Gene gain and loss—overall contraction of gene family size:
The close relationship among the five melanogaster subgroup species made assignments of orthology unambiguous and allowed us to infer the timing of gene duplication and loss events confidently via parsimony (using the phylogeny shown in Figure 1 and assuming loss events to be irreversible). Losses are defined as orthologs that were deleted or exhibited a nonsense mutation in all alleles examined for a particular species. Although they do not include orthologs that we observed to be polymorphic for nonsense mutations (one in erecta, two in melanogaster, and possibly a few in simulans), it is possible that functional alleles are segregating at some lost loci in natural populations. Figures 2 and 3 show the identity of all duplicated/lost genes, and Figure 4 shows the distribution of gain/loss events across all eight lineages. Note that our inferences for the Or family differ substantially from those of Guo and Kim (2007) who report twice as many Or duplications and distribute Or losses differently across the melanogaster subgroup lineages (compare Figure 4 from this study to their Figure 1). These discrepancies are most severe in the subclade including the species with the lowest quality assemblies (simulans and sechellia) and likely result from the previously described differences in annotations (e.g., their acceptance of several spurious nonsense mutations and putative duplicates on identical, short, and/or unassembled contigs).
Starting from a basal set of 64 Or genes present in the MRCA of the subgroup, we observed a total of 4 Or duplication events and 12 Or loss events. Starting from a basal set of 74 Gr genes, we observed 0 Gr duplications and 35 Gr losses. Since two of the Or duplication events and several of both the Or and Gr loss events affected the same genes along different lineages, the numbers of genes that experienced at least 1 duplication or loss are slightly lower. The most striking trend arising from these inferences is the overall contraction of the two families. Using a maximum-likelihood framework implemented in the program Brownie (see appendix), we estimated that the overall rates of loss for Ors and Grs are 0.46 and 1.02 losses per gene per 100 MY, respectively. While a contingency test comparing the proportion of genes lost along at least one lineage to the proportion duplicated showed that the loss rate was only marginally higher than the duplication rate for Ors (Fisher's exact P = 0.05), losses dramatically exceeded duplications for Grs (Fisher's exact P < 10−8). The overall rate of Gr loss, however, masks substantial lineage-specific variation, which we discuss in the Specialists are losing Gr genes approximately five times more rapidly than generalists section.
Divergence 1—Ka/Ks:
Interested in overall rates of divergence, we first examined the ratio of replacement to silent substitution (Ka/Ks) at each locus. We examined a single ratio per gene reflecting substitutions that have occurred across the entire tree in Figure 1. Every Or and Gr was characterized by Ka/Ks less than one (range = 0.01–0.57, median = 0.15; Figure 5; raw data in supplemental Table 6 at http://www.genetics.org/supplemental/), indicating that they are functional and experience purifying selection. Our inferences of Ka/Ks for Ors within the melanogaster subgroup are similar to, but significantly lower than, the values inferred across all 12 Drosophila genomes by Guo and Kim (2007) (paired Wilcoxon P = 0.001, median difference = 0.024). The higher values obtained across all 12 species may reflect saturation at silent sites along long branches.
Figure 5.—

Summaries of the distributions of Ka/Ks, the index of dispersion for both Ka and Ks, and the neutrality index (along the simulans lineage) for the Or and Gr families.
Divergence 2—rate heterogeneity:
To examine the constancy of the rates of silent and replacement divergence among orthologous genes in the Or and Gr families as wholes, we conducted a test of the molecular clock introduced by Langley and Fitch (1974). This method tests whether the observed substitutions (in each gene along each branch) are significantly different than would be expected under a Poisson molecular clock, conditional on the total number of substitutions over a known tree (in all genes across all branches). An advantage of this test is that it can be decomposed into tests of constancy along lineages and across lineages. In addition, we extend the tests to examine silent and replacement substitutions separately.
Conservatively limiting our analysis to the 104 ortholog groups that have not experienced changes in copy number (with the exception of Or19a), we found that both the Or and Gr families are accumulating substitutions in a definitively nonclocklike fashion. Overall tests of heterogeneity in rates of silent and replacement substitution were significant for both families, as were the subtests of heterogeneity within and along branches (all P-values < 10−17; supplemental Table 7 at http://www.genetics.org/supplemental/). A similarly sized data set of randomly chosen protein-coding genes from the same species, however, also led to unequivocal rejection of a neutral Poisson model (supplemental Table 7). This suggests that rate heterogeneity is not unique to the chemoreceptor family and may result from lineage effects such as generation time or from pervasive positive selection. Alternatively, it is also possible that the substitution process in Drosophila is poorly modeled using a Poisson distribution (Gillespie 1994). If true, it would not be surprising that discrepancies summed over many genes would give significant results.
Divergence 3—index of dispersion:
As a complementary approach to the rate-heterogeneity tests, we calculated the index of dispersion [R(t)] for the same subset of proteins, following the procedure outlined by Gillespie (1989, 1994; see materials and methods). Under a strict neutral model, substitutions are Poisson distributed and the variance to mean ratio, R(t), is expected to be 1. Estimates significantly greater than one (overdispersed) have been used to argue against neutral evolution (Gillespie 1989, 1994; Cutler 2000; Kern et al. 2004). These measurements are informative because they pertain to particular proteins, whereas the above tests of rate heterogeneity are descriptive of the families as wholes. Moreover, they provide a test of the Poisson clock that takes uniformly acting lineage effects (e.g., different generation times or population sizes) into account.
Both Or and Gr genes tend to be overdispersed, but to a small degree (Figure 5; raw data in supplemental Table 6 and supplemental Figures 3–6 at http://www.genetics.org/supplemental/). The median R(t) taken over both topologies (see materials and methods) is close to 1.5 for both Or and Gr replacement substitutions and 1.9 for Or and Gr silent substitutions: median Or 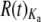 = 1.43, median Gr
= 1.43, median Gr 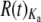 = 1.55, median Or
= 1.55, median Or 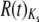 = 1.82, median Gr
= 1.82, median Gr 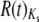 = 1.92. All four of these values are significantly greater than one (Wilcoxon P < 10−6 for each). By simulating empirical null distributions that were based on the scaled trees, we also identified individual genes that were significantly overdispersed. These included 17 Ors and 14 Grs overdispersed for replacement substitutions and 7 Ors and 11 Grs overdispersed for silent substitutions (supplemental Figure 7 at http://www.genetics.org/supplemental/). A contingency test indicated that the proportion of all genes overdispersed for Ka was significantly higher than the proportion overdispersed for Ks (χ2 P = 0.03), particularly within the Or family. Although we did observe several genes that were significantly underdispersed (n = 13), the total number of these cases is close to what we would expect by chance given the number of tests (e.g., ∼5%).
= 1.92. All four of these values are significantly greater than one (Wilcoxon P < 10−6 for each). By simulating empirical null distributions that were based on the scaled trees, we also identified individual genes that were significantly overdispersed. These included 17 Ors and 14 Grs overdispersed for replacement substitutions and 7 Ors and 11 Grs overdispersed for silent substitutions (supplemental Figure 7 at http://www.genetics.org/supplemental/). A contingency test indicated that the proportion of all genes overdispersed for Ka was significantly higher than the proportion overdispersed for Ks (χ2 P = 0.03), particularly within the Or family. Although we did observe several genes that were significantly underdispersed (n = 13), the total number of these cases is close to what we would expect by chance given the number of tests (e.g., ∼5%).
