

Abstract
Functional mapping has emerged as a powerful tool for mapping quantitative trait loci (QTL) that control developmental patterns of complex dynamic traits. Original functional mapping has been constructed within the context of simple interval mapping, without consideration of separate multiple linked QTL for a dynamic trait. In this article, we present a statistical framework for mapping QTL that affect dynamic traits by capitalizing on the strengths of functional mapping and composite interval mapping. Within this so-called composite functional-mapping framework, functional mapping models the time-dependent genetic effects of a QTL tested within a marker interval using a biologically meaningful parametric function, whereas composite interval mapping models the time-dependent genetic effects of the markers outside the test interval to control the genome background using a flexible nonparametric approach based on Legendre polynomials. Such a semiparametric framework was formulated by a maximum-likelihood model and implemented with the EM algorithm, allowing for the estimation and the test of the mathematical parameters that define the QTL effects and the regression coefficients of the Legendre polynomials that describe the marker effects. Simulation studies were performed to investigate the statistical behavior of composite functional mapping and compare its advantage in separating multiple linked QTL as compared to functional mapping. We used the new mapping approach to analyze a genetic mapping example in rice, leading to the identification of multiple QTL, some of which are linked on the same chromosome, that control the developmental trajectory of leaf age.
EVERY biological trait experiences the process of formation and development. For this reason, to better understand the developmental and genetic machinery underlying trait formation, the trait should be repeatedly observed at multiple time points during development and the temporal pattern of genetic control should be compared across different stages of development. Genetic mapping based on molecular linkage maps has been proposed to locate and identify quantitative trait loci (QTL) that affect a complex trait observed at a fixed time point in development (Lander and Botstein 1989). Many important subsequent methodological developments for QTL mapping have been available in the literature (e.g., Jansen and Stam 1994; Zeng 1994; Lynch and Walsh 1998; Kao et al. 1999; Sen and Churchill 2001; Wang et al. 2005). Many of these methods have been instrumental for the identification of QTL in a variety of plants and animals and in humans (Frary et al. 2000; Mackay 2001; C. B. Li et al. 2006; Weiss et al. 2006).
More recently, a series of statistical models, called functional mapping, have been developed to map dynamic QTL for a quantitative trait (Ma et al. 2002; Wu et al. 2003, 2004a,b,c; reviewed in Wu and Lin 2006). Functional mapping integrates mathematical aspects of a biological process into a QTL mapping framework constructed by mixture models. Its fundamental idea originates from the rationale that any biological development follows a universal law that can be described by a mathematical function. For example, growth, i.e., the increase of size, volume, or mass as a function of time, can be described by a logistic curve that is derived from fundamental biophysical and physiological principles (West et al. 2001). Aside from its biological relevance, functional mapping is statistically sensible by capitalizing on the information about the autocorrelation of errors among different time points in development.
Although the merits of functional mapping have been recognized in mapping development-related QTL for several examples (Zhao et al. 2004b,c), its construction within the context of simple interval mapping prevents it from the separation of multiple linked QTL that jointly affect developmental patterns. Composite interval mapping, advocated independently by Jansen and Stam (1994) and Zeng (1994), overcomes the low-resolution limitation of interval mapping. Composite interval mapping integrates the interval test of a putative QTL within a given marker region and partial regression analysis of the markers outside the test interval as cofactors, and it has theoretically proved advantageous for the detection and separation of linked QTL on the same chromosome (Zeng 1993). Thus far, the integration of functional mapping and composite interval mapping to make use of their respective advantages has not been fully explored.
In composite interval mapping, the genetic effects of the markers that do not bracket the putative QTL are fit individually by a partial regression analysis. Such an analysis allows for the separation of linked QTL that are located at a similar region and, thus, greatly increases the precision of QTL mapping (Zeng 1993, 1994; Jansen and Stam 1994). However, the simple use of a mathematical function to model the time-dependent marker effects is not possible because this will need to solve nonlinear equations that define developmental trajectories for all the genotypes at each marker. Gao and Yang (2006) attempted to use a nonparametric approach based on Legendre polynomials to simultaneously model QTL effects and marker effects on a dynamic trait within the context of composite interval mapping. However, this approach does not take advantage of functional mapping to gain access to and visualization of biological relevance through the deployment of mathematical functions.
In this article, we purport to develop a semiparametric approach for composite functional mapping, in which nonparametric smoothing with the Legendre function models the marker effects, while the effect of the tested QTL is modeled by a parametric function. Such a combination of parametric modeling of the QTL effects and nonparametric modeling of the marker effects preserves the biological relevance of functional mapping and, meanwhile, improves the power of QTL detection and the flexibility of the model. We implement a stationary covariance model (Ma et al. 2002) to characterize the structure of the covariance matrix among growth measured at different time points. The statistical behavior of this semiparametric model is investigated through simulations studies. The utility of the model is validated by an example from a rice molecular genetic project.
METHODS
Regression model:
Our model is derived for an F2 population containing n individuals, initiated with two inbred lines. A quantitative trait is measured at multiple time points, say T, for all the individuals. Suppose there is a quantitative trait locus (QTL) with genotypes QQ (2), Qq (1), and qq (0) that controls the temporal expression of the trait in development. A genetic linkage map constructed with molecular markers is used to locate the QTL for the trait. Assume that composite interval mapping is based on m markers. We use a pair of markers to directly estimate and test the QTL and, meanwhile, the other m − 2 markers to serve as cofactors for the test.
