

Abstract
The neuronal microtubule-associated protein tau plays an important role in establishing cell polarity by stabilizing axonal microtubules that serve as tracks for motor-protein–driven transport processes. To investigate the role of tau in intracellular transport, we studied the effects of tau expression in stably transfected CHO cells and differentiated neuroblastoma N2a cells. Tau causes a change in cell shape, retards cell growth, and dramatically alters the distribution of various organelles, known to be transported via microtubule-dependent motor proteins. Mitochondria fail to be transported to peripheral cell compartments and cluster in the vicinity of the microtubule-organizing center. The endoplasmic reticulum becomes less dense and no longer extends to the cell periphery. In differentiated N2a cells, the overexpression of tau leads to the disappearance of mitochondria from the neurites. These effects are caused by tau's binding to microtubules and slowing down intracellular transport by preferential impairment of plus-end–directed transport mediated by kinesin-like motor proteins. Since in Alzheimer's disease tau protein is elevated and mislocalized, these observations point to a possible cause for the gradual degeneration of neurons.
Keywords: mitochondria, transport, endoplasmic reticulum, exocytosis, Alzheimer's disease
The microtubule network plays an important role in maintaining cellular morphology, in mitotic processes, in intracellular trafficking, and in establishing cellular polarity and neurite outgrowth in differentiating neurons (Drubin and Nelson, 1996; Li and Black, 1996; Goodson et al., 1997). Microtubules serve as tracks for the movement of mitochondria (Nangaku et al., 1994; Morris and Hollenbeck, 1995; Rickard and Kreis, 1996), lysosomes (Hollenbeck and Swanson, 1990), peroxisomes (Wiemer et al., 1997) and various other organelles such as endocytotic or exocytotic vesicles (Scales et al., 1997). Furthermore, they are important in establishing and maintaining the functionality of the endoplasmic reticulum (Waterman-Storer et al., 1995). Microtubule (MT)1-dependent transport is achieved through motor proteins such as dynein or kinesin and their relatives (Waterman-Storer and Salmon, 1997; Hirokawa, 1998; Lippincott-Schwartz, 1998). Kinesin is a plus-end–directed motor, whereas dynein-related motor proteins move in the opposite direction (Brady, 1995; Vallee and Sheetz, 1996). The organelles are linked to the motors via membrane/organelle-bound adaptor proteins such as kinectin or dynactin (Kumar et al., 1995; Burkhardt et al., 1997).
The microtubule tracks are stabilized by microtubule-associated proteins (MAPs) such as MAP2, tau, MAP4, and others. These are mostly filamentous proteins that bind to the microtubule surface and promote microtubule assembly (for reviews, see Hirokawa, 1994; Mandelkow and Mandelkow, 1995). The developmental regulation of MAPs, for instance in neuronal differentiation, suggests an important role in organizing the microtubule cytoskeleton (Matus, 1994). Tau is enriched in axons, MAP2 in the somatodendritic compartment of neurons (Binder et al., 1985), whereas MAP4 can be found in many types of tissues (Chapin and Bulinski, 1991; West et al., 1991). MAP2, tau, and MAP4 share a remarkable sequence homology in the conserved repeat regions located in the COOH-terminal microtubule-binding domain (Chapin and Bulinski, 1992). The repeated motifs are preceded by an NH2-terminal projection domain that has been suggested to serve as a spacer between microtubules (Chen et al., 1992) or as an anchor for cellular enzymes (Obar et al., 1989).
Since motor proteins and MAPs both interact with the microtubule surface, the question arises whether MAPs interfere with the movement of motor proteins. Several studies have suggested that this is the case for certain MAPs, especially those of higher molecular weight. Thus, MAP2 can inhibit the motility of dynein or kinesin in vitro (Paschal et al., 1989; Von Massow et al., 1989; Lopez and Sheetz, 1993; Hagiwara et al., 1994), while smaller fragments had only a weak effect. This could be accounted for in several ways: (a) MAPs could have a general steric blocking effect of the extended NH2-terminal projection domain; (b) MAPs could compete with motors for common binding sites, probably on beta-tubulin (Hagiwara et al., 1994); and (c) MAPs could affect transport processes indirectly by altering microtubule dynamics (Waterman-Storer and Salmon, 1997).