A considerable amount of work has focused on the interpretation of overdispersed proteins (Goldman 1994; Nielsen 1997; Zeng et al. 1998; Cutler 2000; Kern et al. 2004; Wilke 2004). There are several interpretations that are unlikely to apply to our data set. As mentioned previously, factors that vary between lineages in a uniform way across loci are removed by the lineage weighting scheme used prior to R(t) calculation (Gillespie 1989) and should not contribute to the observed patterns. Also, while saturation at silent sites has been shown to bias estimates of 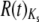 , the mean Ks between the most distant species that we consider is only ∼0.35 for both Ors and Grs. Finally, variation in the strength of selection for major codons (as invoked by Zeng et al. 1998) is unlikely to explain our results; we already reported that Ors and Grs have relatively little codon bias (ENC ∼52), with negligible variation in bias across species (supplemental Table 5 at http://www.genetics.org/supplemental/). In addition, proteins possessing significant
, the mean Ks between the most distant species that we consider is only ∼0.35 for both Ors and Grs. Finally, variation in the strength of selection for major codons (as invoked by Zeng et al. 1998) is unlikely to explain our results; we already reported that Ors and Grs have relatively little codon bias (ENC ∼52), with negligible variation in bias across species (supplemental Table 5 at http://www.genetics.org/supplemental/). In addition, proteins possessing significant  values span the range of ENC values rather than possessing the lowest scores, and there is no correlation between
values span the range of ENC values rather than possessing the lowest scores, and there is no correlation between 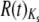 and the absolute value of the deviation of a particular protein's ENC from the family average ENC (supplemental Figures 8 and 9 at http://www.genetics.org/supplemental/).
and the absolute value of the deviation of a particular protein's ENC from the family average ENC (supplemental Figures 8 and 9 at http://www.genetics.org/supplemental/).
The most straightforward interpretation of overdispersion within the Or and Gr families posits that elevated R(t) results from episodic bursts of substitution associated with lineage-specific changes in the strength of positive and/or purifying selection (Gillespie 1989; Kern et al. 2004). This explanation would account for our observation of more frequent overdispersion of Ka than of Ks since selection is likely to be stronger at replacement sites than at silent sites. It would also be compatible with independent evidence of positive selection acting on chemoreceptors in general (see Polymorphism within simulans, below, and Tunstall et al. 2007) and of lineage-specific changes in selection regimes associated with host shifts (see Evolution of Ors and Grs along specific lineages—a role for the chemoreceptor superfamily in host specialization, below). Interestingly, it also provides the best explanation for overdispersion at Or19a. This gene has duplicated along the melanogaster lineage and was therefore excluded from the summary of R(t) results presented above. Nevertheless, the fact that a disproportionate number of replacement substitutions have occurred in this gene along the lineage in which it duplicated (supplemental Table 2 at http://www.genetics.org/supplemental/) and the fact that this burst of substitution appears to have driven the gene's 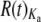 well above one, suggests that changes in the strength of selection have the potential to produce overdispersion on the scale observed in our data set. It also supports previous concerns regarding the use of duplicate genes in R(t) analyses (Ohta 1991).
well above one, suggests that changes in the strength of selection have the potential to produce overdispersion on the scale observed in our data set. It also supports previous concerns regarding the use of duplicate genes in R(t) analyses (Ohta 1991).
Polymorphism within simulans:
Patterns of divergence between species can help reveal nonneutral evolution, but analyses of polymorphism within species typically provide more powerful tests of the specific forces underlying nonneutral evolution. We therefore used available genomic polymorphism data from D. simulans to further characterize the evolutionary forces acting on the Or and Gr families. Our analyses included 54 Or and 61 Gr loci that were covered by the simulans syntenic assemblies and did not show any evidence of polymorphic nonsense mutations. The average number of alleles covered per site at each locus ranged from 0.4 to 5.5 with a mean of 3.5. Although this sample size is too low for analysis of the frequency spectrum, it is sometimes adequate for MK tests of selection (McDonald and Kreitman 1991). MK tests look for a significant difference between the ratio of replacement polymorphism to fixation and the ratio of silent polymorphism to fixation via a contingency test. If all sampled variants and observed fixations are neutral, the two ratios should be the same and the neutrality index—former ratio divided by latter ratio—should equal one (Rand and Kann 1996). Positive selection on replacement sites theoretically lowers this index below one (since it adds to the number of replacement fixations without affecting the number of replacement polymorphisms), while weak purifying selection on replacement sites raises it above one (since it prevents a subset of low-frequency replacement polymorphisms from contributing to the observed replacement fixations).
We inferred the number of silent and replacement polymorphisms and fixations arising along the simulans lineage since this species' MRCA with melanogaster and conducted a polarized MK contingency test at each Or/Gr locus that exhibited a minimum number of variants (see materials and methods). These tests, summarized in Table 2, provided strong evidence for positive selection on both families. First, 6 of 27 Or and 11 of 36 Gr genes were individually significant with NI < 1 at the P < 0.05 level (a larger fraction than expected by chance). Second, the median NI of each family was significantly <1 (Or median NI = 0.61, Wilcoxon P = 0.04; Gr median NI = 0.42, Wilcoxon P < 0.0002). Third, pooled MK tests tallying the total number of silent and replacement polymorphisms and fixations across all loci within each family were significant, with NI < 1 (Or pooled NI = 0.54, Gr pooled NI = 0.38). Interestingly, however, there was no more evidence of positive selection on the Or and Gr families than on 3222 genes scattered across the simulans genome as a whole (Table 2); we found no difference in the proportion of significant tests (contingency P = 0.3), median NI (Wilcoxon P = 0.4), nor pooled NI (three-way contingency test comparing the two pooled MK tables, P = 0.8). Thus either a good portion of the entire simulans genome experiences positive selection (Begun et al. 2007) or some other process such as weak purifying selection on silent sites is driving the pattern. The latter alternative seems unlikely, at least at chemoreceptor loci, which do not show codon bias.
TABLE 2.
McDonald–Kreitman tests for positive selection along the simulans lineage
| Genes | Na | No. significant | P (≤5%) | Median NI | P (=1) | Nb | Pooled NI | Three-way P |
|---|---|---|---|---|---|---|---|---|
| Ors | 27 | 6 | <10−5 | 0.61 | 0.04 | 54 | 0.54 | 0.01 |
| Grs | 36 | 11 | <10−12 | 0.42 | 0.0002 | 61 | 0.38 | |
| Ors + Grsc | 61 | 10 | 0.0003 | 0.48 | <10−5 | 115 | 0.47 | 0.78 |
| Genomec |
3222 |
882 |
0.0000 |
0.55 |
0.0000 |
10,151 |
0.46 |
The results of polarized McDonald–Kreitman (pMK) tests on individual genes is presented in the first five columns, which show the number of genes tested, the number of significant tests, the P-value from a test of whether this number exceeded 5% of genes tested, the median neutrality index (NI) of genes tested, and the P-value from a test of whether the median was significantly less than one. Results from a single pMK test on each pooled gene set are presented in the final three columns, which show the number of genes included in the pool, the NI of the pooled table, and the P-value from a three-way contingency test comparing the pooled NI for one gene set to that from the subsequent gene set (Ors compared to Grs and Ors + Grs compared to the whole genome set).
Genes with any MK table marginal sum <6 were excluded from tests on individual genes.
No genes were excluded from the pooled tests.
Substitutions were polarized using an ancestor inferred via parsimony based on melanogaster and yakuba orthologs only.
The Or and Gr families experience different selection regimes:
Despite evidence of positive selection at both Or and Gr loci, the behavior of the two families is quantitatively different. For one, Gr genes have higher relative rates of replacement divergence (Gr median Ka/Ks = 0.22, Or median Ka/Ks = 0.13; Wilcoxon P = 0.0001; Figure 5). This difference persisted even when excluding genes that have been lost along any lineage (Wilcoxon P = 0.016). High Ka/Ks at Gr loci would traditionally be attributed to stronger positive selection or weaker purifying selection on Gr replacement sites. This interpretation, however, hinges on the idea that Ks reflects only the neutral substitution rate (varying across the genome in concert with local mutation rates). While this assumption is not particularly well supported in Drosophila in general (Akashi 1999), it may be appropriate for chemoreceptors. As previously mentioned, Or and Gr genes show little codon bias. And while Ks did not vary significantly between the Or and Gr families (Wilcoxon P = 0.6), nor among any other discrete functional groups containing genes scattered throughout the genome (data not shown), it did vary significantly among clusters of tandem Ors and Grs (ANOVA P = 0.004, clusters defined as tandem arrays with fewer than 1 kb separating consecutive genes), suggesting that mutation dynamics do vary locally. Finally, regarding this contrast in particular, the fact that Ka by itself was significantly higher at Gr loci than at Or loci (Wilcoxon P = 0.032) supports the idea that Grs experience a different selection regime than do Ors, specifically at replacement sites.