According to the principle of composite interval mapping (Zeng 1993, 1994), the phenotypic value of the trait for individual i measured at time t, yi(t), affected by the putative QTL, is expressed by a linear model,
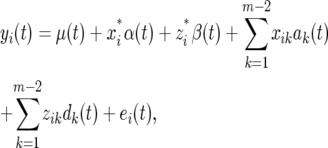 |
(1) |
where μ(t), α(t), and β(t) are the population mean and the additive and dominant effects for the QTL at time t, respectively; 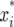 and
and 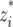 are the indicator variables for individual i that specify the QTL genotypes related to the additive and dominant effects, respectively, which are
are the indicator variables for individual i that specify the QTL genotypes related to the additive and dominant effects, respectively, which are
 |
(2) |
ak(t) and dk(t) are the time-dependent additive and dominant effects associated with marker k (except for the interval constructed by the two markers); xik and zik are the indicator variables that specify the additive and dominant effects of marker k for individual i, respectively, with similar forms indicated by Equation 2; and ei(t) is the time-dependent residual error, normally distributed as N(0, σ2(t)). The covariance between the residual errors at different time points t1 and t2 is denoted as σ(t1, t2). All the variances and covariances form a (T × T) covariance matrix Σ.
Modeling time-dependent genetic effects:
Functional mapping uses a biologically meaningful mathematical function to approximate the genotypic means for an assumed QTL at different time points. If a growth trait is considered, the mathematical function used can be a logistic curve, expressed as
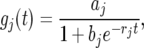 |
(3) |
for QTL genotype j (j = 2, 1, 0), where the set of parameters (aj, bj, rj) defines the shape of the genotypic curve. Thus, by estimating the three curve parameters, the differences in genotypic means over time can be estimated and tested. The time-dependent additive (α(t)) and dominant effects (β(t)) of the QTL shown in Equation 1 can be estimated by
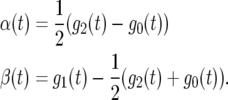 |
(4) |
In composite interval mapping, the markers outside the test interval are used as cofactors for partial regression analysis. Like the QTL effects, the additive and dominant effects of each of these markers can be theoretically fit by logistic curves. But this will be unrealistic because a number of nonlinear functions need to be solved. We utilize a nonparametric approach to approximate the time-dependent marker effects. The advantage of a nonparametric approach lies in its flexibility and, more importantly, the existence of closed-form solutions for the marker effects. Yang et al. (2006) and Gao and Yang (2006) used a Legendre polynomial to model time-dependent genetic effects of a QTL and discussed the utility of this approach.
In this study, Legendre polynomials are implemented to model the genetic effects of individual markers. Let Ls(t) = (L0(τ), L1(τ), ⋯ , Ls(τ)) be a Legendre polynomial of order s, with a general form expressed as
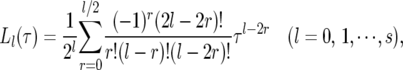 |
where 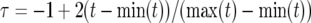 with min(t) and max(t) being the first and last time points, respectively.
with min(t) and max(t) being the first and last time points, respectively.
If the genetic effects of a QTL bracketed by a pair of markers are fit by a parametric model and the genetic effects of the remaining markers (k = 1, · · · , m − 2) are fit by Legendre polynomials of order s, the time-dependent means at different QTL genotypes for individual i are approximated by
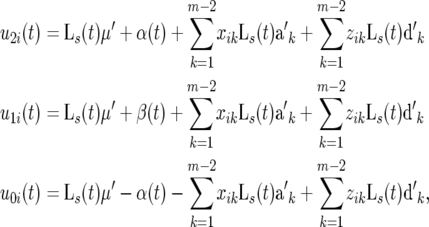 |
(5) |
where 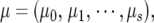 ak = (ak0, ak1, · · · , aks), and dk = (dk0, dk1, · · · , dks) are the base population mean and the base additive and dominant effects for marker k as a cofactor, respectively.
ak = (ak0, ak1, · · · , aks), and dk = (dk0, dk1, · · · , dks) are the base population mean and the base additive and dominant effects for marker k as a cofactor, respectively.
Combining the QTL and marker effects in composite functional mapping, time-dependent genotypic means for different QTL genotypes are modeled by 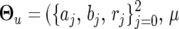 , {ak,
, {ak, 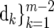 ) if a logistic curve is considered. Any other curve parameters can be included in
) if a logistic curve is considered. Any other curve parameters can be included in 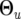 , depending on the nature of a mathematical function.
, depending on the nature of a mathematical function.
Modeling the covariance matrix:
A number of approaches have been proposed to model the covariance structure of serial measurements. A commonly used approach for structuring the covariance is the first-order autoregressive [AR(1)] model (Verbeke and Molenberghs 2000). One advantage of using the AR(1) model is that it provides a general expression for calculating the determinant and inverse of the matrix for any number of time points measured. But it assumes variance stationarity and correlation stationarity; i.e., the residual variance at different time points is the same, expressed as σ2, and the correlation between two different time points t1 and t2 decreases exponentially in ρ with time lag, expressed as corr(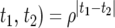 . If the AR(1) model is used, the parameters that model the structure of
. If the AR(1) model is used, the parameters that model the structure of 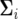 are arrayed by
are arrayed by  . In practice, the AR(1) model may be limited because its underlying two assumptions are violated.
. In practice, the AR(1) model may be limited because its underlying two assumptions are violated.
Two approaches can be used to overcome the heteroscedastic problem of the residual variance for a practical data set. First, the time-dependent residual variance is directly modeled by a parametric function of time (e.g., Pletcher and Geyer 1999). The disadvantage of this approach is that it needs to implement additional parameters for characterizing the time-dependent change of the variance. Second, Carroll and Ruppert's (1984) transform-both-sides (TBS) model can be embedded into the functional finite mixture model (Wu et al. 2004b). This approach does not need any more parameters. As indicated, by empirical analyses with real examples and computer simulations, the TBS-based model can increase the precision of parameter estimation and computational efficiency. Especially, the TBS model preserves original biological means of the curve parameters and, thus, increases its biological relevance, although statistical analyses are based on transformed data.
