

Abstract
Eukaryotic chromosomes are duplicated during S phase and transmitted to progeny during mitosis with high fidelity. Chromosome duplication is controlled at the level of replication initiation, which occurs at cis-acting replicator sequences that are spaced at intervals of ∼40 kb along the chromosomes of the budding yeast Saccharomyces cerevisiae. Surprisingly, we found that derivatives of yeast chromosome III that lack known replicators were replicated and segregated properly in at least 96% of cell divisions. To gain insight into the mechanisms that maintain these “originless” chromosome fragments, we screened for mutants defective in the maintenance of an “originless” chromosome fragment, but proficient in the maintenance of the same fragment that carries its normal complement of replicators (originless fragment maintenance mutants, or ofm). We show that three of these Ofm mutations appear to disrupt different processes involved in chromosome transmission. The OFM1-1 mutant seems to disrupt an alternative initiation mechanism, and the ofm6 mutant appears to be defective in replication fork progression. ofm14 is an allele of RAD9, which is required for the activation of the DNA damage checkpoint, suggesting that this checkpoint plays a key role in the maintenance of the “originless” fragment.
ALL organisms replicate and segregate their chromosomes with high fidelity. A failure to do so can have dire consequences, e.g., cell death and mutation. One of the hallmarks of cancer is a loss of genome stability as a result of high rates of mutation and/or loss of chromosome stability. The budding yeast Saccharomyces cerevisiae provides an excellent model system for studying the molecular mechanisms that contribute to chromosome stability, including DNA replication, DNA repair, and chromosome segregation.
In addition to the inherent accuracy of the chromosome replication and segregation processes themselves, additional conserved mechanisms called checkpoints contribute to chromosome stability. Checkpoints are defined as pathways that promote cell cycle delay or arrest in response to DNA damage or mitotic spindle damage (reviewed by Foiani et al. 2000). The cell cycle delay allows time for the error to be corrected. Checkpoints operate through signal-transduction pathways that involve proteins that sense the damage (sensors), protein kinases that transduce and amplify the signal, and effectors that mediate the response to the signal. The spindle assembly checkpoint delays the G2-M transition in response to improper or impaired kinetochore–microtubule attachments. Two partially redundant checkpoint pathways monitor DNA damage (the DNA damage checkpoint) and slowing or stalling of replication forks (the replication stress checkpoint) during S phase and also delay the G2-M transition.
DNA replication is regulated at the level of replication initiation at individual replicators. While the cis-acting replicators that direct replication initiation in budding yeast, called autonomously replicating sequence (ARS) elements, are smaller than their counterparts in many other eukaryotes, the proteins that are required for replication initiation as well as the proteins that function at the replication fork are highly conserved among eukaryotes (reviewed by Kelly and Stillman 2006). Similarly, DNA repair proteins show a high degree of conservation across eukaryotes, as do proteins involved in sister-chromatid cohesion and chromosome condensation.
In budding yeast, ARS elements, as defined by a plasmid transformation assay, are small (<200 bp). They contain a binding site for the replication initiation protein, origin recognition complex (ORC) (Bell and Stillman 1992); the core of this site is the 11-bp ARS consensus sequence. During G1, ORC, in collaboration with Cdc6p and Cdt1p, loads the six-subunit minichromosome maintenance (Mcm2–7) complex at replication origins to establish prereplication complexes (pre-RCs) that are competent in directing the initiation of DNA replication during S phase (reviewed by Sivaprasad et al. 2006). While all ARS elements function as DNA replication origins on plasmids, some are not normally detectably active as replication initiation sites in their native chromosomal contexts. For example, only 10 of the 19 ARS elements on chromosome III initiate replication in ≥10% of cell cycles (Poloumienko et al. 2001).
One puzzle resulting from the early characterization of ARS elements, and indeed from the earlier characterization of replicating DNA by electron microscopy, is the relatively high density of replication origins. In a recent genomewide study, it was estimated that S. cerevisiae replication forks moved at a mean rate of 2.9 kb/min during an S phase of 55 min, suggesting that a single replication origin that initiates early in S phase should be able to replicate ∼320 kb of DNA (Raghuraman et al. 2001). However, the average spacing of active replication origins was estimated to be 40 kb in the same study, which is similar to the spacing of origins detected by electron microscopy (reviewed by Campbell and Newlon 1991). More recent genomewide analyses based on replication timing, accumulation of single-stranded DNA in hydroxyurea (HU)-treated cells, or chromatin immunoprecipitation of pre-RC proteins show a somewhat closer spacing of potential replication origins (Wyrick et al. 2001; Macalpine and Bell 2005; Feng et al. 2006; Xu et al. 2006). A comparative genomics approach confirmed that the majority of origins are conserved in closely related Saccharomyces species, indicating that there is selection to maintain this short inter-origin spacing (Nieduszynski et al. 2006). These genomewide studies have all been correlated and cataloged in a database of S. cerevisiae replication origins (Nieduszynski et al. 2007) that lists a total of 732 potential replication origins, 325 of which have been confirmed, 275 of which are listed as likely, and 132 of which are listed as dubious. The average spacing between the 600 confirmed or likely replication origins is 20 kb. This list includes replicators that are inefficient or inactive in their normal chromosomal contexts and may also still include some false positives. However, all of these studies support the idea that replicators are present at a high density in yeast chromosomes.
To establish a link between replicators, as defined by ARS elements, and chromosome stability, as well as to examine the effects of increased inter-origin spacing, we began systematically deleting ARS elements from chromosome III. In our initial studies with a 61-kb ring derivative of chromosome III carrying two efficient replicators, ARS307 and ARS309, and one inefficient replicator, ARS308, we found that the deletion of both ARS307 and AR309 severely destabilized the ring chromosome, causing it to be lost in >20% of cell divisions (Dershowitz and Newlon 1993). These findings suggested that replication origins are essential for chromosome maintenance. However, further experiments with the full-length chromosome III revealed that all of the active replication origins within a 164-kb region of the chromosome could be deleted with only a two- to threefold increase in the loss rate (Dershowitz et al. 2007). Surprisingly, we were able to isolate a 142-kb fragment lacking all known replicators, which is lost in <4% of divisions.
In this article we describe our genetic approach toward understanding the replication of this fragment lacking replicators. We isolated mutants defective in the maintenance of a 174-kb “originless” fragment, but proficient in the maintenance of the corresponding fragment with replication origins intact. We identified mutations in five genes that met our initial criteria. We found that the three mutations with the strongest phenotypes are likely to affect originless fragment maintenance through different mechanisms. OFM1-1 appears to disrupt an alternative initiation mechanism on the “originless” fragment and also have a mild initiation defect at normal origins. The ofm6 mutant is likely to have a replication fork progression defect, and ofm14 is a null allele of RAD9, which is required for the activity of the DNA damage checkpoint signal transduction pathway.
MATERIALS AND METHODS
Strains and media:
Yeast strains used in this study are listed in Table 1. All strains were isogenic with YPH499 (Sikorski and Hieter 1989) except the chromosome fragment donor strains, which are in the CF4-16B background (Dershowitz et al. 2007), and the Δydr217c (rad9Δ) strain, which is related to S288C (Brachmann et al. 1998). YKN10 was transformed with a PCR product amplified from the Δydr217c strain to introduce the rad9Δ∷KAN allele. YPD was prepared as described (Rose et al. 1990). Dropout and color assay media were purchased from U.S. Biological (recipes available upon request). Chromoductants were selected on −Leu −Trp −Arg dropout plates containing 60 μg/ml canavanine (Sigma-Aldrich, St. Louis) and 10 μg/ml cycloheximide (Sigma-Aldrich).
TABLE 1.