Grs also differed from Ors in their lower neutrality indices along the simulans lineage. The pooled NI of the Gr family (0.38) was significantly lower than the pooled NI of the Or family (0.54) by a three-way contingency analysis (P = 0.01). Summing across loci with varying levels of constraint can complicate the interpretation of pooled MK tests. The difference between the families proved consistent, however, even when broken down into five contrasts between smaller pools of Or and Gr genes with similar rates of substitution and levels of polymorphism (Gr NI was lower than the Or NI for each of the five smaller pools; supplemental Table 8 at http://www.genetics.org/supplemental/). This difference is also reflected in a trend for the Gr family to have a larger proportion of individually significant genes and a lower median NI than the Or family (Figure 5). Since low neutrality indices are associated with positive selection, the observed difference suggests that Grs experience stronger positive selection than Ors. The alternative explanation of weaker purifying selection, however, still cannot be completely ruled out. It is possible, for example, that Or genes contain a class of sites under weak purifying selection (contributing to observed replacement polymorphisms but not fixations) that are completely neutral in Gr genes (contributing to both polymorphisms and fixations). There was no difference in R(t) between the Or and Gr families at either silent or replacement sites (two-sample Kolmogorov–Smirnov tests P = 0.8 for Ka, P = 0.3 for Ks; Figure 5).
Why might Grs experience a different selection regime than Ors? Despite their similarity, the insect olfactory and gustatory codes may be quite different. The olfactory code is combinatorial with most odorants stimulating multiple olfactory receptor neurons (each expressing a single Or gene) and being represented by the unique assemblage of such neurons transmitting impulses to the antennal lobe. The gustatory system, on the other hand, appears to adhere to a labeled-line model wherein tastants stimulate one or more Gr genes within a single class of gustatory neurons (e.g., sweet, bitter, etc.) that are in turn hard wired to specific behaviors (e.g., attraction, repulsion; Marella et al. 2006). It is possible that this difference in organization makes the olfactory system less flexible than the gustatory system to evolutionary changes in single receptor proteins at the periphery. Moreover, although many coexpressed Grs surely serve independent and unique functions (e.g., Gr5a and Gr64a; Chyb et al. 2003; Jiao et al. 2007), some may be partially redundant, releasing each other from evolutionary constraint. Finally, it is possible that of all the compounds that a fly must be able to recognize, the soluble ones are more variable between environments/mates/hosts than the volatile ones, resulting in more frequent selection on Grs for novel binding affinities or sensitivities. Note that the only Gr known to be involved in mate recognition (Gr68a) has neither a particularly low NI (0.38) nor a particularly high Ka/Ks (0.15) (supplemental Table 6 at http://www.genetics.org/supplemental/; but see Tunstall et al. 2007 for evidence of positive selection on a particular codon site).
Evolution of specific groups of genes within the Or and Gr families
Functional geneticists are rapidly accumulating information on the binding specificities and patterns of expression of individual receptor genes. This information, combined with some of our own inferences, allowed us to assign genes to a priori categories on the basis of function, expression, subfamily, or propensity for loss, and subsequently look for meaningful variation in evolutionary behavior among these categories within the Or and Gr families. The following results describe variation we detected in Ka/Ks only. We found no significant variation in R(t), and the simulans polymorphism data are not sufficient for comparisons of NI among small subgroups of genes.
Or expression groups—genes expressed in large basiconic sensilla are highly conserved:
Or proteins and the dendrites of the olfactory receptor neurons (ORNs) in which they are embedded, are housed in porous sensory hairs found on the antennae and maxillary palpi of adult Drosophila. These sensory hairs, called sensilla, come in five distinct morphological classes: large basiconic, small basiconic, trichoid and ceoloconic (on the antenna only), and thin basiconic (on the antenna and maxillary palp; Shanbhag et al. 2000). We used published data to categorize Or genes according to the class of sensillum in which they are expressed (Couto et al. 2005) and found significant variation among these groups. In particular, Ors expressed in large basiconic sensilla on the adult antenna had significantly lower Ka/Ks than Ors expressed in most other classes (one-way ANOVA P = 0.03; Figure 6A, middle; supplemental Table 9 at http://www.genetics.org/supplemental/; coeloconic sensilla were excluded from the analysis since only one Or gene is known to be expressed therein). This particularly applies to the ab1 and ab2 sensilla, which house a total of six ORNs expressing four of the five most conserved adult Or genes (Or59b, Or85a, Or42b, and Or92a) and, incidentally, two of the most conserved Gr genes (Gr21a and Gr63a). Or59b and Or85a have been physiologically characterized but do not appear unusual in their binding affinities (Hallem and Carlson 2006). Interestingly, Or42b has been implicated in the ability of flies to detect an odor given off by stressed conspecifics (G. Suh, personal communication), and we have no information on Or92a. We found no variation in Ka/Ks among groups of Or genes that respond to aliphatic vs. aromatic compounds (ANOVA P = 0.40), that are expressed during different life stages (ANOVA P = 0.47), or that belong to different subfamilies within the Or tree (ANOVA P = 0.2, Figure 6A, right; supplemental Table 9 at http://www.genetics.org/supplemental/; subfamilies marked on the unrooted phylogram in supplemental Figure 10 at http://www.genetics.org/supplemental/).
Figure 6.—

Variation in mean Ka/Ks among subsets of Or genes (A) and Gr genes (B). The left section in each row contrasts genes that remain intact along all lineages to those that have been lost along at least one lineage. These data were analyzed using nonparametric Wilcoxon rank-sum tests and are summarized by box plots. The middle and right sections in each row contrast functional/expression groups and evolutionary subfamilies, respectively. These data were analyzed using parametric one-way ANOVAs and are summarized by diamond plots. The horizontal line bisecting each diamond and the vertical span of each diamond represent the mean and 95% confidence interval, respectively. The width of each diamond is proportional to the square root of the number of genes in the given category. Two means are significantly different if the central areas of the diamonds, demarcated by short horizontal lines, do not overlap. The three open-circle data points in the group of bitter Grs represent the Ka/Ks values of the intact orthologs of genes that have been lost along one or more lineages. These values were excluded from the statistical analysis (as were all lost genes) but are included here for comparison with the few bitter receptors that remain intact in all lineages.
Gr functional groups and subfamilies—conserved sweet and CO2 receptors, rapidly evolving bitter receptors:
A relative dearth of published functional and expression information for the Gr family made Gr genes more difficult to categorize than Ors (Amrein and Thorne 2005). We did, however, observe highly significant differences in mean Ka/Ks among the following groups: 2 Gr genes that respond to volatile CO2, 8 Grs that putatively recognize sweet compounds, 3 Grs that putatively respond to bitter compounds (another 3 of which were excluded since they have been lost along one or more lineages), and 39 Grs with mostly unknown affinities (one-way ANOVA P = 0.005; Figure 6B, middle; supplemental Table 9 at http://www.genetics.org/supplemental/). In particular, CO2 and sweet responding genes had lower Ka/Ks than bitter and unknown genes.