Likelihood and computational algorithm:
The likelihood function of the observed data including the phenotypic trait, yi = (yi(1), ⋯· · · , yi(T)), and markers, M, is expressed, within the context of a mixture model, as
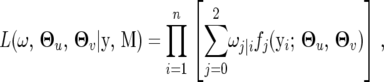 |
(6) |
where the unknown parameters include two parts, ω = (ωj|i) and 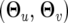 . In statistics, the parameters ωj|i determine the proportions of different mixture normals and actually reflect the segregation of the QTL in the population, which can be inferred in terms of the recombination fractions among the markers and QTL. For an F2 mapping population, n progeny can be classified into nine different groups on the basis of known genotypes of a pair of markers. Thus, in each of such marker–genotype groups, the mixture proportion or frequency of a joint QTL genotype is progeny specific and can be expressed as the conditional probability of QTL genotype j for progeny i given its marker genotype (Lynch and Walsh 1998; Wu et al. 2007), symbolized by ωj|i. The parameters
. In statistics, the parameters ωj|i determine the proportions of different mixture normals and actually reflect the segregation of the QTL in the population, which can be inferred in terms of the recombination fractions among the markers and QTL. For an F2 mapping population, n progeny can be classified into nine different groups on the basis of known genotypes of a pair of markers. Thus, in each of such marker–genotype groups, the mixture proportion or frequency of a joint QTL genotype is progeny specific and can be expressed as the conditional probability of QTL genotype j for progeny i given its marker genotype (Lynch and Walsh 1998; Wu et al. 2007), symbolized by ωj|i. The parameters 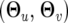 determine the QTL genotype-specific distribution density
determine the QTL genotype-specific distribution density 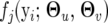 that is assumed to be multivariate normal, expressed as
that is assumed to be multivariate normal, expressed as
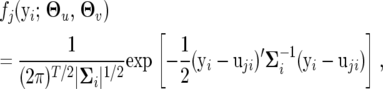 |
(7) |
where the mean vector uji = (uji(1), · · · , uji(T)) and covariance matrix  for individual i are modeled by
for individual i are modeled by 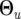 and
and 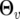 , respectively. The mean vector and covariance matrix are considered to be individual specific, because different individuals may receive different patterns of measurement, e.g., different time points and unevenly spaced time intervals.
, respectively. The mean vector and covariance matrix are considered to be individual specific, because different individuals may receive different patterns of measurement, e.g., different time points and unevenly spaced time intervals.
The EM algorithm is implemented to obtain the maximum-likelihood estimates (MLEs) of unknown parameters, 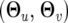 , for the likelihood expressed by Equation 6. In the E step, the posterior probabilities of each QTL genotype for individual i are calculated by
, for the likelihood expressed by Equation 6. In the E step, the posterior probabilities of each QTL genotype for individual i are calculated by
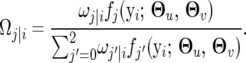 |
In the M step, the log-likelihood equations are derived in terms of Ωj|i to estimate the parameters associated with the QTL and marker effects and the covariance matrix. It is possible to derive the closed forms for the base population means and base additive and dominant effects for h = m − 2 markers as cofactors, which are expressed as
 |
with  , and β = (β(1),⋯, β(T)).
, and β = (β(1),⋯, β(T)).
It is difficult to derive the closed forms for the parameters that are used to model the time-dependent QTL effects and covariance matrix. These parameters can be estimated by implementing the simplex or Newton–Raphson algorithm in the estimation process with the EM algorithm (Zhao et al. 2004a; H. Y. Li et al. 2006). Modeled by the AR(1) (Ma et al. 2002), the closed forms for estimating the determinant and inverse of the covariance matrix can be derived, which will increase the computational efficiency of functional mapping.
Hypothesis tests:
One of the most significant advantages of functional mapping is that it can ask and address biologically meaningful questions at the interplay between gene actions and trait dynamics by formulating a series of hypothesis tests. Wu et al. (2004a); described several general hypothesis tests for different purposes. Although all these general tests can be directly used in this study, we here propose the test about the existence of a QTL that affects the process and shape of a dynamic trait.
Testing whether a specific QTL is associated with a dynamic trait is a first step toward the understanding of the genetic architecture of the trait. The genetic control over the entire dynamic process of a trait can be tested by formulating the following hypotheses:
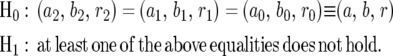 |
(8) |
H0 states that there are no QTL affecting growth curves (the reduced model), whereas H1 proposes that such QTL do exist (the full model). The test statistic for testing the hypotheses (8) is calculated as the log-likelihood ratio of the reduced to the full model,
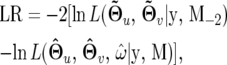 |
(9) |
where the tildes and circumflexes denote the MLEs of the unknown parameters under H0 and H1, respectively, and 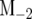 is the marker information excluding the two tested markers. The critical value of the likelihood-ratio (LR) test statistic can be determined by estimating its behavior under the null hypothesis for a whole genome. An empirical approach based on permutation tests by destroying the relationships between the phenotypic values and tested marker-interval genotypes (Doerge and Churchill 1996; Zou et al. 2004; Jin et al. 2007) is usually used to determine the critical threshold of the LR for interval mapping. But this approach cannot be directly used for composite interval mapping in which additional markers (excluding the two tested markers) serve as cofactors to be associated with the phenotypic values. Zeng (1994) proposed a simulation approach to examine the distribution of the LR values under the null hypothesis. The phenotypic values simulated under the null hypothesis should reflect the effects of the markers as cofactors. This can be done by assuming that the time-dependent phenotypic values follow a multivariate normal distribution with mean vector
is the marker information excluding the two tested markers. The critical value of the likelihood-ratio (LR) test statistic can be determined by estimating its behavior under the null hypothesis for a whole genome. An empirical approach based on permutation tests by destroying the relationships between the phenotypic values and tested marker-interval genotypes (Doerge and Churchill 1996; Zou et al. 2004; Jin et al. 2007) is usually used to determine the critical threshold of the LR for interval mapping. But this approach cannot be directly used for composite interval mapping in which additional markers (excluding the two tested markers) serve as cofactors to be associated with the phenotypic values. Zeng (1994) proposed a simulation approach to examine the distribution of the LR values under the null hypothesis. The phenotypic values simulated under the null hypothesis should reflect the effects of the markers as cofactors. This can be done by assuming that the time-dependent phenotypic values follow a multivariate normal distribution with mean vector
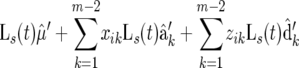 |
and the covariance matrix with the AR(1) structure. The threshold for the tested interval is estimated as the 5% percentile of the LR values from 1000 simulation replicates. A genomewide critical threshold is determined by scanning through the entire linkage map, although this process is computationally extensive.