Yeast strains used in this study
| Strain name | Genotype | Source |
|---|---|---|
| YKN10 | 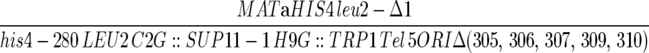 |
This work |
| ura3-52 lys2-801 trp1-Δ63 his3-Δ200 ade2-101 cyh2 can1 kar1-Δ15 ARO7 | ||
| F510 | 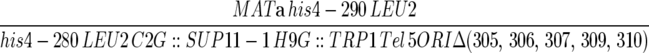 |
Dershowitz et al. (2007) |
| ura3-52 trp1-Δ63 ade2-101 | ||
| F510-3-16 | 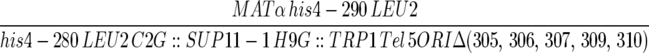 |
This work |
| ura3-52 trp1-Δ63 ade2-101 | ||
| F013 | 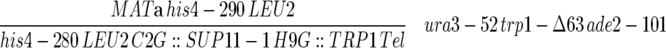 |
Dershowitz et al. (2007) |
| F013-1-24 | 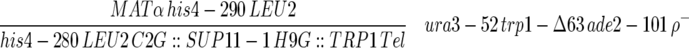 |
This work |
| YIC129 |  |
This work |
| ura3-52 trp1-Δ63 ade2-101 | ||
| YDN293 | 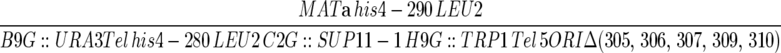 |
Dershowitz et al. (2007) |
| ura3-52 trp1-Δ63 ade2-101 | ||
| YDN108 | 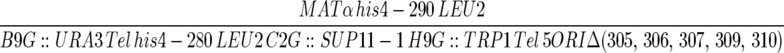 |
This work |
| ura3-52 trp1-Δ63 ade2-101 | ||
| F510α4A1-4 | 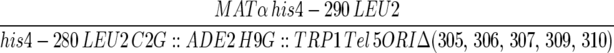 |
This work |
| ura3-52 trp1-Δ63 ade2-101 kar1-Δ15 | ||
| F510a6A6 | 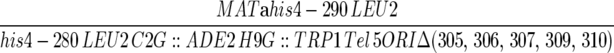 |
This work |
| ura3-52 trp1-Δ63 ade2-101 kar1-Δ15 | ||
| F013αB2C-1C | 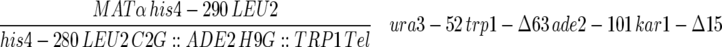 |
This work |
| YJT3 | MATα leu2-Δ1 ura3-52 lys2-801 trp1-Δ63 HIS3 ade2-101 cyh2 can1 kar1-Δ15 aro7Δ∷KAN | This work |
| YJT123 | MATaleu2-Δ1 ura3-52 lys2-801 trp1-Δ63 HIS3 ade2-101 cyh2 can1 kar1-Δ15 aro7Δ∷KAN OFM1-1 | This work |
| YJT132 | MATα leu2-Δ1 ura3-52 lys2-801 trp1-Δ63 his3-Δ200 ade2-101 cyh2 can1 kar1-Δ15ARO7 OFM1-1 | This work |
| YJT380 | 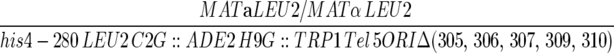 |
This work |
| ura3-52/ura3-52 lys2-801/lys2-801 trp1-Δ63/trp1-Δ63 HIS3/his3-Δ200 | ||
| ade2-101/ade2-101 cyh2/cyh2 can1/can1 kar1-Δ15/kar1-Δ15 ARO7/aro7Δ∷KAN OFM1/OFM1-1 | ||
| YJT436 | 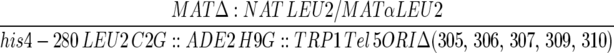 |
This work |
| ura3-52/ura3-52 lys2-801/lys2-801 trp1-Δ63/trp1-Δ63 HIS3/his3-Δ200 | ||
| ade2-101/ade2-101 cyh2/cyh2 can1/can1 kar1-Δ15/kar1-Δ15 ARO7/aro7Δ∷KAN OFM1/OFM1-1 | ||
| YJT378 | 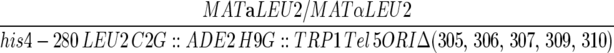 |
This work |
| ura3-52/ura3-52 lys2-801/lys2-801 trp1-Δ63/trp1-Δ63 HIS3/his3-Δ200 | ||
| ade2-101/ade2-101 cyh2/cyh2 can1/can1 kar1-Δ15/kar1-Δ15 ARO7/aro7Δ∷KAN OFM1-1/OFM1-1 | ||
| YJT380 | 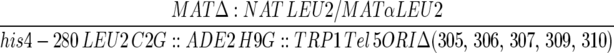 |
This work |
| ura3-52/ura3-52 lys2-801/lys2-801 trp1-Δ63/trp1-Δ63 HIS3/his3-Δ200 | ||
| ade2-101/ade2-101 cyh2/cyh2 can1/can1 kar1-Δ15/kar1-Δ15 ARO7/aro7Δ∷KAN OFM1-1/OFM1-1 | ||
| YJT446 | MATaleu2-Δ1 ura3-52 lys2-801 trp1-Δ63 his3-Δ200 ade2-101 cyh2 can1 kar1-Δ15 ARO7 ofm2 | This work |
| YJT142 | MATaleu2-Δ1 ura3-52 lys2-801 trp1-Δ63 his3-Δ200 ade2-101 cyh2 can1 kar1-Δ15 ARO7 ofm5 | This work |
| YJT129 | MATα leu2-Δ1 ura3-52 lys2-801 trp1-Δ63 his3-Δ200 ade2-101 cyh2 can1 kar1-Δ15 ARO7 ofm6 | This work |
| YJT128 | MATaleu2-Δ1 ura3-52 lys2-801 trp1-Δ63 HIS3 ade2-101 cyh2 can1 kar1-Δ15 aro7Δ∷KAN ofm6 | This work |
| YIC117 | 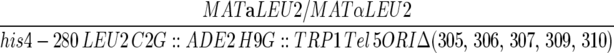 |
This work |
| ura3-52/ura3-52 lys2-801/lys2-80 trp1-Δ63/trp1-Δ6 HIS3/his3-Δ200 | ||
| ade2-101/ade2-10 cyh2/cyh can1/can1 kar1-Δ15/kar1-Δ1 ARO7/aro7Δ∷KAN OFM6/ofm6 | ||
| YIC116 | 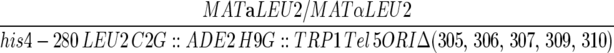 |
This work |
| ura3-52/ura3-52 lys2-801/lys2-801 trp1-Δ63/trp1-Δ63 HIS3/his3-Δ200 | ||
| ade2-101/ade2-101 cyh2/cyh2 can1/can1 kar1-Δ15/kar1-Δ15 ARO7/aro7Δ∷KAN ofm6/ofm6 | ||
| YJT90 | MATα leu2-Δ1 ura3-52 lys2-801 trp1-Δ63 his3-Δ200 ade2-101 cyh2 can1 kar1-Δ15 ARO7 rad9ofm14 | This work |
| YJT83 | MATaleu2-Δ1 ura3-52 lys2-801 trp1-Δ63 HIS3 ade2-101 cyh2 can1 kar1-Δ15 aro7Δ∷KAN rad9ofm14 | This work |
| YJT386 |  |
This work |
| ura3-52/ura3-52 lys2-801/lys2-801 trp1-Δ63/trp1-Δ63 HIS3/his3-Δ200 | ||
| ade2-101/ade2-101 cyh2/cyh2 can1/can1 kar1-Δ15/kar1-Δ15 ARO7/aro7Δ∷KAN RAD9/rad9ofm14 | ||
| YJT384 |  |
This work |
| ura3-52/ura3-52 lys2-801/lys2-801 trp1-Δ63/trp1-Δ63 HIS3/his3-Δ200 | ||
| ade2-101/ade2-101 cyh2/cyh2 can1/can1 kar1-Δ15/kar1-Δ15 ARO7/aro7Δ∷KAN rad9ofm14/rad9ofm14 | ||
| YJT383 |  |
This work |
| ura3-52/ura3-52 lys2-801/lys2-801 trp1-Δ63/trp1-Δ63 HIS3/his3-Δ200 | ||
| ade2-101/ade2-101 cyh2/cyh2 can1/can1 kar1-Δ15/kar1-Δ15 ARO7/aro7Δ∷KAN RAD9/rad9ofm14 OFM6/ofm6 | ||
| YIC120 |  |
This work |
| ura3-52/ura3-52 lys2-801/lys2-801 trp1-Δ63/trp1-Δ63 HIS3/his3-Δ200 | ||
| ade2-101/ade2-101 cyh2/cyh2 can1/can1 kar1-Δ15/kar1-Δ15 ARO7/aro7Δ∷KAN | ||
| Δydr217c | MATα his3-Δ1 leu2-Δ0 lys2-Δ0 ura3-Δ0 ydr217c(rad9)Δ∷KAN | Open Biosystems |
| YJT117 | MATaleu2-Δ1/MATα leu2-Δ0 ura3-52/ura3-Δ0 lys2-801/lys2-Δ0 ade2-101/ADE2 | This work |
| trp1-Δ63/TRP1 HIS3/his3-Δ1 can1/CAN1 cyh2/CYH2 kar1-Δ15/KAR1 aro7Δ∷KAN/ARO7 | ||
| ofm14/rad9Δ∷KAN | ||
| YJT135 | MATα leu2-Δ1 ura3-52 lys2-801 trp1-Δ63 his3-Δ200 ade2-101 cyh2 can1 kar1-Δ15 | This work |
| ARO7 rad9Δ∷KAN |
Plasmids:
pRS316RAD9 (a gift from N. Lowndes, National University of Ireland, Galway) was used to complement the ofm14 mutation. This plasmid was also used to construct pRS316RAD9gaprepair, which was used to rescue the rad9 allele from the ofm14 mutant. pRS316RAD9 was partially digested with NdeI, completely digested with BstEII, and filled in, and BglII linkers were added prior to recircularization. A plasmid was identified in which the 4.3-kb fragment containing the Rad9p open reading frame was deleted. Oligonucleotide synthesis and DNA sequencing were provided by the Molecular Resource Facility of UMDNJ-New Jersey Medical School (Newark, NJ).