We decided to further categorize Gr genes according to their phylogenetic history by dividing our unrooted Bayesian tree into eight clades/subfamilies of closely related paralogs (labeled A–H in Figure 7). These groupings revealed another level of significant variation in Ka/Ks (one-way ANOVA P < 0.0001, Figure 6B, right). In accordance with the results of the previous analysis, the subfamily comprising the sweet and CO2 receptor sister clades (A in Figure 7) had one of the lowest mean Ka/Ks ratios, and the subfamily including five of the six putative bitter receptors (E in Figure 7) had the highest mean Ka/Ks (Figure 6B). The phylogenetic groupings also revealed remarkable heterogeneity within the large group of Grs with unknown functions. Most strikingly, the alternative splice forms of Gr28b and their four closest relatives (subfamily H in Figure 7) are at least as conserved as the sweet/CO2 receptors (mean Ka/Ks = 0.06). Also, subfamily B, interesting by virtue of its position as sister to the sweet/CO2 responders, had a relatively low mean ratio (Ka/Ks of Gr10a = 0.05, mean of the rest = 0.21). The fact that the honeybee genome appears to contain genes in both of these subfamilies (B and H), as well as genes in the sweet/CO2 clade (A), suggests that they play fundamental roles common to many different types of insects (Robertson and Wanner 2006).
Interestingly, the Gr subfamilies themselves can be grouped into pairs or trios with similar Ka/Ks. For example, as one moves counterclockwise around the tree in Figure 7 from subfamily A to H, the mean Ka/Ks of each consecutive clade grows larger, peaking with subfamily E, and then waning through F and G to the highly conserved subfamily H (resulting in an arc in the right section of Figure 6B). This nonrandom pattern boosts our confidence in the deep bifurcations within the Bayesian Gr family tree and supports the idea that subfamilies comprise functionally related genes.
Chemoreceptors that have been lost at least once have high Ka/Ks:
Our final analysis of heterogeneity within the Or and Gr families contrasts genes that have been lost in at least one species (and were therefore excluded from the previous analyses) with genes that remain intact in all five species. We found that the intact orthologs of the former have significantly higher median Ka/Ks than the latter within both families (Wilcoxon P = 0.04 and 0.0002 for Ors and Grs, respectively; Figure 6; supplemental Table 9 at http://www.genetics.org/supplemental/). Interestingly, however, this effect becomes weak when Gr genes are blocked by subfamily (two-way ANOVA including subfamily and loss as factors, P for effect of loss = 0.03), suggesting that high Ka/Ks and high rates of loss tend to characterize specific subfamilies rather than individual genes within diverse subfamilies. For example, subfamily E, which contains almost all of the putative bitter receptors, has the highest mean Ka/Ks (Figure 6B) and has sustained the highest number of losses (Figure 7); the lost genes within this group do not have higher Ka/Ks than their intact relatives (one-way ANOVA P = 0.7). The observed relationship between loss and Ka/Ks may thus reflect their shared association with particular functions (e.g., detection of bitter compounds; see Specialists are losing a nonrandom set of Grs, below).
Evolution of Ors and Grs along specific lineages—a role for the chemoreceptor superfamily in host specialization
Our investigation of patterns of divergence and polymorphism showed that the Or and Gr families are not evolving neutrally and suggested that they experience positive selection along at least the simulans lineage. These analyses, however, say nothing about the biological phenomena underlying these patterns. One possible cause of nonneutral evolution is ecological adaptation. The five sequenced species vary greatly in their biogeography and ecology, and their ancestors must have therefore undergone several evolutionary transitions from one ecological niche to another. McBride (2007) previously described a clear difference in the evolutionary behavior of Or and Gr genes along the lineages of D. sechellia and D. simulans and hypothesized that the evolutionary transition from a host generalist to a host specialist occurring along the sechellia lineage contributed to the difference. We now have data from three additional species and six additional lineages (Figure 1). Since a second, independent transition to host specialization occurred along one of these additional lineages (that leading to erecta), we can now ask whether the association between host specialization and rapid Or/Gr evolution holds up under closer scrutiny.
Specialists are losing Grs approximately five times more rapidly than generalists:
McBride (2007) showed that the strict host specialist sechellia has lost both Or genes (6 losses) and Gr genes (13 losses) faster than its generalist sister species simulans (χ2 P = 0.0001; Figure 4). We can now see that the same is true for the other specialist/generalist species pair within the melanogaster subgroup. Namely, erecta is losing Or/Gr genes more rapidly than its generalist sister species yakuba: D. erecta has lost 11 and yakuba has lost 0 of 133 genes present in their MRCA (χ2 P = 0.0007; Figure 4). Note, however, that erecta has lost only Grs, and that there are four additional generalist lineages in the five-species tree ignored by these sister-species comparisons (Figure 4).
To examine lineage-specific variation in the rate of gene loss in a more powerful and inclusive way, we used a maximum-likelihood framework newly implemented in the program Brownie (O'Meara et al. 2006; see appendix). Unlike methods that examine the evolution of gene family size by tallying the total number of intact genes in extant lineages without regard to orthology and paralogy (e.g., Hahn et al. 2005), this new framework traces the status of individual genes across a phylogeny and is thereby able to isolate gene loss from gene gain. We compared the following five alternative models for Ors and Grs separately: (1) “null” model, the entire subgroup has a single rate of gene loss; (2) “host ecology” model, generalists and specialists have different rates; (3) “sechellia” model, sechellia has a different rate from all other lineages; (4) “erecta” model, erecta has a different rate from all other lineages; and (5) “biogeography” model, cosmopolitan species (melanogaster and simulans) have a different rate from endemic African species/ancestors. The relative fit of the models to the observed data was assessed using the AICc. The AICc summarizes the log likelihood of a model minus a penalty for each parameter estimated. It is examined on a relative scale with the best model having the lowest AICc (ΔAICc = 0) and all other models being judged by how much larger their scores are than that of the best model (ΔAICc > 0). As a rule of thumb, models with ΔAICc ≤ 2 are considered to have substantial support, models where 4 ≤ ΔAICc ≤ 7 have low support, and models with ΔAICc > 10 have essentially no support (Burnham and Anderson 1998). The estimated rates of loss and the AICc for each model are reported in Table 3.
TABLE 3.
Comparisons of five models of lineage-specific Or and Gr gene loss
|
Ors |
Grs |
|||||||
|---|---|---|---|---|---|---|---|---|
| Modela | No. rates | Lineage(s) | Rateb | ΔAICcc | ωid | Rateb | ΔAICcc | ωid |
| Null | 1 | All | 0.46 | 2.79 | 0.158 | 1.02 | 8.97 | 0.010 |
| Host ecology | 2 | Generalist | 0.48 | 4.87 | 0.056 | 0.61 | 0.00 | 0.867 |
| Specialist | 0.37 | 2.89 | ||||||
| sechellia | 2 | Generalist + ere | 0.32 | 0.00 | 0.636 | 0.82 | 5.51 | 0.055 |
| sechellia | 4.24 | 6.59 | ||||||
| erecta | 2 | Generalist + sec | 0.50 | 4.26 | 0.076 | 0.82 | 5.51 | 0.055 |
| erecta | 0.21 | 2.21 | ||||||
| Biogeography | 2 | African | 0.46 | 4.29 | 0.075 | 1.17 | 8.42 | 0.013 |
| Cosmopolitan |
0.43 |
0.0003 |
||||||
The null model assigns a single rate of loss to all branches in Figure 4, while the other four models divide the branches into two groups according to ecology or biogeography and assign an independent rate to each group.
Rates of gene loss are maximum-likelihood estimates (see appendix) and are reported as number of losses per gene per 100 million years.
Relative corrected Akaike information criteria. Better fitting models have lower values. Values for best model are in italics.
Akaike weights. Better fitting models have higher values. Values for best model are in italics.
For the Or family, the host ecology model, wherein specialists and generalists lose Or genes at different rates, was actually the worst fitting of all five models (having the highest ΔAICc = 4.87). Instead, either sechellia stands out alone as losing Or genes more rapidly than all other lineages (the sechellia model was the best fit with ΔAICc = 0) or all lineages are losing Or genes at the same rate (null model ΔAICc = 2.79). These results suggest that rapid Or loss is not a general characteristic of host specialization in vinegar flies.