MONTE CARLO SIMULATION
We carried out simulation studies to investigate the statistical properties of functional mapping integrated with the idea of composite interval mapping. An F2 population with 150 individuals was simulated for a chromosome segment of length 120 cM covered by 13 evenly spaced markers (10 cM per interval). Four QTL were placed at positions 15 (1), 36 (2), 62 (3), and 104 cM (4) from the left end of the chromosome. Three genotypes at a QTL, Q rQ r, Q r qr, and qrqr (r = 1, · · · , 4), were assumed to follow a logistic growth curve, as described by Equation 5, with different sets of given growth parameters, from which the time-dependent additive and dominance effects of the QTL are calculated with Equation 6. By defining an overall mean logistic curve (u(t)), 81 genotypic mean curves contributed by the four assumed QTL are expressed as
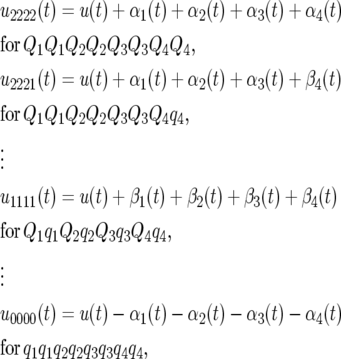 |
under the assumption that there is no epistasis among the four QTL, where the time-dependent additive (αr(t)) and dominant effects (βr(t)) at the rth QTL are specified by Equation 6. Assume that the F2 individuals are measured for a dynamic trait at eight even-spaced time points. The time-dependent phenotypic values of the trait were simulated to follow a multivariate normal distribution with the four-QTL genotypic mean vector and the residual covariance matrix fit by the AR(1) model. The values of the variance σ2 and correlation ρ were empirically determined, assuring a modest heritability for the dynamic trait at the middle of the measurement period. The simulated phenotypic data for individual F2 progeny may also be logistic curves because of the underlying logistic mean curve.
The assumed phenotypic and marker data were analyzed by composite functional mapping. To obtain the best fit of the data, the optimal number of markers involved in the partial regression analysis of composite functional mapping and the optimal order of the Legendre polynomial to model the marker effects should be determined. We used the Bayesian information criterion (BIC) (Schwarz 1978) as the model selection criterion of the optimal marker number and polynomial order. The BIC is defined as
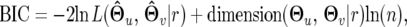 |
where  and
and 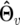 are the MLEs of parameters under the Legendre polynomial of order r, dimension(
are the MLEs of parameters under the Legendre polynomial of order r, dimension(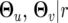 ) represents the number of independent parameters under order r, and n is the total number of observations at a particular time point. The optimal model is one that displays the minimum BIC value.
) represents the number of independent parameters under order r, and n is the total number of observations at a particular time point. The optimal model is one that displays the minimum BIC value.
An important issue for composite interval mapping is to determine the optimal number and combination of markers as cofactors on the basis of model selection criteria. We empirically chose different numbers of markers, zero, three, four, five, and six, that bracket the marker interval containing the tested QTL, as the cofactors of partial regression analysis, and then evaluated each of these choices with the BIC values calculated for different orders of the Legendre polynomial (Table 1). At a given number of cofactors, order 4 gives the minimum BIC value, whereas for a given order, five cofactors best fit the data. Overall, the Legendre function of order 4 and five markers as cofactors are incorporated into composite functional mapping for this simulated data set. For the test of the presence of a QTL, we simulated 500 additional samples under the null model to obtain the empirical critical values of the LR test statistic. Under the alternative model, the simulation was replicated 100 times to calculate empirical power by counting the number of runs in which the test statistics were greater than the critical values. Calculating the averaged LRs at every scanning point over the segment of chromosome, we depicted the statistic profiles for the five models with the number of cofactors, zero, three, four, five, and six.
TABLE 1.
BIC values under different models of composite functional mapping by choosing zero, three, four, five, and six markers as cofactors
| Order of Legendre polynomial
|
||||||
|---|---|---|---|---|---|---|
| Model | 0 | 1 | 2 | 3 | 4 | 5 |
| C0 | 3417.4 | |||||
| C3 | 3380.2 | 3285.2 | 3139.5 | 3088.4 | 3096.1 | |
| C4 | 3361.8 | 3270.2 | 3098.6 | 3023.4 | 3031.6 | |
| C5 | 3351.6 | 3261.0 | 3088.9 | 3012.6 | 3023.1 | |
| C6 | 3354.7 | 3265.3 | 3091.4 | 3014.4 | 3026.9 | |
Different orders of the Legendre polynomial are considered under each model.
Composite functional mapping without cofactor corresponds to traditional functional mapping, which detects one flat peak, plus a small peak, on the whole chromosome segment (Figure 1). This indicates that the traditional approach has no power to separate individual QTL on a chromosome. When markers as cofactors are involved in composite functional mapping under the Legendre polynomial of order 4, more peaks above the critical threshold are detected, suggesting the detection of more QTL. When three cofactors are involved, four QTL can be identified, but the first QTL is not correctly located. The involvement of four cofactors leads to the detection of five QTL, with one between the second and the third QTL being spurious. When the number of cofactors increases to five, all of the four assumed QTL can be accurately detected. The further increase of cofactor number to six gives a similar LR profile to that for four cofactors. On the basis of the profile analysis (Figure 1), the best number of markers as cofactors in composite functional mapping is consistent with the BIC result.