Ofm mutant screen:
YKN10 was grown to log phase in −Leu medium. Cells were harvested, washed in water, and resuspended in 50 mm sodium phosphate buffer, pH 7.0. Ethyl methanesulfonate (EMS; Sigma-Aldrich) was added to 3% v/v for 10 min and then neutralized with 10% sodium thiosulfate (Sigma-Aldrich). This treatment resulted in 30% survival. Cells were diluted, plated on color assay medium, and incubated at 30° for 5 days prior to visually scoring the sectoring phenotype. Colonies with elevated sectoring were streaked on color assay plates; 45 showed a heritable increase in sectoring. Cells from a red colony from each of these streaks were mated to the fragment donor strains F510-3-16 (5ORIΔ) and F013-1-24 (0ORIΔ), and chromoductants were selected as described above. The kar1-Δ15 mutation in strain YKN10 prevents efficient nuclear fusion upon mating, causing the formation of transient heterokaryons and a few diploids. In the plates used to select chromoductants, the absence of Leu and Trp selects against the recipient strains, i.e., the mutants lacking the 5ORIΔ fragment. The presence of canavanine and cycloheximide selects against both the donor strains and against diploids because the can1 and cyh2 mutations are recessive. Rare cells with single chromosome transfers of the LEU2- and TRP1-marked 5ORIΔ or 0ORIΔ fragments from the donor strains into the can1 cyh2 (canavanine- and cycloheximide-resistant) recipient strain grow into colonies. Independent single colonies from these chromoduction plates were then streaked on color assay plates to score the sectoring phenotype. Of the 45 mutants, 11 showed elevated sectoring with the 5ORIΔ fragment compared to the 0ORIΔ fragment.
Genetic analysis:
Cells from red colonies of the original Ofm mutant isolates were crossed to YJT3, and diploids were selected on −His −Tyr plates. The resulting diploids were sporulated as described by Brachmann et al. (1998), and the 5ORIΔ fragment was introduced by chromoduction into each of the spores from either F510 or F510-3-16, depending on mating type. Later crosses made use of F510α4A1-4 or F510a6A6 as donor strains, which carry ADE2 rather than SUP11-1 on the 174-kb 5ORIΔ fragment. When the heterozygous diploid derived from the original OFM1-1 isolate was sporulated, the spore viability was so poor that 2:2 segregation of the mutant phenotype could not be scored. OFM1-1 spores were identified, and subsequent backcrosses showed good spore viability and 2:2 segregation of OFM1-1.
Sensitivity to DNA-damaging agents:
To assay UV sensitivity, serial dilutions of stationary cultures were spotted on YPD and exposed to ultraviolet light using either a Spectrolinker XL-1000 (Spectronics) UV source at doses of 50, 100, 150, or 200 J/m2 or timed exposures to a germicidal lamp (General Electric, Fairfield, CT). To assay sensitivity to the other agents, either 1:5 or 1:10 serial dilutions of stationary cultures were spotted on YPD plates containing hydroxyurea (25, 50, 100, 150, or 200 mm), methyl methanesulfonate (MMS; 0.0025, 0.005, 0.01, 0.015, 0.02, or 0.03%), phleomycin (4, 8, 10, or 12 μg/ml), or camptothecin (1, 3, 10, or 20 μg/ml). All agents were purchased from Sigma-Aldrich. Plates were incubated at 30° in the dark and scored daily.
Photography:
Images were acquired as TIFF files with a Nikon D-100 camera fitted with an AF Micro-Nikkor 60 mm f/2.8 D lens. Images were processed (cropped and then brightness/contrast and color balance adjusted) in Photoshop.cs v8.0 (Adobe Systems).
Analysis of replication intermediates:
Cells were harvested from asynchronous cultures grown in YPD to a density of 1.5–2.0 × 106/ml, and DNA was prepared as described (Theis and Newlon 1994). Approximately 60 μg of DNA was digested with EcoRI (New England Biolabs, Beverly, MA); replication intermediates were enriched by BND-cellulose (Sigma-Aldrich) chromatography, electrophoresed on two-dimensional (2D) gels, and blotted to Nytran SuperCharge (Schleicher & Schuell, Keene, NH) as described (Theis and Newlon 2001). [α-32P]dATP-labeled probes were prepared by random priming (Amersham Biosciences) and hybridized as described. Images were acquired on a Molecular Dynamics Typhoon 9410 scanner. Images were cropped, the gray-scale levels adjusted, and files saved in TIFF format using ImageQuant 5.2 (GE Healthcare). Photoshop.cs v8.0 was used to adjust the size of the images.
Plasmid stability assays:
Plasmids pDK243 (one copy of ARS1), pDK412 (7 copies of ARS1), pDK368-7 (ARS1 plus seven copies of wild-type histone H4 ARS, ARS209), pDK398-7 (ARS1 plus seven copies of an inactive mutant version of ARS209) were generously provided by Doug Koshland (Carnegie Institution, Baltimore) (Hogan and Koshland 1992). Transformants of wild type and OFM1-1 strains were grown in −Leu medium to log phase. The plasmid stability was determined as the percentage of plasmid-bearing cells in the culture by plating appropriate dilutions on −Leu and YPD plates and counting the resulting colonies. The average and standard deviations were calculated using data from three independent transformants.
Loss rate determinations:
Fluctuation analyses were performed using the colony isolation method described previously (Dershowitz and Newlon 1993). Red colonies were tested for leucine and tryptophan auxotrophy to distinguish chromosome loss events from gene conversions and mitotic recombinations. The loss rate per division and standard deviation was calculated by the method of Lea and Coulson (1949).
Checkpoint activation assays:
Cultures were processed for Western blot analysis as described by Pellicioli et al. (1999). α-Rad53 antibody was obtained from C. Santocanale and J. Diffley. α-Rad9 antibody was a gift from N. Lowndes. Following treatment with the primary antibody, secondary peroxidase-conjugated antibody was incubated with the Protran membrane for 1 hr and the filter was then exposed for a few minutes to Kodak XR film. To examine the DNA damage checkpoint, cells were arrested with 20 μg/ml α-factor (PRIMM, Milano, Italy); while arrested, 4-nitroquinoline 1-oxide (4-NQO; Sigma-Aldrich) was added to 2 μg/ml. To examine the replication stress checkpoint, cells were first synchronized with 2 μg/ml α-factor and then released into fresh medium containing 200 mm HU. FACS analysis was performed on aliquots from each of the time points as described by Foiani et al. (1994) using a Becton Dickinson FACScan.
RESULTS
The Ofm mutant screen:
We initiated a study to systematically delete all the known replicators on a eukaryotic chromosome, focusing eventually on a 174-kb fragment of chromosome III containing five efficient replicators, ARS305, ARS306, ARS307, ARS309, and ARS310, and seven inefficient or inactive ones, ARS300, ARS301, ARS302, ARS303, ARS320, ARS304, and ARS308 (Dershowitz et al. 2007). All the chromosome fragments were present in strains that were otherwise haploid; i.e., the strains are partially disomic for chromosome III (Figure 1). We used a colony color assay to visually screen for loss events (Hieter et al. 1985). The basis of the assay is that ade2-101 mutants accumulate a red pigment. The ade2-101 mutation can be suppressed by the ochre-suppressor tRNA, SUP11-1, or complemented by a wild-type copy of ADE2, one or the other of which we inserted into particular chromosome III fragments ∼7.5 kb to the left of CEN3. Potential chromosome fragment loss events are indicated by the appearance of a red sector in a colony. Chromosome loss events are confirmed by scoring the Trp and Leu phenotypes of cells from red sectors or colonies. The deletion allele leu2Δ1 is present on the balancer chromosome, while the wild-type allele resides on the fragment in its natural context. The TRP1 gene, which complements the trp1-Δ63 mutation in the strain background, was inserted at the fragmentation site to the right of ARS310. Therefore, fragment loss events yield red sectors that are Trp− and Leu−, while loss of the SUP11-1 or ADE2 gene from the fragment by gene conversion or mitotic recombination yields red sectors that are either Trp+ Leu+ or Trp+ Leu−.
Figure 1.—

Chromosome loss assay: line diagrams of chromosome III derivatives carried by the partially disomic strain with the 174-kb chromosome III fragment and the haploid strain following loss of the fragment. Phenotypes of colonies produced by these strains are indicated.