When MAP4 is overexpressed in mouse Ltk− cells, it interferes with motor-protein–dependent organelle motility and trafficking in vivo (Bulinski et al., 1997). Furthermore, overexpression of tau or MAP2c in COS-fibroblasts indicates that both MAPs might function as phosphorylation-dependent regulators of organelle transport (Sato-Harada et al., 1996). The phosphorylation and dephosphorylation of microtubule-associated proteins is believed to be a key factor regulating the affinity to microtubules, thereby facilitating and supporting vital cellular processes such as proliferation, differentiation, and trafficking through stabilization of the tracks for motor proteins (Lopez and Sheetz, 1995; Mandell and Banker, 1996; Sato-Harada et al., 1996; Drewes et al., 1997). Indeed, MAPs isolated from cells are phosphorylated and it is well documented that this phosphorylation interferes with binding to microtubules in vitro and in vivo (Drechsel et al., 1992; Biernat et al., 1993; Illenberger et al., 1996).
In axonal growth, where a dynamic microtubule array is indispensable for differentiation, the phosphorylation and distribution of tau are correlated (Mandell and Banker, 1996; Li and Black, 1996), suggesting a regulated balance between kinases and phosphatases, as well as a finely tuned gradient of tau concentration. In Alzheimer's disease (AD), this balance between kinases and phosphatases seems to be disturbed (Trojanowski and Lee, 1995; Mandelkow et al., 1995). Neurofibrillary tangles, one of the hallmarks of this disease, consist of paired helical filaments, which are mainly composed of hyperphosphorylated microtubule-associated protein tau (Wischik et al., 1988). Furthermore, tau in AD mislocalizes to the somatodendritic compartment of neurons (Braak et al., 1994) and tau concentration has been reported to be elevated in AD brain to a significant extent (Khatoon et al., 1992). This further strengthens the importance of proper control of posttranslational modifications, localization, and intracellular concentration of microtubule-associated proteins in vivo.
These observations lead us to investigate whether the overexpression of tau protein had an effect on organelle and vesicle trafficking in cultured cells. This approach had been useful for studying the distribution of exogenous MAPs (Barlow et al., 1994; Olson et al., 1995; Bulinski et al., 1997; Kaech et al., 1996). In previous studies, we had shown that CHO and neuroblastoma N2a cells can serve as models for the state of MAP phosphorylation during the cell cycle, and that the phosphorylation of tau by the kinase MARK leads to microtubule breakdown (Preuss et al., 1995; Drewes et al., 1997; Illenberger et al., 1998). Here we demonstrate that the overexpression of tau leads to a change in cell shape, loss of polarization, and the retardation of cell growth. One of the most visible consequences is the dramatically altered distribution of mitochondria. Instead of being dispersed throughout the cytoplasm, the mitochondria become clustered near the nucleus at the microtubule organizing center (MTOC). In differentiated neuroblastoma cells, the neurites become nearly devoid of mitochondria. The endoplasmic reticulum becomes less dense and retracts from the cell periphery or from neurites. Transferrin endocytosed by clathrin-coated vesicles accumulates near the MTOC and recycling is significantly slowed down. These effects can be explained by assuming that tau interferes with MT-driven transport, in the case of mitochondria preferentially with the plus-end–directed transport (by kinesin-like motors) so that the minus-end– directed transport (by dynein) predominates. This results in a net inward transport of mitochondria and other organelles such as peroxisomes towards the MTOC, where the microtubule minus-ends are organized. The perturbations of the balance in microtubule-dependent transport, especially in neurites, would be consistent with the observed impairment of axonal traffic in Alzheimer's disease neurons, their loss of polarity, and their subsequent degeneration.