Figure 1.—

The profiles of the LR values across a simulated linkage group under different models of composite functional mapping by choosing zero, three, four, five, and six markers as cofactors. The vertical dashed lines indicate the locations of four hypothesized QTL. The critical thresholds calculated are indicated as horizonal lines.
Table 2 tabulates the MLEs of the QTL positions and the power of QTL detection for the simulated data set obtained by traditional and composite functional mapping. Traditional functional mapping fails to detect the first and third QTL, whereas composite functional mapping can correctly detect all the assumed QTL. Because the first three QTL are relatively clustered, far from the fourth QTL, they were detected as a “big” one by traditional functional mapping with 100% power. These three clustered QTL can be individually detected by composite functional mapping although the power of detecting each QTL is a little bit reduced. However, for an unlinked QTL (i.e., fourth QTL), composite functional mapping displays much more detection power than traditional functional mapping (Table 2). The growth curves for each QTL genotype were estimated under the optimal order of the Legendre polynomial (4) and optimal number of cofactors (five) (Table 3). Basically, all these parameters can be accurately estimated, each with reasonably high estimation precision.
TABLE 2.
The MLEs of the QTL positions, sampling errors (in parentheses) of the MLEs, and power to detect significant QTL by traditional functional mapping (FM) and composite functional mapping (CFM) under the optimal order of the Legendre polynomial (4) and the optimal number of cofactors (five)
| QTL
|
||||
|---|---|---|---|---|
| Model | 1 | 2 | 3 | 4 |
| True position | 15 | 36 | 62 | 104 |
| FM | ||||
| MLE | — | 33.28 | — | 101.15 |
| — | (3.12) | — | (15.78) | |
| 95% power | — | 100 | — | 81 |
| CFM | ||||
| MLE | 15.10 | 34.97 | 67.89 | 103.78 |
| (3.38) | (4.43) | (4.86) | (5.41) | |
| 95% power | 83 | 86 | 91 | 96 |
TABLE 3.
The MLEs of genotypic curve parameters at the four assumed QTL and sampling errors (in parentheses) of the estimates by composite functional mapping under the optimal order of the Legendre polynomial (4) and the optimal number of cofactors (five)
|
Q rQ r
|
Q rq r
|
qrqr
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| QTL | a2 | b2 | r2 | a1 | b1 | r1 | a0 | b0 | r0 |
| 1 | |||||||||
| True | 41.32 | 3.12 | 0.86 | 38.52 | 3.05 | 0.93 | 43.04 | 4.65 | 1.12 |
| MLE | 39.82 | 4.31 | 0.83 | 38.48 | 4.15 | 0.89 | 42.33 | 5.78 | 1.34 |
| (1.03) | (0.69) | (0.05) | (0.92) | (0.28) | (0.08) | (0.98) | (0.43) | (0.02) | |
| 2 | |||||||||
| True | 41.50 | 3.86 | 1.00 | 40.99 | 3.70 | 1.17 | 42.96 | 4.84 | 1.40 |
| MLE | 38.75 | 6.02 | 0.79 | 40.74 | 4.03 | 1.00 | 44.25 | 5.15 | 1.39 |
| (0.69) | (1.15) | (0.04) | (0.56) | (0.51) | (0.06) | (0.62) | (0.25) | (0.05) | |
| 3 | |||||||||
| True | 38.32 | 3.19 | 1.05 | 37.86 | 3.48 | 0.99 | 42.82 | 4.67 | 1.49 |
| MLE | 38.11 | 3.44 | 0.79 | 37.13 | 4.29 | 0.82 | 40.66 | 4.84 | (0.04) |
| (0.97) | (0.35) | (0.05) | (0.65) | (0.97) | (0.09) | (0.94) | (0.35) | (0.04) | |
| 4 | |||||||||
| True | 42.63 | 3.42 | 1.45 | 41.43 | 4.17 | 1.20 | 38.77 | 3.78 | 0.70 |
| MLE | 42.74 | 3.25 | 1.16 | 40.30 | 3.69 | 0.95 | 36.11 | 4.58 | 0.75 |
| (0.49) | (0.46) | (0.08) | (1.00) | (0.60) | (0.11) | (0.680) | (0.88) | (0.06) | |
Additional simulation studies were carried out to investigate the effects of different heritabilities and sample sizes on the precision of parameter estimation and the power of QTL detection. Because all the results are expected, they are not presented here. The expected results are that the behavior of the model is better with increased heritability and sample size. It seems that the heritability and sample size interact to influence the statistical performance of the model.
EXAMPLE
Weng et al. (2000) reported a doubled-haploid (DH) population with 111 lines generated by crossing an indica rice variety Gui-630 and a japonica rice variety Taiwanjing. A linkage map composed of 175 RFLP markers was constructed for the DH population, covering a total length of 1225 cM (Figure 2). This DH population was grown with replicates in a field trial (Zhou et al. 2001). For each plant, the number of developed leaves on the main stem was counted, and the length of the developing leaf was measured every 5–7 days from day 30 after sowing until the full development of the leaf. These measured data were used to estimate the leaf age of a plant (y), using
 |
Zhou et al. (2001) showed that the growth of leaf age of a plant obeys a parabolic curve with time, described by a function
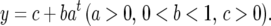 |
(10) |
For a DH population, there are two homozygous genotypes at a QTL, denoted by QQ (2) and qq (0). Thus, by testing the differences of curve parameters between the two QTL genotypes, we can estimate whether and how a QTL affects the growth trajectories of leaf age in the DH population. In composite functional mapping, the equation of leaf age growth was incorporated to specify the QTL effect, whereas the Legendre polynomial was used to model the marker effects by choosing different numbers of markers (three, four, five, and six), as cofactors, that are close to the test interval of QTL. The best number of cofactors is determined under different orders of the Legendre polynomial using the BIC criterion. It is found that the optimal number of cofactors and optimal order of the polynomial are 4 and 2, respectively, that best explain the rice data. The critical threshold at the 5% significance level for declaring the presence of QTL for the growth trajectories of leaf age was calculated by simulation studies with 500 replicates.