The 174-kb chromosome fragment with all replicators intact (the 0ORIΔ fragment) was lost at a rate of less than once in 10,000 divisions (Figure 2A). Deleting the five efficient replicators on the 174-kb fragment increased the loss rate ∼20-fold, but this fragment was lost only about once in 700 cell divisions (Figure 2B). The inactive replicators associated with HML become weakly active when the early, efficient replicators ARS305 and ARS306 are deleted (Vujcic et al. 1999), so one explanation for the stability of the 5ORIΔ fragment is that the normally inefficient/inactive replicators become active once the efficient replicators are removed. We tested this idea by using 2D gels to examine replication intermediates of ARS301, ARS302, ARS303/ARS320, and ARS304 in the 174-kb 5ORIΔ fragment and by removing the leftmost 32 kb of the 5ORIΔ fragment, deleting ARS300, ARS301, ARS302, ARS303, ARS320, and ARS304. We found that ARS301 and ARS303 were partially activated in the 174-kb 5ORIΔ fragment and that the loss rate of the doubly truncated, 142-kb 5ORIΔ fragment was increased ∼15-fold compared to the 174-kb 5ORIΔ fragment (Dershowitz et al. 2007). However, the 142-kb 5ORIΔ fragment was still replicated and segregated properly 98% of the time (Figure 2C). ARS308 is closely associated with CEN3, so it was removed by swapping CEN4 for CEN3; this substitution increased the loss rate <2-fold (Figure 2D). This 142-kb chromosome fragment lacks all known replicators, yet is still replicated and segregated properly in >96% of cell divisions. This result was completely unanticipated. Furthermore, it suggests that there are at least two mechanisms contributing to the replication of the 174-kb 5ORIΔ fragment (Figure 2B): activation of the inefficient/inactive replicators and a second, unanticipated mechanism that also allows replication of the 142-kb 6ORIΔ fragment (Figure 2D).
Figure 2.—

Line diagrams of chromosome III fragments and their loss rates. The positions of ARS elements or their deletions are indicated by vertical lines. Landmark genes are indicated by arrowheads. Loss rates were determined in the CF4-16B33 strain background and taken from Dershowitz et al. (2007); they differ slightly although not significantly from those measured in the YKN10 background (see Table 2). (A) 174-kb 0ORIΔ fragment. (B) 174-kb 5ORIΔ fragment. (C) 142-kb 5ORIΔ fragment. (D) 142-kb 6ORIΔ fragment.
We took a genetic approach to elucidate the mechanisms of replication of the 174-kb 5ORIΔ fragment, conducting a screen for mutants defective in the maintenance of the “originless” chromosome fragment. We chose to start the screen with the 174-kb 5ORIΔ fragment (Figure 2B) for two reasons. First, utilizing this fragment should allow the isolation of mutants that affect both mechanisms of replication: the activation of the normally inefficient/inactive replicators and the alternative mechanism(s) that maintains the 142-kb 6ORIΔ fragment lacking these replicators. Second, the 174-kb 5ORIΔ fragment has a relatively low loss rate, giving rise to colonies with few or no red sectors indicative of loss events; this phenotype makes a practical starting point for a visual screen for identifying colonies with increased sectoring.
Our screen consisted of two parts as outlined in Figure 3. The first was similar to that employed by Spencer et al. (1990) to identify chromosome transmission fidelity (ctf) mutants. Our starting strain, YKN10 (Table 1), was mutagenized with EMS, and 142,000 colonies carrying the fragment were examined for increased sectoring. A total of 45 isolates that had a heritable increase in sectoring were found. To identify those mutants that had a specific defect in the maintenance of the 5ORIΔ fragment, as opposed to a defect in the maintenance of any chromosome, e.g., ctf mutants, we performed a secondary screen using a single chromosome transfer technique called chromoduction (Ji et al. 1993) to introduce either the original 5ORIΔ fragment or a replicator-intact (0ORIΔ) fragment into each of the 45 isolates. The kar1-Δ15 mutation prevents normal karyogamy during mating, which decreases the efficiency of diploid formation ∼30-fold (Vallen et al. 1992). The mating partners form a transient heterokaryon, and, at a low frequency, single chromosomes are transferred from one nucleus to the other. With appropriate markers in the strain background and on the chromosome of interest, these rare single chromosome transfers can be selected (see materials and methods). Of the original 45 isolates, 11 had a greater sectoring rate with the 5ORIΔ fragment compared to the 0ORIΔ fragment (Figure 3). We refer to these as originless fragment maintenance (ofm) mutants. An example of the Ofm− phenotype is shown in Figure 4.
Figure 3.—

Summary of the Ofm mutant screen.
Figure 4.—

Ofm phenotype. Cells from a single colony of the ofm14 mutant lacking the 174-kb 5ORIΔ fragment were mated to donor strains carrying either the 174-kb 5ORIΔ or the 174-kb 0ORIΔ fragment. Individual chromoductants were selected and then streaked on color assay plates to score their sectoring phenotypes. Both fragments are marked with SUP11-1, which suppresses the ade2-101 mutation carried by the strain. Chromosome loss events were detected as red sectors on a white colony background. Representative colonies were photographed after 5 days of growth at 30°. Note the high rate of sectoring with the 5ORIΔ fragment compared to the 0ORIΔ fragment.
Segregation analysis showed that in 9 of the 11 isolates the Ofm phenotype resulted from single-gene mutations. Of these 9 mutations, 8 were recessive and 1 was dominant. The 8 recessive mutations fell into four complementation groups (ofm2, ofm5, ofm6, and ofm14), and segregation analysis showed that the dominant mutation is distinct from these (OFM1-1). The loss rates for the 174-kb 5ORIΔ and 0ORIΔ fragments were determined by fluctuation analysis (Dershowitz and Newlon 1993) in each of the mutants (Table 2). The loss rates of these fragments in the YKN10 parent strain were similar to the loss rates that we measured for these fragments in a different strain background in our initial study (Dershowitz et al. 2007). The loss rates of the 5ORIΔ fragment in the ofm2 and ofm5 mutants were only 3- to 5-fold higher than the loss rate of the 0ORIΔ fragment, indicating that these mutants have a rather weak Ofm phenotype. A further characterization of these two mutants will be presented elsewhere. The remaining three mutants have a robust Ofm phenotype with 80- to 400-fold higher loss rates for the 5ORIΔ fragment than for the 0ORIΔ fragment. The loss rate of the 5ORIΔ fragment was elevated ∼7-fold relative to wild type in the ofm6 and ofm14 mutants and ∼50-fold in the OFM1-1 mutant. As illustrated in Table 3, the ofm6 and ofm14 mutants are recessive because their heterozygous diploids have loss rates for the 5ORIΔ fragment similar to the wild-type diploid. Furthermore, the two mutants complement because the loss rate of the 5ORIΔ fragment in the double heterozygote was also similar to wild type. The loss rate of the 5ORIΔ fragment in the heterozygous OFM1-1 diploid was elevated relative to the wild-type diploid, indicating that the OFM1-1 mutation is semidominant (Table 3); the loss rate in the homozygous diploid was too high for us to measure.
TABLE 2.
Loss rates of various test chromosomes in Ofm mutants
| Loss rate per generation ± SD × 105
|
||||
|---|---|---|---|---|
| Strain | 174-kb 5ORIΔ fragmenta | 174-kb 0ORIΔ fragmentb | 142-kb 5ORIΔ fragmentc | Full-length 5ORIΔ chromosomed |
| Wild type | 210 ± 30 | 2.7 ± 1.6 | 3,800 ± 600 | 9.9 ± 3 |
| OFM1-1 | 10,000 ± 1000 | 23 ± 4 | Not recoverede | 240 ± 40 |
| ofm2 | 6,100 ± 1500 | 1,700 ± 200 | ND | ND |
| ofm5 | 5,000 ± 1000 | 930 ± 210 | ND | ND |
| ofm6 | 1,400 ± 100 | 4.2 ± 1.2 | 7,700 ± 600 | 530 ± 90 |
| ofm14 | 1,500 ± 100 | 19 ± 4 | 9,600 ± 800 | 39 ± 11 |
| rad9Δ | 2,100 ± 400 | 32 ± 6 | ND | 53 ± 9 |
ND, not determined.
Shown in Figure 2B; introduced by chromoduction from F510, F510-3-16, F510α4A1-4, or F510a6A.
Shown in Figure 2A; introduced by chromoduction from F013, F013-1-24, or F013αB2C-1C.
Shown in Figure 2C; introduced by chromoduction from YDN293 or YDN108.
Full-length chromosome deleted for ARS305, ARS306, ARS307, ARS309, and ARS310; introduced by chromoduction from YIC129.
Rare chromoductants were either Ura− or Trp−, indicating chromosome rearrangement, e.g., break-induced replication using the balancer chromosome to restore the sequences that had been removed by the fragmentation or had restored an origin by gene conversion.
TABLE 3.