For the Gr family, in contrast, specialists do appear to be losing genes significantly faster than generalists. The host ecology model fit the observed Gr data much better than any other, with an AICc 5.51 units smaller than the next best model (Table 3). Although the host ecology model and the next best sechellia and erecta models are not nested, a traditional likelihood-ratio test can be used to compare the host ecology model with the null model (one rate for all taxa), which is nested; this test indicates that the former is significantly better than the latter with P = 0.0009. The current data thus support the idea that specialization on a novel host plant is generally associated with a contraction of the Gr family. We estimate that the two specialist lineages have lost Grs approximately five times more rapidly than the six generalist lineages (2.89 losses/gene/100 MY compared to 0.61 losses/gene/100 MY; Table 3; see appendix for details of calculation). Note that Gr loss in specialists is unlikely to be an artifact of adaptation to the laboratory since pseudogenes were verified in two independent strains of both sechellia and erecta.
Specialists are losing a nonrandom set of Grs:
Both sechellia and erecta have experienced a surprisingly rapid contraction of the Gr family. We tested whether the Gr genes lost along specialist lineages comprise a nonrandom subsample of the Gr family as a whole in several different ways. First, we asked whether they are phylogenetically clustered—more closely related to each other than a random set of Grs of the same size. Since it is unclear where the Gr family tree should be rooted, we examined the position of lost genes on an unrooted version of our tree (Figure 7, specialist losses highlighted in red). We found that the smallest subtree including all 19 lost genes (the portion of the tree falling to the left side of the dotted black line in the center of Figure 7) was significantly smaller than the smallest subtrees including 10,000 random samples of 19 Grs (P = 0.001). This confirms the visual impression that Gr losses in specialists are phylogenetically clustered. We then wondered whether the close relationship of Gr genes lost in specialists is associated with a common function. Although almost nothing is known about most of the Grs lost in specialists, 3 of them are among a small set of six putative bitter receptors (all six named in orange in Figure 7), and many others are found in the part of the tree that contains these bitter receptors (subfamily E in Figure 7). None of the lost genes, on the other hand, fall within the slightly larger set of eight putative sweet receptors (named in blue in Figure 7). A post hoc contingency test suggests that this difference in overlap between the lost genes and bitter vs. sweet receptors may be meaningful (Fisher's exact P = 0.05).
The fact that Grs lost along specialist lineages are phylogenetically clustered in one part of the tree suggests that some may be functionally related, and the fact that they partially overlap with a group of six putative bitter receptors, hints at what this shared function might be. Bitter compounds tend to be deterrents, and the Grs that recognize them are important because they warn insects about potentially harmful toxins and/or pathogens present in potential resources. There are at least two reasons why specialized flies may lose such genes. For one, specialists may lose, via adaptive evolution, bitter Grs recognizing plant compounds that deterred their generalist ancestors from their newly acquired host. For example, sechellia's loss of repulsion to the main toxins in M. citrifolia appears to be associated with the lack of expression of a peripheral chemosensory gene (an odorant-binding protein) in gustatory hairs on the foretarsi (Matsuo et al. 2007). Note that this hypothesis applies to the evolution of preference for a novel host, but would not necessarily apply to flies that specialize on one of many ancestral hosts. Alternatively, specialists may lose, via relaxed constraint, Gr genes recognizing food-borne pathogens to which they are no longer exposed. Specialists are likely exposed to a narrower array of such pathogens than generalists because they attack only one or a few hosts and/or because their hosts sometimes contain toxins that limit the growth of harmful microorganisms. For example, octanoic acid has antifungal properties that may inhibit the growth of molds on M. citrifolia (Viegas et al. 1989; Hilgren and Salverda 2000). This idea that specialist flies need fewer bitter Grs because they are exposed to fewer harmful compounds is similar to a hypothesis proposed to explain the surprisingly small size of the honeybee Gr family (n = 10). Robertson and Wanner (2006) suggested that honeybees may not need many Grs because they have mutualistic relationships with plants, which have evolved to attract and reward them rather than deter them with toxins.
A second seemingly nonrandom quality of the Gr genes lost in specialists is that 5 of a total of 19 have been lost independently along both the erecta and sechellia lineages (Gr22c, Gr28bA, Gr39aA, Gr93d, and Gr93dL1). Simulations that take the size of the Gr family, the total number of loss events along each lineage, and the structure of the phylogeny into account suggest that this number of overlapping losses is unlikely to be explained by chance (P = 0.02). Similarly, of a total of only 7 Grs lost in generalists, 2 appear to have been lost independently along two different generalist lineages (Gr59dL1 and Gr98dL1), and 1 appears to have been lost three different times (Gr59aL2). This pattern of overlap is even less likely to be explained by chance than that observed among specialist lineages (P = 0.003). There is also a somewhat nonrandom pattern of overlap between the two sets of lineages (specialists and generalists, P = 0.02; see dashed red branches in Figure 7). Interestingly, while the fact that lost Gr genes are clustered in one part of the phylogeny may account for the observed overlap within specialist lineages and between specialist and generalist lineages (P-values for a second set of simulations wherein losses are restricted to one part of the tree = 0.07 and 0.1, respectively), it cannot explain the extreme overlap observed among the generalist lineages (P = 0.007). One possible explanation for this overlap is that generalist losses are not completely independent, but rather result from separate fixations of ancestral nonsense variants.
Specialist lineages are characterized by unusually high Ka/Ks at chemoreceptor loci:
McBride (2007) demonstrated that Or and Gr genes that remain intact have higher Ka/Ks in sechellia than in simulans. To examine the generality of this phenomenon in our expanded data set, we conducted three related analyses. We first compared the Ka/Ks ratios characterizing the tip lineages (leading to each of the five species) to those inferred for the subgroup as a whole and found that both sechellia and erecta have experienced a significant increase in Ka relative to Ks at chemoreceptor loci. This can be seen in Table 4, which shows that the mean deviation of sechellia and erecta Ka/Ks ratios from the subgroup values is significantly positive, while that of the three generalists is either significantly negative (simulans) or insignificant (melanogaster and yakuba; see also supplemental Figure 11 at http://www.genetics.org/supplemental/). The consistency of these species-specific results suggests that it would be valid to implement a simpler model with just two Ka/Ks ratios per locus—one for the two specialist lineages and one for remaining generalist lineages. Implementation of this second model confirmed that Ka/Ks is significantly elevated along specialist lineages at both Or and Gr loci (paired Wilcoxon P = 0.0003 and 0.003, respectively; Figure 8, Table 5). Last, to gain insight into the proximal cause of the observed difference in Ka/Ks, we examined Ka and Ks individually by contrasting each specialist with its generalist sister species. As previously reported, both Ka and Ks are higher in sechellia than in simulans, but the increase in the former is relatively greater than the increase in the latter (McBride 2007; median parameter estimates and P-values for Ors and Grs are separately found in supplemental Table 10 at http://www.genetics.org/supplemental/). Interestingly, Ks was lower in erecta than in yakuba (P = 0.001), despite equal Ka (P = 0.7; supplemental Table 10).
TABLE 4.
Deviations of species-specific Ka/Ks values from those of the whole subgroup
|
Ors |
Grs |
Ors + Grs |
||||
|---|---|---|---|---|---|---|
| Species | Deviation | Pa | Deviation | Pa | Deviation | Pa |
| melanogaster | −0.018 | 0.83 | −0.014 | 0.19 | −0.017 | 0.062 |
| simulans | -0.045 | 0.62 | −0.038 | 0.14 | −0.041 | 0.019 |
| sechellia | 0.011 | 0.038 | 0.051 | 0.00003 | 0.035 | 0.000006 |
| yakuba | −0.0063 | 0.88 | −0.004 | 0.67 | −0.006 | 0.96 |
|
erecta |
0.015 |
0.001 |
0.010 |
0.062 |
0.014 |
0.0003 |
P-values are from paired Wilcoxon rank-sum tests that ask whether the median deviation is significantly different from zero. Significant values are in italic.