Figure 2.—

Linkage map in rice constructed of 175 RFLP markers (Weng et al. 2000).
Figure 3 illustrates the profile of the LR values for testing the presence of QTL across the entire rice genome. Significant QTL for leaf age growth were detected on chromosomes 1, 3, 6, 10, and 11 with composite functional mapping. It is interesting to note that composite functional mapping can detect three different QTL on chromosome 3, as indicated by three steep peaks of the LR profile (Figure 3). The MLEs of curve parameters for different QTL genotypes and the parameters that model the residual covariance matrix, along with the marker intervals of the detected QTL, were tabulated in Table 4. The estimates of the standard errors of the MLEs obtained by a bootstrapping approach suggest that these curve and matrix-structuring parameters can be precisely estimated. The growth curves of QTL genotypes at each QTL (Figure 4) drawn with the estimates of curve parameters suggest that all the QTL detected are expressed increasingly with time, but display different directions of allelic effects. Favorable alleles that increase leaf age growth are contributed by parent Gui-630 for the three QTL detected on chromosomes 1, 5, and 6 and by parent Taiwanjing for the three QTL on chromosome 3 and one QTL on chromosome 7 (Figure 4).
Figure 3.—

The profiles of the log-likelihood ratios (LR) between the full and the reduced (no QTL) model estimated from composite functional mapping (solid curves) and functional mapping models (shaded curves) for leaf age growth trajectories across the entire genome. The genomic positions corresponding to the peaks of the curves are the MLEs of the QTL positions. Tick marks on the x-axis represent the positions of markers on the linkage group, whose names are given in Figure 1. The threshold values for claiming the existence of a QTL are given as the horizonal lines. To save computational time, the threshold was determined on the basis of those chromosomes that possibly contain significant QTL in terms of high LR peaks.
TABLE 4.
The MLEs of curve parameters and sampling errors (in parentheses) of the estimates for the QTL detected on different chromosomes by composite functional mapping
| Position
|
QQ
|
qq
|
Residual
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| QTL | Chromosome | Marker interval | a2 | b2 | c2 | a0 | b0 | c0 | σ2 | ρ |
| 1 | 1 | C178–RZ776 | 1.22 | 0.55 | 5.69 | 0.84 | 0.61 | 5.48 | 0.97 | 0.13 |
| (0.09) | (0.01) | (0.13) | (0.07) | (0.01) | (0.08) | (0.01) | (0.02) | |||
| 2 | 3 | C6C–C814A | 0.83 | 0.60 | 5.10 | 1.06 | 0.58 | 5.84 | 0.98 | 0.30 |
| (0.33) | (0.04) | (0.11) | (0.21) | (0.03) | (0.11) | (0.02) | (0.02) | |||
| 3 | 3 | RZ993–C825B | 0.90 | 0.58 | 5.29 | 1.06 | 0.58 | 5.71 | 0.99 | 0.39 |
| (0.07) | (0.01) | (0.09) | (0.11) | (0.01) | (0.16) | (0.02) | (0.04) | |||
| 4 | 3 | C1032A–RZ142 | 0.85 | 0.60 | 5.33 | 1.19 | 0.55 | 5.74 | 0.98 | 0.25 |
| (0.09) | (0.02) | (0.13) | (0.11) | (0.01) | (0.18) | (0.02) | (0.03) | |||
| 5 | 6 | R276–R2071 | 1.00 | 0.60 | 6.03 | 1.02 | 0.55 | 4.91 | 0.99 | 0.37 |
| (0.28) | (0.04) | (0.17) | (0.29) | (0.04) | (0.15) | (0.02) | (0.02) | |||
| 6 | 10 | RZ892–RZ561 | 1.10 | 0.57 | 5.80 | 1.02 | 0.58 | 5.29 | 0.98 | 0.28 |
| (0.14) | (0.02) | (0.18) | (0.07) | (0.01) | (0.10) | (0.02) | (0.02) | |||
| 7 | 11 | C189–C6A | 0.96 | 0.58 | 5.35 | 1.28 | 0.54 | 5.53 | 0.98 | 0.33 |
| (0.06) | (0.01) | (0.08) | (0.10) | (0.01) | (0.14) | (0.03) | (0.02) | |||
Figure 4.—

Two growth curves each presenting a group of genotypes, QQ (solid curves) and qq (shaded curves), at each of the seven QTL, detected by composite functional mapping on chromosomes 1, 3, 6, 10, and 11. Alleles Q and q are inherited for the three QTL (1, 5, and 6) detected on chromosomes 1, 6, and 10 from parents Gui-630 and Taiwanjing, respectively. The inverse is true for the four QTL (2, 3, 4, and 7) detected on chromosomes 3 and 11.
The same data set was analyzed by traditional functional mapping. As compared to composite functional mapping, the traditional model is found to have less power to detect significant QTL. For example, it detects four QTL on chromosomes 1, 3, 10, and 11, which are all detected by composite functional mapping, but it fails to detect the QTL on chromosome 6 detected by composite functional mapping (Figure 2). Also, composite functional mapping can simultaneously detect three linked QTL on the same chromosome, whereas the traditional approach confounds the effects of different linked QTL. The two QTL, bracketed by marker intervals C6C–C814A and RZ993–C825B and separated by ∼25 cM, can be detected individually by composite functional mapping.
DISCUSSION
The genetic architecture of complex traits can be well understood by incorporating their developmental features. Functional mapping that integrates genetics, statistics, and developmental biology has proven to be useful for deciphering the ontogenetic development of the genetic control of a complex trait (Wu and Lin 2006). Original models for functional mapping have been based on simple interval mapping of QTL in which two flanking markers that bracket a QTL are assumed to be independent of all the markers outside the marker interval tested. Although such interval mapping has power for QTL mapping, it is often limited when more than one QTL is located on the same chromosome. Zeng (1993, 1994) and Jansen and Stam (1994) proposed a joint approach for simultaneously modeling the two flanking markers for testing the existence of a QTL by interval mapping and the markers outside the interval by a partial regression analysis. This so-called composite interval mapping, powerful for the separation of multiple linked QTL, has been instrumental in the genetic mapping of complex traits (Zeng et al. 2000).