Loss rates of the 174-kb 5ORIΔ fragment in homozygous and heterozygous diploids
| Loss rate ± SD × 105
|
||
|---|---|---|
| Strain | Homozygous | Heterozygous |
| Wild type | 300 ± 60 | NA |
| OFM1-1 a/α | NDa | 870 ± 130 |
| OFM1-1 Δ/α | NDa | 1800 ± 300 |
| ofm6 | 1700 ± 300 | 390 ± 70 |
| ofm14 | 950 ± 110 | 310 ± 60 |
| Doubly heterozygous | ||
| ofm6 ofm14 | 280 ± 50 | |
NA, not applicable. ND, not determined.
While chromoductants were obtained, the loss rate was too high to determine a reliable loss rate.
Ofm mutants disrupt different mechanisms of 5ORIΔ fragment maintenance:
One of the rationales for performing the screen with the 174-kb 5ORIΔ fragment (Figure 2B) was the potential of isolating mutants defective in either of the two mechanisms contributing to the replication of this fragment, the activation of the inefficient/inactive replicators, or the alternative mechanism(s) that maintains the 142-kb 6ORIΔ fragment (Figure 2D) lacking these replicators. If a particular Ofm mutant were defective in the activation of the inefficient/inactive replicators, then the replication of the 174-kb 5ORIΔ fragment would be dependent solely on the alternative mechanism. Deleting the inefficient/inactive replicators from the fragment should have no effect on its maintenance since these replicators are rendered nonfunctional by the mutation. Therefore, the loss rate of the 142-kb 5ORIΔ fragment (Figure 2C) in such a mutant should be the same as that of the 174-kb 5ORIΔ fragment. Conversely, if a particular Ofm mutant is defective in an alternative mechanism, then the replication of the 174-kb 5ORIΔ fragment would be solely dependent on the activation of the inefficient/inactive replicators. Therefore, in such a mutant, it should not be possible to recover the 142-kb 5ORIΔ fragment, or the loss rate of this fragment would be substantially greater than the 2% rate seen in the wild-type strain.
As shown in Table 2, we failed to recover OFM1-1 chromoductants with the 142-kb 5ORIΔ fragment, suggesting that the OFM1-1 mutation might interfere with an alternative mechanism for fragment maintenance. While we favor the hypothesis that OFM1-1 interferes with an alternative mechanism, we cannot rule out the trivial possibility that a modest increase in the loss rate of the 142-kb 5ORIΔ fragment on top of the already high loss rate (10%) of the 174-kb 5ORIΔ fragment in the mutant might preclude the recovery of the 142-kb 5ORIΔ fragment.
We also found that the loss rate of the 142-kb 5ORIΔ fragment was increased relative to the 174-kb 5ORIΔ fragment in the ofm6 and ofm14 mutants. Therefore, the inefficient/inactive replicators contribute to the stability of the 174-kb 5ORIΔ fragment in each of the mutants. However, the increases in loss rates were modest, three- and sevenfold in ofm6 and ofm14, respectively, resulting in loss rates of 7.7 × 10−2 and 9.6 × 10−2 losses/division. These modest increases in loss rate of the 142-kb 5ORIΔ fragment make it unlikely that the primary defect of the ofm6 and ofm14 mutants is in an alternative initiation mechanism.
The average spacing between active replication origins in S. cerevisiae is ∼40 kb. Assuming similar fork rates and initiation times, the forks initiated at adjacent origins would converge after traveling ∼20 kb on average. The deletion of the five efficient origins from the 174-kb chromosome III fragment creates a situation in which a replication fork would have to travel a much longer distance than average. A single initiation event in the center of the 174-kb 5ORIΔ fragment would require each fork to traverse 86.5 kb, while a single initiation event near one end would require a single fork to traverse nearly 174 kb. Therefore, it seems plausible that mutants with a fork progression defect would show an Ofm phenotype. It should be possible to distinguish mutants that impair the ability of the 174-kb 5ORIΔ fragment to initiate replication from those that impair replication elongation. Mutants of the former class should be rescued by adding an efficient origin at one end while those of the latter class should not be. We chose to examine this possibility by introducing the full-length 5ORIΔ chromosome into the Ofm mutants. This chromosome carries the same five origin deletions as the 174-kb 5ORIΔ fragment, but also carries the origins distal to the fragmentation point: the inefficient origin, ARS313, is located at 192 kb, ∼20 kb distal to the fragmentation point, and the efficient origin, ARS315, is located at 225 kb (Poloumienko et al. 2001). The full-length 5ORIΔ chromosome was 40-fold more stable than the 174-kb 5ORIΔ fragment in the OFM1-1 and ofm14 mutants (Table 2), suggesting that the presence of efficient replication origins suppresses the fragment maintenance defect in these two mutants. In contrast, in the ofm6 mutant, the full-length 5ORIΔ chromosome was only threefold more stable than the 174-kb 5ORIΔ fragment, and >50-fold less stable than the full length 5ORIΔ chromosome in the wild-type strain, suggesting that the defect in ofm6 is in fork progression.
The conclusion that ofm6 has a defect in fork progression rests on the assumption that initiation at ARS313 and ARS315 is not impaired on the full-length 5ORIΔ chromosome. While initiation on the 174-kb 5ORIΔ chromosome fragment is difficult to address in this strain background due to the presence of identical sequences on the wild-type balancer chromosome, we did examine initiation at several origins in haploid—i.e., lacking a test fragment/chromosome—strains. Initiation at the origins ARS305, ARS315, and ARS1421 was unaffected in the ofm6 and ofm14 mutants (data not shown).
OFM1-1 mutants, however, do have a slight initiation defect. This defect is seen in neutral–neutral 2D gels (Brewer and Fangman 1987) probed for ARS305 and ARS315 (Figure 5). Small Y-shaped replication intermediates (arrowheads) were detected in the OFM1-1 mutant, but not in the wild-type strain, indicating that these origins fail to fire some fraction of the time in the mutant. While both ARS elements are affected, the defect is more pronounced at ARS315. This initiation defect was also seen as a decrease in plasmid stability in OFM1-1 mutants that could be partially suppressed by the presence of multiple ARS elements on the plasmid (Table 4). The activity of ARS1 is decreased as reflected in the lower mitotic stabilities of pDK243 and pDK398-7 in the OFM1-1 strain compared to wild type. Having multiple copies of either ARS1 or ARS209 increased the stability of the plasmid, consistent with an initiation defect (compare pDK412 to pDK243 and pDK368-7 to pDK398-7); multiple copies of ARS209 rescued better than multiple copies of ARS1 (compare pDK368-7 to pDK412). In summary, the OFM1-1 mutation decreases initiation efficiency at all three origins examined, but the strength of the effect varies.
Figure 5.—

Two-dimensional gel analysis of ARS305 and ARS315 replication intermediates in the OFM1-1 mutant. (Top) 2D gels (Brewer and Fangman 1987) probed to detect replication intermediates from ARS305 and ARS315. Abundant bubble-shaped intermediates are detected from both ARS elements in the wild-type and OFM1-1 strains. The arrowheads point to small Y-shaped intermediates (solid arrowhead, ARS305; half-solid arrowhead, ARS315). DNA isolated from the OFM1-1 strain has more small Y-shaped intermediates than DNA from the wild-type strain, especially for ARS315, indicating a slight initiation defect. (Bottom) Diagrams, drawn to scale, of the EcoRI fragments detected by the probes used; note that the ARS elements are near the center of each fragment.
TABLE 4.
Plasmid stabilities
| Strain | pDK243 (ARS1) | pDK412 (ARS1 × 7) | pDK368-7 (ARS1 + ARS209 × 7) | pDK398-7 (ARS1 + mut ARS209 × 7) |
|---|---|---|---|---|
| Wild type | 91 ± 3 | 96 ± 6 | 85 ± 1 | 92 ± 1 |
| OFM1-1 | 31 ± 6 | 76 ± 7 | 96 ± 3 | 31 ± 1 |
Values shown are the percentage of plasmid-bearing cells (mean ± standard deviation) in a log-phase culture grown under selection.
Some of the Ofm mutants are sensitive to DNA damage:
Many mutations that disrupt aspects of DNA metabolism cause sensitivity to DNA damage (DePamphilis 2006). Therefore, we examined whether our Ofm mutants were sensitive to a variety of DNA-damaging agents (Table 5), including MMS (alkylation of bases), UV light (pyrimidine dimer and 6-4 photo-product formation), phelomycin (double-strand break formation), camptothecin (trapped topoisomerase II intermediates), and HU (depletion of dNTP pools). The OFM1-1 mutant was slightly sensitive to MMS, while the ofm5 mutant was very sensitive to both MMS and HU and somewhat sensitive to UV. The ofm6 mutant was slightly sensitive to UV and HU. The ofm14 mutant was very sensitive to UV light, while the ofm2 mutant was somewhat sensitive. None of the mutants was sensitive to phleomycin or camptothecin.
TABLE 5.