Figure 8.—

Distribution of the pairwise difference in Ka/Ks between specialist and generalist lineages across all Or/Gr genes (hatched bars) and the whole-genome set (shaded bars).
TABLE 5.
Ka/Ks values characterizing generalist and specialist lineages
| Genes | Generalist mediana | Specialists examined | Specialist median | Paired Wilcoxon Pb |
|---|---|---|---|---|
| Ors + Grs | 0.1331 | Both | 0.1635 | 0.000004 |
| erecta | 0.1595 | 0.0002 | ||
| sechellia | 0.1736 | 0.000001 | ||
| Ors | 0.1049 | Both | 0.1556 | 0.0003 |
| erecta | 0.1478 | 0.0013 | ||
| sechellia | 0.1554 | 0.0087 | ||
| Grs | 0.1658 | Both | 0.1874 | 0.003 |
| erecta | 0.1827 | 0.027 | ||
| sechellia | 0.2284 | 0.00003 | ||
| Whole genome | 0.0639 | Both | 0.0790 | 0.0000 |
| erecta | 0.0746 | 0.0000 | ||
|
sechellia |
0.0726 |
0.0000 |
The two specialist lineages are examined jointly and then individually.
Paired Wilcoxon rank-sum tests examine the null hypothesis that the median pairwise difference in Ka/Ks between the generalist lineages and the given specialist lineage(s) equals zero.
Our analyses clearly demonstrate that Or and Gr genes have higher Ka/Ks along the sechellia and erecta lineages than along all examined generalist lineages within the melanogaster subgroup. McBride (2007) suggested that demography may contribute to this phenomenon since a similar pattern was observed in a set of 190 random genes (sechellia vs. simulans only). Indeed, we find that even when examining all five species and their ancestors simultaneously, sechellia and erecta have higher Ka/Ks than generalist lineages in sets of >7000 genes scattered throughout the genome (N = 7622, P < 10−16; Table 5; Figure 8). One demographic factor that may drive an increase in Ka/Ks by weakening the strength of purifying selection relative to drift is low effective population size (Ne). D. sechellia is known to have unusually low Ne (Kliman et al. 2000), and although there is little information available regarding the population size of erecta, its narrow geographic range along the Gulf of Guinea coast suggests that it too may have low Ne (Lachaise et al. 2003). Interestingly, however, population size itself may reflect specialization. For example, strong selection for adaptation to M. citrifolia could explain why sechellia's Ne is much lower than that of the closely related generalist island-endemic D. mauritiana (Kliman et al. 2000). And the dependence of erecta on a highly seasonal host that, at least in the Ivory Coast, appears to provide ripe fruit for only 2–3 months per year may reduce erecta's Ne further than would its limited geographic range alone (Rio et al. 1983).
If demographics could completely account for the elevation of Ka/Ks along specialist lineages, we would expect the magnitude of the elevation at Or/Gr genes to be equivalent to that observed in the whole-genome set. Instead, we found that Ka/Ks is significantly more elevated at Or/Gr loci than in the rest of the genome (Wilxocon P = 0.005; Table 6). This result is illustrated in Figure 8; although the distribution of the difference in Ka/Ks between the two types of lineages is shifted to the right of zero for both sets of genes, the distribution for chemoreceptors (hatched bars) is shifted further to the right than the distribution for the whole genome (shaded bars). This result also holds for comparisons of the generalists to each specialist individually and for comparisons of each species tip lineage to the whole subgroup (supplemental Table 11 and supplemental Figure 11 at http://www.genetics.org/supplemental/). Thus, although small Ne probably contributes to elevated Ka/Ks at chemoreceptor loci, other factors are also likely to play a role. These include relaxed purifying and/or positive selection associated with specialization on a novel host plant.
TABLE 6.
Comparisons of the deviation of specialist Ka/Ks from generalist Ka/Ks at Or/Gr loci to that in the whole-genome set
| Genome |
Ors |
Grs |
Ors + Grs |
||||
|---|---|---|---|---|---|---|---|
| Contrasta | Median difference | Median difference | Pb | Median difference | Pb | Median difference | Pb |
| gen–spec | 0.0091 | 0.0294 | 0.029 | 0.0261 | 0.076 | 0.0292 | 0.0052 |
| gen–ere | 0.0066 | 0.0238 | 0.021 | 0.0115 | 0.31 | 0.0186 | 0.018 |
| gen–sec |
0.0043 |
0.0166 |
0.17 |
0.0531 |
0.0002 |
0.0416 |
0.0004 |
Specialists are examined jointly (gen–spec) and individually (gen–ere, gen–sec).
P-values are from Wilcoxon tests that ask whether Ka/Ks along specialist lineages is more elevated (greater median difference) at Ors, Grs, or Ors + Grs than across the genome as a whole.
Specialist Ka/Ks is particularly elevated among Or genes expressed in larvae:
We investigated the potential biological basis of relaxed purifying selection and/or positive selection acting on Or genes along specialist lineages by determining whether elevation in Ka/Ks was consistent across groups of genes expressed during different life stages. Interestingly, it was not. The Ka/Ks ratios of the 12 Or genes expressed only in larvae (Couto et al. 2005; Kreher et al. 2005) were dramatically elevated in specialists (mean difference = 0.088, paired t-test P = 0.00007) while those of the 32 Or genes expressed only in adults were slightly, but insignificantly, elevated (mean difference = 0.013, paired t-test P = 0.3). Elevation in Ka/Ks for 8 Ors expressed in both larvae and adults was intermediate (mean difference = 0.065, paired t-test P = 0.11). This heterogeneity is manifest in a significant interaction between host ecology and expression in a nested ANOVA (interaction term P = 0.002; supplemental Table 12A at http://www.genetics.org/supplemental/) and is illustrated in Figure 9A. Moreover, the trend is displayed by both sechellia and erecta individually (supplemental Table 13 at http://www.genetics.org/supplemental/).
Since demographic phenomena should affect different types of Or genes equally, these data provide further evidence that variation in the strength of purifying/positive selection contribute to the high Ka/Ks ratios characterizing specialist lineages. Moreover, the fact that Or genes expressed in larvae have the most elevated ratios is consistent with the idea that such a change in selection is related to specialization on a novel host. Drosophila larvae are immersed in the heterogeneous environment of their decaying host and are faced with the principal challenge of converting this host into body mass as quickly and efficiently as possible. It is therefore likely that nearly all of the 12 Or genes they express detect host-related compounds. Adults, on the other hand, must not only eat and lay eggs, but also find mates and navigate through nonhost environments. The Or genes they express are therefore likely to be more diverse in function than those of larvae, and we would not expect all of them to experience a change in selection regime during host specialization. Note that the degree to which adult Or Ka/Ks ratios are elevated in specialists is not noticeably greater than that to which the genome wide Ka/Ks ratios are elevated (slopes of solid and shaded lines approximately equal in Figure 9A), suggesting that demography may account for the difference at the majority of adult loci.
Ka/Ks is particularly elevated in erecta for Gr genes that respond to sweet compounds:
A similar analysis to that described above revealed that elevation of Ka/Ks along specialist lineages is also heterogeneous within the Gr family. In particular, the Ka/Ks ratios of putative sweet receptors were twice as elevated as those of putative bitter receptors and Gr genes with unknown functions (Figure 9B; CO2/pheromone receptors lumped with unknowns since there are so few). The ANOVA table and raw means for these analyses are provided in supplemental Tables 12B and 13 at http://www.genetics.org/supplemental/. We wondered whether this effect could have been an artifact of log transformation since sweet Grs have low Ka/Ks in general and log transformations accentuate differences between observations with low values. Comparison of sweet receptors to the most conserved genes in the unknown set, however, indicated that it was not (Figure 9B). Even so, the relatively great increase in Ka/Ks at sweet Grs traces to the erecta lineage only, which contributes more to the inferred specialist Ka/Ks ratios than does the sechellia lineage because it is much longer; Ka/Ks along the sechellia lineage appears to be consistently elevated across Gr genes with different functions (supplemental Table 13). Moreover, we can think of no obvious reason why sweet Grs would experience an unusually large change in selective environment during host specialization.