In this article, we have integrated the principles of functional mapping and composite interval mapping within a unifying framework for QTL mapping, aimed at increasing the resolution of multiple QTL on the same region of a chromosome. The new model allows us to approximate the ontogenetic changes of the genetic effects triggered by a QTL and the markers outside the test interval. Because many biological processes, such as growth, follow a particular pattern of development (West et al. 2001), the ontogenetic control of a QTL can be mathematically described and, thereby, tested by estimating the parameters that define a biological process (Ma et al. 2002). If a parametric approach is implemented to model the time-dependent marker effects, this would lead to tremendous computational burden. We instead fit marker effects by capitalizing on the linear property of a Legendre polynomial. In fact, the Legendre polynomial is flexible in the modeling of various forms of curves and has been used to model genetic and environmental effects in quantitative genetic studies (Kirkpatrick and Heckman 1989; Schaeffer 2004; Meyer 2005a,b) and QTL mapping (Gao and Yang 2006; Yang et al. 2006). One important aspect of functional mapping is to model the structure of the covariance matrix by a stationary or nonstationary approach. Because of its computational simplicity, AR(1) is advantageous for structuring the covariance although it needs the variance and covariance stationarity assumptions. The TBS-based model can relax the assumption of variance stationarity (Wu et al. 2004b), but it has not resolved the covariance stationarity issue when embedded into the AR(1) model. A so-called structured antedependence (SAD) model, advocated by Zimmerman and Núñez-Antón (2001), can be used to simultaneously model the time-dependent changes of and variance and correlation in the analysis of longitudinal traits. The SAD model is found to display many favorable properties (Zimmerman and Núñez-Antón 2001). More recently, Zhao et al. (2005) applied this model to functional mapping and further explored its robustness in modeling the covariance structure through a comparison with the AR(1) model.
The advantage of our composite functional mapping lies in the combination of biologically sensible functions that enhance biological relevance of QTL detection and the flexibility of a nonparametric approach that is aimed at increasing the computational efficiency. The model is used to analyze a published data set on the growth of leaf age in rice (Zhou et al. 2001). As compared to traditional functional mapping, the new model has tremendous power to detect and separate linked QTL located on the similar regions of a chromosome. The model has been found to be robust in that it provides reasonable estimates of QTL effects and positions in a wide range of parameter space, as demonstrated by simulation studies.
It is possible that our model can be modified in several areas. First, a mathematical function is used to model the ontogenetic change of genetic effects of a QTL, considering that the trait studied undergoes a certain developmental pattern. There may also be many traits that do not follow a mathematical function. Yang et al. (2006) used the Legendre polynomial to model the time-dependent QTL effects. A similar idea was employed in Cui et al. (2006) and Lin and Wu (2006), who incorporated the Legendre-based transformation to model some particular stage of growth or one aspect of a joint longitudinal and time-to-event analysis. Second, composite functional mapping can be extended to explore the effects of interaction between different QTL (Kao and Zeng 2002) and QTL and environments (Zhao et al. 2004b,c) on variation in a dynamic trait by expanding Equation 4 to interaction terms with quantitative genetic theory (Lynch and Walsh 1998). Beyond composite interval mapping, Zeng and colleagues (Kao and Zeng 1997; Kao et al. 1999) extended a so-called multiple-interval mapping approach to a genomewide search for the distribution of QTL throughout the genome. The integration of functional mapping and multiple-interval mapping will help to improve the statistical behavior of functional mapping. All the modified models will certainly prove their value in elucidating the genetic architecture of dynamic traits and will probably be the beginning of detecting the driving forces behind dynamic genetics and their relationship to the organism as a whole. The computer code for the method proposed can be requested from the corresponding author.
Acknowledgments
The preparation of this manuscript was partially supported by National Science Foundation grant no. 0540745 and the National Natural Science Foundation of China grant no. 30471236.
References
- Carroll, R. J., and D. Ruppert, 1984. Power-transformations when fitting theoretical models to data. J. Am. Stat. Assoc. 79: 321–328. [Google Scholar]
- Cui, Y. H., J. Zhu and R. L. Wu, 2006. Functional mapping for genetic control of programmed cell death. Physiol. Genomics 25: 458–469. [DOI] [PubMed] [Google Scholar]
- Doerge, R. W., and G. A. Churchill, 1996. Permutation tests for multiple loci affecting a quantitative character. Genetics 142: 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frary, A., T. C. Nesbitt, A. Frary, S. Grandillo, E. van der Knaap et al., 2000. fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289: 85–88. [DOI] [PubMed] [Google Scholar]
- Gao, H. J., and R. Q. Yang, 2006. Composite interval mapping of QTL for dynamic traits. Chin. Sci. Bull. 51: 1857–1862. [Google Scholar]
- Kao, C.-H., and Z.-B. Zeng, 1997. General formulas for obtaining the MLEs and the asymptotic variance-covariance matrix in mapping quantitative trait loci when using the EM algorithm. Biometrics 53: 653–665. [PubMed] [Google Scholar]
- Kao, C.-H., and Z.-B. Zeng, 2002. Modeling epistasis of quantitative trait loci using Cockerham's model. Genetics 160: 1243–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C.-H., Z.-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, M., and N. Heckman, 1989. A quantitative genetic model for growth, shape, reaction norms, and other infinite-dimensional characters. J. Math. Biol. 27: 429–450. [DOI] [PubMed] [Google Scholar]