Sensitivities of Ofm mutants to DNA-damaging agents
| Strain | UVa | MMSb | HUc | PHLEOd | CPTe |
|---|---|---|---|---|---|
| Wild type | + | + | + | + | + |
| OFM1-1 | + | ++ | + | + | + |
| ofm2 | +++ | + | + | ND | ND |
| ofm5 | +++ | ++++ | ++++ | ND | ND |
| ofm6 | ++ | + | ++ | + | + |
| ofm14 | ++++ | + | + | ND | ND |
The number of “+” signs reflects a semiquantitative summary of the sensitivities of the different strains to a series of doses for each agent as described in materials and methods. Increased sensitivity is indicated by an increased number of “+” signs. ND, not determined.
Ultraviolet light.
Methyl methanesulfonate.
Hydroxyurea.
Phleomycin.
Camptothecin.
DNA damage sensitivity can result either from an inability to repair the damaged DNA or from a failure to activate the DNA damage checkpoint (Weinert and Hartwell 1990). In S. cerevisiae, DNA damage activates a signal transduction cascade, which results in the phosphorylation of Rad53p, a homolog of the mammalian effector kinase, Chk2p (Sanchez et al. 1996; Sun et al. 1996). To determine if the DNA damage checkpoint was intact, the phosphorylation status of Rad53p was assessed in cells that were arrested in G1 with α-factor and then treated with the UV mimetic, 4-NQO. Rad53p phosphorylation is indicated by a decrease in the mobility of the protein on a Western blot. The slower migrating form of Rad53p was detected after 15 min of 4-NQO treatment in wild-type cells and at all subsequent time points; the same was true of the OFM1-1, ofm2, ofm5, and ofm6 mutants, indicating that the signal transduction pathway leading to Rad53p activation is intact in these mutants (Figure 6). However, the observation that no change in the mobility of Rad53p was detected at any of the time points in the ofm14 mutant indicates that this mutant failed to activate Rad53p under these conditions. As a control, we also monitored the phosphorylation of Rad53p in response to the replication stress checkpoint, which functions through a signal transduction pathway that is partially redundant with the DNA damage checkpoint pathway. In this case, cells were synchronized in G1 with α-factor and then released into medium containing the ribonucleotide reductase inhibitor, hydroxyurea. Rad53p was phosphorylated in the ofm14 mutant, as it was in the wild-type strain and the other mutants, indicating that the replication stress checkpoint was activated in response to HU (Figure 6).
Figure 6.—

Western blots of Rad53p in Ofm mutants. Portions of Western blots probed to detect Rad53p are shown. The hyperphosphorylation of Rad53p, indicating activation of the checkpoint, is detected as a decrease in electrophoretic mobility. (Left, 4-NQO) Log-phase cells were arrested in G1 with 20 μg/ml α-factor; after arrest, the UV mimetic, 4-nitroquinoline 1-oxide, was added to a final concentration of 2 μg/ml, and samples were taken at 0, 15, 30, 45, and 60 min; FACS analysis confirmed that cells remain blocked in G1. Note that only the ofm14 mutant failed to hyperphosphorylate Rad53p under these conditions, indicating that the signal transduction cascade is blocked prior to Rad53p activation. (Right, HU) log-phase cells were first synchronized in G1 with α-factor and then released from the G1 block into medium containing 0.2 m hydroxyurea, and samples were taken at 0, 30, 60, 120, and 180 min, except for the ofm2 time course in which samples were taken at 20, 40, 60, 90, and 120 min. Note that Rad53p was hyperphosphorylated by all the strains, indicating that the replication stress signal transduction cascade is intact.
ofm14 is an allele of RAD9:
We initially attempted to complement the sectoring phenotype of the ofm14 mutant using a yeast genomic library. This was unsuccessful because the rate of mis-segregation of the 5ORIΔ fragment (a cell with two copies of the fragment gives rise to a colony with few sectors) was greater than the probability of obtaining a complementing clone from the library. The UV sensitivity of the ofm14 mutant was tightly linked to the Ofm sectoring phenotype; the two phenotypes cosegregated in 20 tetrads dissected from a heterozygous diploid strain. To identify the gene responsible for the ofm14 phenotype, we examined a number of candidate genes for their inability to complement the ofm14 UV sensitivity. A strong candidate gene was RAD9 because it is known that Rad9p is required for Rad53p activation in response to DNA damage but is dispensable for Rad53p activation in HU-treated cells (Pellicioli et al. 1999). The ofm14 mutant was crossed to a number of strains from the systematic deletion collection (Giaever et al. 2002), and the resulting diploids were tested for UV sensitivity. The ofm14/Δydr217c∷KAN diploid was UV sensitive (Figure 7A), suggesting that ofm14 is an allele of RAD9. This hypothesis was confirmed by showing that RAD9 is linked to ofm14, that the UV sensitivity and the sectoring phenotype of the 174-kb 5ORIΔ fragment of ofm14 can be complemented by a plasmid carrying RAD9 (Figure 7, B and C), and that the deletion of RAD9 causes an Ofm phenotype (Table 2). We cloned the rad9ofm14 allele by gap repair (Orr-Weaver et al. 1983) and sequenced it. The sequence showed a single G-to-A transition at position 3191 of the open reading frame, converting codon TRP-1064, within the BRCT repeats, to a stop codon. This premature stop codon appears to cause a null allele on the basis of observations that the mutant failed to phosphorylate Rad53p in G1-arrested cells in response to the UV mimetic 4-NQO (Figure 6), that no Rad9p was detected by polyclonal antibodies on Western blots (data not shown), and that the deletion allele showed a UV sensitivity and 5ORIΔ fragment loss rate similar to that of the ofm14 allele (Figure 7B and Table 2).
Figure 7.—

UV sensitivity of ofm14, rad9Δ, and ofm14/rad9Δ strains and complementation of both the sectoring phenotype and UV sensitivity of ofm14 by pRS316RAD9. (A) Serial fivefold dilutions of the indicated strains were spotted on YPD plates. The wild-type and ofm14 strains are in the YKN10 background, the rad9Δ strain is from the yeast deletion collection (S288C-related background), and the diploid is the product of the mating of the two. The plate on the right was UV-irradiated while that on the left was not. As seen in the second row, rad9Δ fails to complement the UV sensitivity of ofm14. (B, top) Serial fivefold dilutions of the haploid wild-type, ofm14, and rad9Δ strains in the YKN10 background. Plate on the right was UV irradiated while that on the left was not. Note that the ofm14 and rad9Δ mutants show similar UV sensitivities when in the same strain background. (Bottom) Serial fivefold dilutions of the ofm14 haploid transformed with pRS316RAD9 or pRS416 alone. pRS416 is a derivative of pRS316 (Sikorski and Hieter 1989) with minor modifications in the polylinker and the lacZ gene (Brachmann et al. 1998). (C) Complementation of the ofm14 sectoring phenotype by pRS316RAD9. Representative colonies of an ofm14 strain carrying the 174-kb 5ORIΔ fragment transformed with pRS316RAD9 or pRS416 were photographed after 5 days of growth at 30° on −uracil limiting adenine plates.
DISCUSSION
We undertook a genetic approach to understanding the unanticipated mitotic stability of a fragment of chromosome III lacking all known efficient replicators. We isolated five different mutants in our screen. While all the mutations destabilize the 174-kb 5ORIΔ fragment, they differ in the extent to which they also destabilize the 174-kb 0ORIΔ fragment and, we infer, all chromosomes. Three mutations preferentially destabilize the 5ORIΔ fragment: OFM1-1, ofm6, and ofm14 (Table 2). Because the ofm14 mutant was sensitive to UV irradiation in addition to destabilizing the 5ORIΔ fragment, we were able to cross the ofm14 mutant to various strains known to be UV sensitive from the yeast knockout collection and score UV sensitivity, i.e., noncomplementation, in the resulting diploids. The rad9Δ (ydr217CΔ) failed to complement the ofm14 mutation as shown by the UV sensitivity of the diploid. Linkage analysis and complementation of both the UV sensitivity and 174-kb 5ORIΔ fragment loss phenotypes of the ofm14 mutant by a RAD9 plasmid confirmed that ofm14 is an allele of RAD9 (Figure 7 and Table 2).
How might rad9 mutations result in an Ofm phenotype? RAD9 is part of the DNA damage response signal transduction pathway. RAD9 is required for the MEC1/TEL1-dependent autophosphorylation of Rad53p in response to DNA damage (Gilbert et al. 2001; Sweeney et al. 2005; Ma et al. 2006). RAD53 is homologous to the effector kinases in the DNA damage response pathways of other organisms, e.g., CHK2 in humans and cds1 in Schizosaccharomyces pombe (reviewed by Melo and Toczyski 2002). Rad53p kinase activity is required for cell cycle arrest and the induction of DNA damage repair activities (Allen 1994; Weinert et al. 1994). RAD53 is also required to stabilize replication forks that encounter template damage (Lopes et al. 2001; Sogo et al. 2002) and to delay the activation of late-firing or dormant origins in response to both DNA damage and hydroxyurea-induced nucleotide depletion (Santocanale and Diffley 1998; Shirahige et al. 1998; Santocanale et al. 1999).