CONCLUSIONS
We have conducted the first comprehensive analysis of the molecular evolution of the insect chemoreceptor superfamily over short timescales. We find that orthologous Or and Gr genes have moderate Ka/Ks ratios, experience strong purifying selection, and do not evolve rapidly at the sequence level. Nonetheless, variability in substitution rates across lineages reveals nonneutral evolution, and a comparison of divergence to polymorphism along the simulans lineage provides the best evidence to date of positive selection at chemoreceptor loci.
We also document variation in evolutionary behavior within the superfamily. Or genes experience different selection regimes from Gr genes. Distinct subfamilies within the Gr tree have characteristic rates of divergence that appear to be associated with different functions. And the intact orthologs of Or and Gr genes that have been lost along one or more lineages diverge more rapidly than the orthologs of genes that remain intact along all lineages. Although much of this variation may simply reflect differences in constraint, it provides insight into chemoreceptor functions and suggests interesting candidates for further research.
Finally, our investigation of lineage-specific evolution suggests that ecological adaptation may underlie much of the nonneutral evolution characterizing the chemoreceptor superfamily. Specialization on novel host plants along both the sechellia and erecta lineages coincides with a dramatic contraction of the Gr family and rapid rates of amino acid substitution in both families. The identities of the genes most affected by these two phenomena support the idea that they are driven by host adaptation. Moreover, the speed of Gr loss indicates that the large differences in family size previously observed among distantly related insects (Robertson and Wanner 2006) may develop over surprisingly short periods of time.
Acknowledgments
We give thanks for technical help to Brian O'Meara for suggesting the test of phylogenetic clustering and for the new implementation of Brownie, to Andy Kern for advice on parsing silent and replacement polymorphisms and fixations, to J. J. Emerson for providing scripts that implement Jim Kent's chaining software, to Jake Byrnes for advice and help with the MCMC procedure, to Jun Wang and Maria Vibranovski for statistical help, and to Hugh Robertson for consultation on the simulans and yakuba annotations. Alisha Holloway kindly provided whole-genome coding gene alignments for simulans, yakuba, and melanogaster. We are grateful to Michael Turelli, Sergey Nuzhdin, Dave Begun, Marty Kreitman, Richard Hudson, and Chuck Langley for several long and very helpful discussions. Manyuan Long, Michael Turelli, Sergey Nuzhdin, and two anonymous reviewers provided helpful comments on earlier drafts of this manuscript. C.S.M. is supported by a National Science Foundation Predoctoral Fellowship. J.R.A. is supported by a Department of Education Graduate Assistance in Areas of National Need genomics grant awarded to University of Chicago's Ecology and Evolution Department.
References
- Akashi, H., 1994. Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. Genetics 136 927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., 1999. Inferring the fitness effects of DNA mutations from polymorphism and divergence data: statistical power to detect directional selection under stationarity and free recombination. Genetics 151 221–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altekar, G., S. Dwarkadas, J. P. Huelsenbeck and F. Ronquist, 2004. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 20 407–415. [DOI] [PubMed] [Google Scholar]
- Altschul, S., T. Madden, A. Schaffer, J. Zhang, Z. Zhang et al., 1998. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrein, H., and N. Thorne, 2005. Gustatory perception and behavior in Drosophila melanogaster. Curr. Biol. 15 R673–R684. [DOI] [PubMed] [Google Scholar]
- Begun, D. J., A. K. Holloway, K. S. Stevens, L. W. Hiller, Y. Poh et al., 2007. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 5 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney, E., M. Clamp and R. Durbin, 2004. GeneWise and genomewise. Genome Res. 14 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham, K. P., and D. R. Anderson, 1998. Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach. Springer-Verlag, New York.
- Chevenet, F., C. Brun, A. L. Banuls, B. Jacq and R. Christen, 2006. TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics 7 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chyb, S., A. Dahanukar, A. Wickens and J. R. Carlson, 2003. Drosophila Gr5a encodes a taste receptor tuned to trehalose. Proc. Natl. Acad. Sci. USA 100 14526–14530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyne, P. J., C. G. Warr and J. R. Carlson, 2000. Candidate taste receptors in Drosophila. Science 287 1830–1834. [DOI] [PubMed] [Google Scholar]
- Clyne, P. J., C. G. Warr, M. R. Freeman, D. Lessing, J. Kim et al., 1999. A novel family of divergent seven-transmembrane proteins: candidate odorant receptors in Drosophila. Neuron 22 327–338. [DOI] [PubMed] [Google Scholar]
- Couto, A., M. Alenius and B. J. Dickson, 2005. Molecular, anatomical, and functional organization of the Drosophila olfactory system. Curr. Biol. 15 1535–1547. [DOI] [PubMed] [Google Scholar]
- Cutler, D. J., 2000. Understanding the overdispersed molecular clock. Genetics 154 1403–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium, 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450 203–218. [DOI] [PubMed] [Google Scholar]
- Drosophila Odorant Receptor Nomenclature Committee, 2000. A unified nomenclature system for the Drosophila odorant receptors. Cell 102 145–146. [DOI] [PubMed] [Google Scholar]
- Gao, Q., and A. Chess, 1999. Identification of candidate Drosophila olfactory receptors from genomic DNA sequence. Genomics 60 31–39. [DOI] [PubMed] [Google Scholar]
- Geweke, J., 1992. Evaluating the Accuracy of Sampling-Based Approaches to the Calculation of Posterior Moments (With Discussion). Oxford University Press, Oxford.
- Gillespie, J. H., 1989. Lineage effects and the index of dispersion of molecular evolution. Mol. Biol. Evol. 6 636–647. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. H., 1994. The Causes of Molecular Evolution. Oxford University Press, New York.