- Jansen, R. C., and P. Stam, 1994. High resolution mapping of quantitative traits into multiple loci via interval mapping. Genetics 136: 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, C., J. P. Fine and B. S. Yandell, 2007. A unified semiparametric framework for QTL analyses, with application to spike phenotypes. J. Am. Stat. Assoc. 102: 56–67. [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. B., A. L. Zhou and T. Sang, 2006. Rice domestication by reducing shattering. Science 311: 1936–1939. [DOI] [PubMed] [Google Scholar]
- Li, H. Y., B.-R. Kim and R. L. Wu, 2006. Identification of quantitative trait nucleotides that regulate cancer growth: a simulation approach. J. Theor. Biol. 242: 426–439. [DOI] [PubMed] [Google Scholar]
- Lin, M., and R. L. Wu, 2006. A joint model for nonparametric functional mapping of longitudinal trajectories and time-to-events. BMC Bioinformatics 7: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Ma, C.-X., G. Casella and R. L. Wu, 2002. Functional mapping of quantitative trait loci underlying the character process: a theoretical framework. Genetics 161: 1751–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, T. F. C., 2001. Quantitative trait loci in Drosophila. Nat. Rev. Genet. 2: 11–20. [DOI] [PubMed] [Google Scholar]
- Meyer, K., 2005. a Estimates of genetic covariance functions for growth of Angus cattle. J. Anim. Breed. Genet. 122: 73–85. [DOI] [PubMed] [Google Scholar]
- Meyer, K., 2005. b Random regression analyses using B-splines to model growth of Australian Angus cattle. Genet. Sel. Evol. 37: 473–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher, S. D., and C. J. Geyer, 1999. The genetic analysis of age-dependent traits: modeling the character process. Genetics 153: 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer, L. R., 2004. Application of random regression models in animal breeding. Livestock Prod. Sci. 86: 35–45. [Google Scholar]
- Schwarz, G., 1978. Estimating the dimension of a model. Ann. Stat. 6: 461–464. [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke, G., and G. Molenberghs, 2000. Linear Mixed Models for Longitudinal Data. Springer-Verlag, New York.
- Wang, H., Y. M. Zhang, X. M. Li, G. Masinde, S. Mohan et al., 2005. Bayesian shrinkage estimation of quantitative trait loci parameters. Genetics 170: 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng, Q., W. Wu, W. Li, H. Liu, D. Tang et al., 2000. Construction of an RFLP linkage map of rice using DNA probes from two different sources. J. Fujian Agric. Univ. 29: 129–133. [Google Scholar]
- Weiss, L. A., L. Pan, M. Abney and C. Ober, 2006. The sex-specific genetic architecture of quantitative traits in humans. Nat. Genet. 38: 218–222. [DOI] [PubMed] [Google Scholar]
- West, G. B., J. H. Brown and B. J. Enquist, 2001. A general model for ontogenetic growth. Nature 413: 628–631. [DOI] [PubMed] [Google Scholar]
- Wu, R. L., and M. Lin, 2006. Functional mapping—how to map and study the genetic architecture of dynamic complex traits. Nat. Rev. Genet. 7: 229–237. [DOI] [PubMed] [Google Scholar]
- Wu, R. L., C.-X. Ma, W. Zhao and G. Casella, 2003. Functional mapping of quantitative trait loci underlying growth rates: a parametric model. Physiol. Genomics 14: 241–249. [DOI] [PubMed] [Google Scholar]
- Wu, R. L., C.-X. Ma, M. Lin and G. Casella, 2004. a A general framework for analyzing the genetic architecture of developmental characteristics. Genetics 166: 1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, R. L., C.-X. Ma, M. Lin, Z. H. Wang and G. Casella, 2004. b Functional mapping of growth QTL using a transform-both-sides logistic model. Biometrics 60: 729–738. [DOI] [PubMed] [Google Scholar]
- Wu, R. L., Z. H. Wang, W. Zhao and J. M. Cheverud, 2004. c A mechanistic model for genetic machinery of ontogenetic growth. Genetics 168: 2383–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, R. L., C.-X. Ma and G. Casella, 2007. Statistical Genetics of Quantitative Traits: Linkage, Maps, and QTL. Springer-Verlag, New York.
- Yang, R. Q., Q. Tian and S. Z. Xu, 2006. Mapping quantitative trait loci for longitudinal traits in line crosses. Genetics 173: 2339–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z.-B., 1993. Theoretical basis for separation of multiple linked gene effects in mapping quantitative trait loci. Proc. Natl. Acad. Sci. USA 90: 10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z.-B., 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z.-B., J. Liu, L. F. Stam, C.-H. Kao, J. M. Mercer et al., 2000. Genetic architecture of a morphological shape difference between two Drosophila species. Genetics 154: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, W., R. L. Wu, C.-X. Ma and G. Casella, 2004. a A fast algorithm for functional mapping of complex traits. Genetics 167: 2133–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, W., C.-X. Ma, J. M. Cheverud and R. L. Wu, 2004. b A unifying statistical model for QTL mapping of genotype-sex interaction for developmental trajectories. Physiol. Genomics 19: 218–227. [DOI] [PubMed] [Google Scholar]
- Zhao, W., J. Zhu, M. Gallo-Meagher and R. L. Wu, 2004. c A unified statistical model for functional mapping of genotype × environment interactions for ontogenetic development. Genetics 168: 1751–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, W., Y. Q. Chen, G. Casella, J. M. Cheverud and R. L. Wu, 2005. A non-stationary model for functional mapping of longitudinal quantitative traits. Bioinformatics 21: 2469–2477. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., W. Li, W. Wu, Q. Chen, D. Mao et al., 2001. Genetic dissection of heading time and its components in rice. Theor. Appl. Genet. 102: 1236–1242. [Google Scholar]
- Zimmerman, D. L., and V. Núñez-Antón, 2001. Parametric modeling of growth curve data: an overview (with discussion). Test 10: 1–73. [Google Scholar]
- Zou, F., J. P. Fine, J. Hu and D. Y. Lin, 2004. An efficient resampling method for assessing genome-wide statistical significance in mapping quantitative trait loci. Genetics 168: 2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]