One hypothesis that could account for the Ofm phenotype of the rad9ofm14 mutant is that Rad9p stabilizes stalled forks or aids in the restart of collapsed forks. When a replication fork is blocked upon encountering endogenous DNA damage, a fork initiated at an adjacent origin can complete replication of the region beyond the damage; as the number of active origins decreases, the probability becomes low that there will be an active origin beyond the damage, making DNA damage checkpoint-dependent fork stabilization and restart increasingly important for the completion of replication. This model is consistent with the observation that deletion of RAD9 caused destabilization of a replicator-deficient yeast artificial chromosome (van Brabant et al. 2001).
However, this simple arrest/restart model does not explain one important phenotype of the ofm14 mutant. The arrest/restart model predicts that the 174-kb 5ORIΔ fragment and the full-length 5ORIΔ chromosome should have similar loss rates because the originless region in the full-length 5ORIΔ chromosome is slightly longer than the 174-kb 5ORIΔ fragment. In both cases, the replication of a region distal to a blocked fork would depend on a fork initiated at the inefficient/inactive ARS elements flanking HML or by some other mechanism. In contrast to this expectation, the loss rate of the full-length 5ORIΔ chromosome in the ofm14 mutant was only fourfold higher than the loss rate of this chromosome in the wild-type strain (Table 2). Therefore, even in the ofm14 mutant, it seems likely that a fork can successfully traverse the 225 kb from ARS315, or the 192 kb from ARS313, to the left end of the chromosome. This finding is also consistent with our failure to detect the phosphorylation-dependent mobility shift of Rad53p on Western blots from a wild-type strain carrying the 174-kb 5ORIΔ fragment (M. Lopes, M. Foiani and C. Newlon, unpublished observations), although if Rad53p were activated only in those cells that lose the fragment (1 loss/700 cell divisions), it possible that the mobility shift could not have been detected. A second inconsistency with the arrest/restart model is that Rad9p is not required for activation of Rad53p in response to HU treatment, which inhibits ribonucleotide reductase and is widely used to induce replication stress (Pellicioli et al. 1999). This inconsistency depends on the extent to which the slow-moving forks induced by HU treatment actually mimic replication forks stalled by physiological processes or DNA damage. It remains a possibility that the forks initiated at an efficient origin like ARS315 are of better “quality” than those initiated on the 5ORIΔ fragment; i.e., they are less prone to collapse. This possibility could be tested by examining the components moving with the replication fork by a chromatin immunoprecipitation (ChIP)-on-chip experiment utilizing a strain carrying the 5ORIΔ fragment with a balancer chromosome III from Saccharomyces carlsbergensis.
In addition to its clearly defined role in activation of the DNA damage checkpoint, recent work has strongly suggested a role for Rad9p in the recombinational repair of double-strand breaks during G2 in S. cerevisiae (Toh et al. 2006). These findings are consistent with experiments in S. pombe that indicate a similar role for crb2, the ortholog of RAD9 (Caspari et al. 2002). RAD52 is required for homologous recombination in S. cerevisiae. On the basis of our finding that deletion of rad52 had very little effect on the loss rate of the 174-kb 5ORIΔ fragment (Dershowitz et al. 2007), it is unlikely that the loss of Rad9p activity in recombinational repair is the basis of the Ofm phenotype of the rad9ofm14 mutation.
We have not yet identified the mutant gene in either ofm6 or OFM1-1. The defect in ofm6 appears to be in fork progression. This conclusion is based on the observation that the loss rate of the full-length 5ORIΔ chromosome is only threefold lower than that of the 5ORIΔ fragment in the ofm6 strain. The defect in OFM1-1 appears to be in an alternative initiation mechanism on the basis of the observation that we failed to recover the 142-kb 5ORIΔ fragment in this mutant (Table 2). We have also shown by 2D gel analysis that the OFM1-1 mutant has a slight initiation defect at normal origins (Figure 5). The defect appears to be more severe at ARS315 than at ARS305. In addition, the plasmid stability assay indicates an initiation defect at ARS1 (Table 4). Because there is an initiation defect in all three origins examined, we suggest that the OFM1-1 mutation confers a general initiation defect at normal origins. As expected for an initiation defect, we found that an ARS1 plasmid carrying seven copies of ARS209 (Hogan and Koshland 1992) was stable in the mutant, while a plasmid carrying seven copies of a mutant derivative of ARS209 was as unstable as a plasmid carrying only ARS1 (Table 4).
At this point, it is not clear what the relationship is between the defect in the alternative initiation mechanism and the mild initiation defect at normal origins. It is interesting to note, however, that the orc2-1 mutant behaves similarly to the OFM1-1 mutant. At the permissive temperature, orc2-1 strains show decreased stability of ARS1- and ARS209-based plasmids (Dillin and Rine 1997; Dershowitz et al. 2007) and an initiation defect at ARS1 (Fox et al. 1995; Liang et al. 1995; Dillin and Rine 1997). In addition, the loss rate of the 174-kb 5ORIΔ fragment is elevated fivefold in the orc2-1 mutant, and the 142-kb 5ORIΔ fragment could not be recovered (Dershowitz et al. 2007). These observations are consistent with the hypothesis that the normal replication machinery plays a role in the alternative initiation mechanism, although at this point we cannot rule out the possibility that the elevated fragment loss rates in the orc2-1 mutant are due to ORC's role in cohesin-independent sister-chromatid cohesion (Shimada and Gasser 2007).
In summary, we have utilized a genetic screen to try to understand the replication of a fragment of chromosome III lacking known replicators. We isolated mutations in three different genes that preferentially destabilize the 5ORIΔ fragment compared to the 0ORIΔ fragment: OFM1-1, ofm6, and ofm14. Taking advantage of the UV sensitivity of the ofm14 mutant, we were able to show that ofm14 is an allele of rad9, implicating the DNA damage checkpoint in the maintenance of the 5ORIΔ fragment. Additional mutants in the DNA damage checkpoint pathway also show an Ofm phenotype (our unpublished results). We are currently focusing on identifying the genes mutated in the ofm6 and OFM1-1 strains. These mutations appear to affect different aspects of fragment maintenance. The ofm6 mutation appears to affect fork progression on the basis of similar loss rates for the 174-kb 5ORIΔ fragment and the full-length 5ORIΔ chromosome. While the OFM1-1 mutation does increase the loss rate of the full-length 5ORIΔ chromosome, it increases the loss rate of the 174-kb 5ORIΔ fragment much more dramatically. We believe that further characterization of these mutants and the “originless” chromosome fragments will lead to interesting insights into links between DNA replication and genome stability.
Acknowledgments
We are grateful to Noel Lowndes for providing the RAD9 plasmid and Rad9p antibodies, to John Diffley and C. Santocanale for Rad53p antibodies, to Marco Foiani for help with Rad53p activation assays and for comments on the manuscript, and to A. Spaisman, K. Carroll, F. DiSanzo, and S. Sajous for assistance with the analysis of the Ofm mutants. This work was supported by National Institutes of Health grant GM35679 to C.S.N. and by a fellowship from Istituto Pasteur Fondazione Cenci Bolognetti to C.I. L.F. was supported by the Ministero Universitá Ricerca Scientifica Tecnologica-Progetti Ateneo, C.M. was supported by a Pasteurian Sciences graduate fellowship, and G.L. was supported by the Associazione Italiana per la Ricerca sul Cancro.