- Goldman, N., 1994. Variance to mean ratio, R(t), for Poisson processes on phylogenetic trees. Mol. Phylogenet. Evol. 3 230–239. [DOI] [PubMed] [Google Scholar]
- Guo, S., and J. Kim, 2007. Molecular evolution of Drosophila odorant receptor genes. Mol. Biol. Evol. 24 1198–1207. [DOI] [PubMed] [Google Scholar]
- Hahn, M. W., T. De Bie, J. E. Stajich, C. Nguyen and N. Cristianini, 2005. Estimating the tempo and mode of gene family evolution from comparative genomic data. Genome Res. 15 1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallem, E. A., and J. R. Carlson, 2006. Coding of odors by a receptor repertoire. Cell 125 143–160. [DOI] [PubMed] [Google Scholar]
- Hallem, E. A., A. Dahanukar and J. R. Carlson, 2006. Insect odor and taste receptors. Annu. Rev. Entomol. 51 113–135. [DOI] [PubMed] [Google Scholar]
- Hallem, E. A., M. G. Ho and J. R. Carlson, 2004. The molecular basis of odor coding in the Drosophila antenna. Cell 117 965–979. [DOI] [PubMed] [Google Scholar]
- Hey, J., and R. M. Kliman, 1993. Population genetics and phylogenetics of DNA sequence variation at multiple loci within the Drosophila melanogaster species complex. Mol. Biol. Evol. 10 804–822. [DOI] [PubMed] [Google Scholar]
- Hilgren, J. D., and J. A. Salverda, 2000. Antimicrobial efficacy of a peroxyacetic/octanoic acid mixture in fresh-cut-vegetable process waters. J. Food Sci. 65 1376–1379. [Google Scholar]
- Hill, C. A., A. N. Fox, R. J. Pitts, L. B. Kent, P. L. Tan et al., 2002. G protein-coupled receptors in Anopheles gambiae. Science 298 176–178. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1983. Testing the constant-rate neutral allele model with protein sequence data. Evolution 37 203–217. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck, J. P., and F. Ronquist, 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17 754–755. [DOI] [PubMed] [Google Scholar]
- Hurvich, C. M., and C.-L. Tsai, 1989. Regression and time series model selection in small samples. Biometrika 76 297–307. [Google Scholar]
- Jiao, Y., S. J. Moon and C. Montell, 2007. A Drosophila gustatory receptor required for the responses to sucrose, glucose, and maltose identified by mRNA tagging. Proc. Natl. Acad. Sci. USA 104 14110–14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W. J., R. Baertsch, A. Hinrichs, W. Miller and D. Haussler, 2003. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. USA 100 11484–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern, A. D., C. D. Jones and D. J. Begun, 2004. Molecular population genetics of male accessory gland proteins in the Drosophila simulans complex. Genetics 167 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliman, R. M., P. Andolfatto, J. A. Coyne, F. Depaulis, M. Kreitman et al., 2000. The population genetics of the origin and divergence of the Drosophila simulans complex species. Genetics 156 1913–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreher, S. A., J. Y. Kwon and J. R. Carlson, 2005. The molecular basis of odor coding in the Drosophila larva. Neuron 46 445–456. [DOI] [PubMed] [Google Scholar]
- Lachaise, D., P. Capy, M.-L. Cariou, D. Joly, F. Lemeunier et al., 2003. Nine relatives from one African ancestor: population biology and evolution of the Drosophila melanogaster subgroup species, pp. 315–343 in The Evolution of Population Biology, edited by R. S. Singh and M. K. Uyenoyama. Cambridge University Press, Cambridge, UK.
- Lachaise, D., and L. Tsacas, 1974. Les drosophilidae des savanes preforestieres de la region tropicale de Lamto (Cote d'Ivoire). II. Le peuplement des fruits de Pandanus candelabrum (Pandanacees). Ann. Univ. Abidjan Ser. E Ecol. 7 153–192. [Google Scholar]
- Langley, C. H., and W. M. Fitch, 1974. An examination of the constancy of the rate of molecular evolution. J. Mol. Evol. 3 161–177. [DOI] [PubMed] [Google Scholar]
- Louis, J., and J. R. David, 1986. Ecological specialization in the Drosophila melanogaster species subgroup: a case study of Drosophila sechellia. Acta Oecol. 7 215–230. [Google Scholar]
- Marella, S., W. Fischler, P. Kong, S. Asgarian, E. Rueckert and K. Scott, 2006. Imaging taste responses in the fly brain reveals a functional map of taste category and behavior. Neuron 49 285–295. [DOI] [PubMed] [Google Scholar]
- Matsuo, T., S. Sugaya, J. Yasukawa, T. Aigaki and Y. Fuyama, 2007. Odorant-binding proteins OBP57d and OBP57e affect taste perception and host-plant preference in Drosophila sechellia. PLoS Biol. 5 e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride, C. S., 2007. Rapid evolution of smell and taste receptor genes during host specialization in Drosophila sechellia. Proc. Natl. Acad. Sci. USA 104 4996–5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh locus In Drosophila. Nature 351 652–654. [DOI] [PubMed] [Google Scholar]
- Munzner, T., F. Guimbretiere, S. Tasiran, L. Zhang and Y. Zhou, 2003. TreeJuxtaposer: scalable tree comparison using focus+context with guaranteed visibility. Proc. ACM SIGGRAPH 22 453–462. [Google Scholar]
- Nielsen, R., 1997. Robustness of the estimator of the index of dispersion for DNA sequences. Mol. Phylogenet. Evol. 7 346–351. [DOI] [PubMed] [Google Scholar]
- Nozawa, M., and M. Nei, 2007. Evolutionary dynamics of olfactory receptor genes in Drosophila species. Proc. Natl. Acad. Sci. USA 104 7122–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara, B. C., C. Ane, M. J. Sanderson and P. C. Wainwright, 2006. Testing for different rates of continuous trait evolution using likelihood. Evolution 60 922–933. [PubMed] [Google Scholar]
- Ohta, T., 1991. Multigene families and the evolution of complexity. J. Mol. Evol. 33 34–41. [DOI] [PubMed] [Google Scholar]
- Plummer, M., N. Best, K. Cowles and K. Vines, 2006. CODA: convergence diagnosis and output analysis for MCMC. R News 6 7–11. [Google Scholar]
- Rand, D. M., and L. M. Kann, 1996. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol. Biol. Evol. 13 737–748. [DOI] [PubMed] [Google Scholar]
- Rio, B., G. Couturier, F. Lemeunier and D. Lachaise, 1983. Evolution d'une specialisation saisonniere chez Drosophila erecta (Diptera, Drosophilidae). Ann. Entomol. Soc. Fr. 19 235–248. [Google Scholar]
- Robertson, H. M., and K. W. Wanner, 2006. The chemoreceptor superfamily in the honey bee, Apis mellifera: expansion of the odorant, but not gustatory, receptor family. Genome Res. 16 1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, H. M., C. G. Warr and J. R. Carlson, 2003. Molecular evolution of the insect chemoreceptor gene superfamily in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 100 14537–14542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist, F., and J. P. Huelsenbeck, 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19 1572–1574. [DOI] [PubMed] [Google Scholar]
- Shanbhag, S. R., B. Mueller and R. A. Steinbrecht, 2000. Atlas of olfactory organs of Drosophila melanogaster 2. Internal organization and cellular architecture of olfactory sensilla. Arthropod Struct. Dev. 29 211–229. [DOI] [PubMed] [Google Scholar]
- Swofford, D. L., 2002. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Sinauer Associates, Sunderland, MA.
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsacas, L., and G. Bachli, 1981. Drosophila sechellia n. sp., huiteme espece du sous-group melanogaster des iles Sechelles (Diptera, Drosophilidae). Rev. Fr. Entomol. 3 146–150. [Google Scholar]
- Tunstall, N. E., T. Sirey, R. D. Newcomb and C. G. Warr, 2007. Selective pressures on Drosophila chemosensory receptor genes. J. Mol. Evol. 64 628–636. [DOI] [PubMed] [Google Scholar]
- Viegas, C. A., M. F. Rosa, I. Sa-Correia and J. M. Novais, 1989. Inhibition of yeast growth by octanoic and decanoic acids produced during ethanolic fermentation. Appl. Environ. Microbiol. 55 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira, F. G., A. Sanchez-Gracia and J. Rozas, 2007. Comparative genomic analysis of the odorant-binding protein family in 12 Drosophila genomes: purifying selection and birth-and-death evolution. Genome Biol. 8 R225.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosshall, L. B., H. Amrein, P. S. Morozov, A. Rzhetsky and R. Axel, 1999. A spatial map of olfactory receptor expression in the Drosophila antenna. Cell 96 725–736. [DOI] [PubMed] [Google Scholar]
- Watson, L., and M. J. Dallwitz, 1992. The families of flowering plants: descriptions, illustrations, identification, and information retrieval. http://delta-intkey.com.
- Wilke, C. O., 2004. Molecular clock in neutral protein evolution. BMC Genet. 5 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, F., 1990. The “effective number of codons” used in a gene. Gene 87 23–29. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13 555–556. [DOI] [PubMed] [Google Scholar]
- Zeng, L.-W., J. W. Comeron, B. Chen and M. Kreitman, 1998. The molecular clock revisited: the rate of synonymous vs. replacement change in Drosophila. Genetica 102–103: 369–382. [PubMed] [Google Scholar]