References
- Allen, J. B., Z. Zhou, W. Siede, E. C. Friedberg and S. J. Elledge, 1994. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 8: 2416–2428. [DOI] [PubMed] [Google Scholar]
- Bell, S. P., and B. Stillman, 1992. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357: 128–134. [DOI] [PubMed] [Google Scholar]
- Brachmann, C. B., A. Davies, G. J. Cost, E. Caputo, J. Li et al., 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132. [DOI] [PubMed] [Google Scholar]
- Brewer, B. J., and W. L. Fangman, 1987. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51: 463–471. [DOI] [PubMed] [Google Scholar]
- Campbell, J. L., and C. S. Newlon, 1991. Chromosomal DNA replication, pp. 41–146 in The Molecular and Cellular Biology of the Yeast Saccharomyces: Genome Dynamics, Protein Synthesis, and Energetics, edited by J. R. Broach, J. R. Pringle and E. Jones. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Caspari, T., J. M. Murray and A. M. Carr, 2002. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 16: 1195–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePamphilis, M. L., 2006. DNA Replication and Human Disease. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Dershowitz, A., and C. S. Newlon, 1993. The effect on chromosome stability of deleting replication origins. Mol. Cell. Biol. 13: 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dershowitz, A., M. Snyder, M. Sbia, J. H. Skurnick, L. Y. Ong et al., 2007. Linear derivatives of Saccharomyces cerevisiae chromosome III can be maintained in the absence of autonomously replicating sequence elements. Mol. Cell. Biol. 27: 4652–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin, A., and J. Rine, 1997. Separable functions of ORC5 in replication initiation and silencing in Saccharomyces cerevisiae. Genetics 147: 1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, W., D. Collingwood, M. E. Boeck, L. A. Fox, G. M. Alvino et al., 2006. Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat. Cell Biol. 8: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foiani, M., F. Marini, D. Gamba, G. Lucchini and P. Plevani, 1994. The B subunit of the DNA polymerase alpha-primase complex in Saccharomyces cerevisiae executes an essential function at the initial stage of DNA replication. Mol. Cell. Biol. 14: 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foiani, M., A. Pellicioli, M. Lopes, C. Lucca, M. Ferrari et al., 2000. DNA damage checkpoints and DNA replication controls in Saccharomyces cerevisiae. Mutat. Res. 451: 187–196. [DOI] [PubMed] [Google Scholar]
- Fox, C. A., S. Loo, A. Dillin and J. Rine, 1995. The origin recognition complex has essential functions in transcriptional silencing and chromosomal replication. Genes Dev. 9: 911–924. [DOI] [PubMed] [Google Scholar]
- Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles et al., 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391. [DOI] [PubMed] [Google Scholar]
- Gilbert, C. S., C. M. Green and N. F. Lowndes, 2001. Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol. Cell 8: 129–136. [DOI] [PubMed] [Google Scholar]
- Hieter, P., C. Mann, M. Snyder and R. W. Davis, 1985. Mitotic stability of yeast chromosomes: a colony color assay that measures nondisjunction and chromosome loss. Cell 40: 381–392. [DOI] [PubMed] [Google Scholar]
- Hogan, E., and D. Koshland, 1992. Addition of extra origins of replication to a minichromosome suppresses its mitotic loss in cdc6 and cdc14 mutants of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 89: 3098–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, H., D. P. Moore, M. A. Blomberg, L. T. Braiterman, D. F. Voytas et al., 1993. Hotspots for unselected Ty1 transposition events on yeast chromosome III are near tRNA genes and LTR sequences. Cell 73: 1007–1018. [DOI] [PubMed] [Google Scholar]
- Kelly, T. J., and B. Stillman, 2006. Duplication of DNA in eukaryotic cells, pp. 1–29 in DNA Replication and Human Disease, edited by M. L. DePamphilis. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Lea, D., and C. Coulson, 1949. The distribution of numbers of mutants in bacterial population. J. Genet. 49: 264–285. [DOI] [PubMed] [Google Scholar]
- Liang, C., M. Weinreich and B. Stillman, 1995. ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell 81: 667–676. [DOI] [PubMed] [Google Scholar]
- Lopes, M., C. Cotta-Ramusino, A. Pellicioli, G. Liberi, P. Plevani et al., 2001. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561. [DOI] [PubMed] [Google Scholar]
- Ma, J. L., S. J. Lee, J. K. Duong and D. F. Stern, 2006. Activation of the checkpoint kinase Rad53 by the phosphatidyl inositol kinase-like kinase Mec1. J. Biol. Chem. 281: 3954–3963. [DOI] [PubMed] [Google Scholar]
- MacAlpine, D. M., and S. P. Bell, 2005. A genomic view of eukaryotic DNA replication. Chromosome Res. 13: 309–326. [DOI] [PubMed] [Google Scholar]
- Melo, J., and D. Toczyski, 2002. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 14: 237–245. [DOI] [PubMed] [Google Scholar]
- Nieduszynski, C. A., Y. Knox and A. D. Donaldson, 2006. Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 20: 1874–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieduszynski, C. A., S. Hiraga, P. Ak, C. J. Benham and A. D. Donaldson, 2007. OriDB: a DNA replication origin database. Nucleic Acids Res. 35: D40–D46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver, T. L., J. W. Szostak and R. J. Rothstein, 1983. Genetic applications of yeast transformation with linear and gapped plasmids. Methods Enzymol. 101: 228–245. [DOI] [PubMed] [Google Scholar]
- Pellicioli, A., C. Lucca, G. Liberi, F. Marini, M. Lopes et al., 1999. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 18: 6561–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poloumienko, A., A. Dershowitz, J. De and C. Newlon, 2001. Completion of replication map of Saccharomyces cerevisiae chromosome III. Mol. Biol. Cell 12: 3317–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman, M. K., E. A. Winzeler, D. Collingwood, S. Hunt, L. Wodicka et al., 2001. Replication dynamics of the yeast genome. Science 294: 115–121. [DOI] [PubMed] [Google Scholar]
- Rose, M. D., F. Winston and P. Hieter, 1990. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sanchez, Y., B. A. Desany, W. J. Jones, Q. Liu, B. Wang et al., 1996. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271: 357–360. [DOI] [PubMed] [Google Scholar]
- Santocanale, C., and J. F. Diffley, 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395: 615–618. [DOI] [PubMed] [Google Scholar]
- Santocanale, C., K. Sharma and J. F. Diffley, 1999. Activation of dormant origins of DNA replication in budding yeast. Genes Dev. 13: 2360–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada, K., and S. M. Gasser, 2007. The origin recognition complex functions in sister-chromatid cohesion in Saccharomyces cerevisiae. Cell 128: 85–99. [DOI] [PubMed] [Google Scholar]
- Shirahige, K., Y. Hori, K. Shiraishi, M. Yamashita, K. Takahashi et al., 1998. Regulation of DNA-replication origins during cell-cycle progression. Nature 395: 618–621. [DOI] [PubMed] [Google Scholar]
- Sikorski, R. S., and P. Hieter, 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaprasad, U., A. Dutta and S. P. Bell, 2006. Assembly of pre-replication complexes, pp. 63–88 in DNA Replication and Human Disease, edited by M. L. DePamphilis. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sogo, J. M., M. Lopes and M. Foiani, 2002. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602. [DOI] [PubMed] [Google Scholar]
- Spencer, F., S. L. Gerring, C. Connelly and P. Hieter, 1990. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics 124: 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Z., D. S. Fay, F. Marini, M. Foiani and D. F. Stern, 1996. Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev. 10: 395–406. [DOI] [PubMed] [Google Scholar]
- Sweeney, F. D., F. Yang, A. Chi, J. Shabanowitz, D. F. Hunt et al., 2005. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr. Biol. 15: 1364–1375. [DOI] [PubMed] [Google Scholar]
- Theis, J. F., and C. S. Newlon, 1994. Domain B of ARS307 contains two functional elements and contributes to chromosomal replication origin function. Mol. Cell. Biol. 14: 7652–7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis, J. F., and C. S. Newlon, 2001. Two compound replication origins in Saccharomyces cerevisiae contain redundant origin recognition complex binding sites. Mol. Cell. Biol. 21: 2790–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh, G. W., A. M. O'Shaughnessy, S. Jimeno, I. M. Dobbie, M. Grenon et al., 2006. Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair 5: 693–703. [DOI] [PubMed] [Google Scholar]
- Vallen, E. A., M. A. Hiller, T. Y. Scherson and M. D. Rose, 1992. Separate domains of KAR1 mediate distinct functions in mitosis and nuclear fusion. J. Cell Biol. 117: 1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Brabant, A. J., C. D. Buchanan, E. Charboneau, W. L. Fangman and B. J. Brewer, 2001. An origin-deficient yeast artificial chromosome triggers a cell cycle checkpoint. Mol. Cell 7: 705–713. [DOI] [PubMed] [Google Scholar]
- Vujcic, M., C. A. Miller and D. Kowalski, 1999. Activation of silent replication origins at autonomously replicating sequence elements near the HML locus in budding yeast. Mol. Cell. Biol. 19: 6098–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, T. A., and L. H. Hartwell, 1990. Characterization of RAD9 of Saccharomyces cerevisiae and evidence that its function acts posttranslationally in cell cycle arrest after DNA damage. Mol. Cell. Biol. 10: 6554–6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, T. A., G. L. Kiser and L. H. Hartwell, 1994. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 8: 652–665. [DOI] [PubMed] [Google Scholar]
- Wyrick, J. J., J. G. Aparicio, T. Chen, J. D. Barnett, E. G. Jennings et al., 2001. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science 294: 2357–2360. [DOI] [PubMed] [Google Scholar]
- Xu, W., J. G. Aparicio, O. M. Aparicio and S. Tavare, 2006. Genome-wide mapping of ORC and Mcm2p binding sites on tiling arrays and identification of essential ARS consensus sequences in S. cerevisiae. BMC Genomics 7: 276. [DOI] [PMC free article] [PubMed] [Google Scholar]