Materials and Methods
Cell Culture
CHO cells were grown in HAM's F12 medium supplemented with 10% fetal calf serum and 2 mM glutamine (Biochrom, Berlin, Germany). For immunofluorescence, cells were seeded onto coverslips at a density of 2 × 104 cells/cm2 in 24-well culture dishes and grown overnight at 37°C with 5% CO2. N2a neuroblastoma cells were grown in MEM Earle supplemented with 10% fetal calf serum, 2 mM glutamine (Biochrom) and 1% nonessential amino acids (Sigma Chemical Co., Deisenhofen, Germany). Differentiation of N2a cells was achieved by addition of 1 μM retinoic acid (Sigma Chemical Co.) for 24–48 h in the presence of 0.1% fetal calf serum. For stable transfection, plasmids derived from pcDNA3 (Invitrogen Corp., Leek, The Netherlands) coding for the longest human tau-isoform htau40 under the control of the cytomegalovirus promoter were introduced into the cells with the DOTAP-method (Boehringer Mannheim, Mannheim, Germany) according to the manufacturer's instructions. Stable transfectants were selected in the presence of 800 μg/ml (CHO) or 1 mg/ml geneticin (N2a) as described (Preuss et al., 1995), and subsequently recloned.
Antibodies and Dyes
The following antibodies were used: rat monoclonal antitubulin antibodies YL1/2 and mouse monoclonal antibody DM1A were purchased from Serotec (Oxford, UK) and Sigma Chemical Co., respectively. Polyclonal rabbit anti–tau antibody K9JA was from DAKOPATTS (Hamburg, Germany), polyclonal rabbit antibody used as an ER marker was a gift from Dr. D. Louvard (Institut Curie, Paris; Louvard et al., 1982) and polyclonal anticatalase antibody was from Dr. W. Just (University of Heidelberg, Heidelberg, Germany). Antivimentin antibody V9 was from Boehringer Mannheim. All fluorescently (TRITC, FITC, and AMCA) labeled secondary antibodies were from DIANOVA (Hamburg, Germany). Fluorescent dyes DiOC6(3), MitoTracker™ Green, MitoTracker™ Red, and rhodamine-labeled transferrin were purchased from Molecular Probes, Inc. (Eugene, OR). DiOC6(3) was used as a stain for in vivo observation of the ER at 2.5 μg/ml.
Immunofluorescence
Cells were fixed with 2% paraformaldehyde or methanol, and incubation with antibodies was performed essentially as described (Preuss et al., 1995). Tau isoforms tightly bound to microtubules can be visualized by methanol fixation (which allows unbound protein to diffuse out, see Fig. 2 c). By contrast, weakly bound constructs (e.g., K10, K12) cannot be visualized by methanol fixation (since they would diffuse away). Instead, it is necessary to use paraformaldehyde, which fixes all cellular proteins but tends to dissociate tau from microtubules (see Fig. 3, for details see Illenberger et al., 1998). To visualize mitochondria, MitoTracker™ Green was added at a concentration of 400 nM 30 min before fixation. CHO cells stably transfected with human tau40 or mock-transfected cells were incubated for 1 h with 5 μM nocodazole (Sigma Chemical Co.) or 6 h with 1–10 μM taxotere (Rhone-Poulence Rorer, Vitry, France). Cells were fixed before and 1 or 4 h after nocodazole, and 6 h after taxotere treatment, respectively. Cells were examined with an Axioplan fluorescence microscope (Carl Zeiss Jena GmbH, Jena, Germany) equipped with a 100× oil-immersion objective and filters optimized for triple-label experiments. Pictures were taken with a cooled CCD camera (Visicam; Visitron, Puchheim, Germany) and analyzed using the MetaMorph® software package (Visitron). The fraction of the cell area containing mitochondria was quantified as follows: after fixation and immunofluorescence labeling, the pictures were recorded and the areas containing mitochondria were circumscribed manually (omitting the nucleus) and calculated. The same was done with the total cell area visible in the microtubule stain. A minimum of 30 cells were measured per experiment and the ratios plotted (e.g., see Fig. 11).
Figure 2.
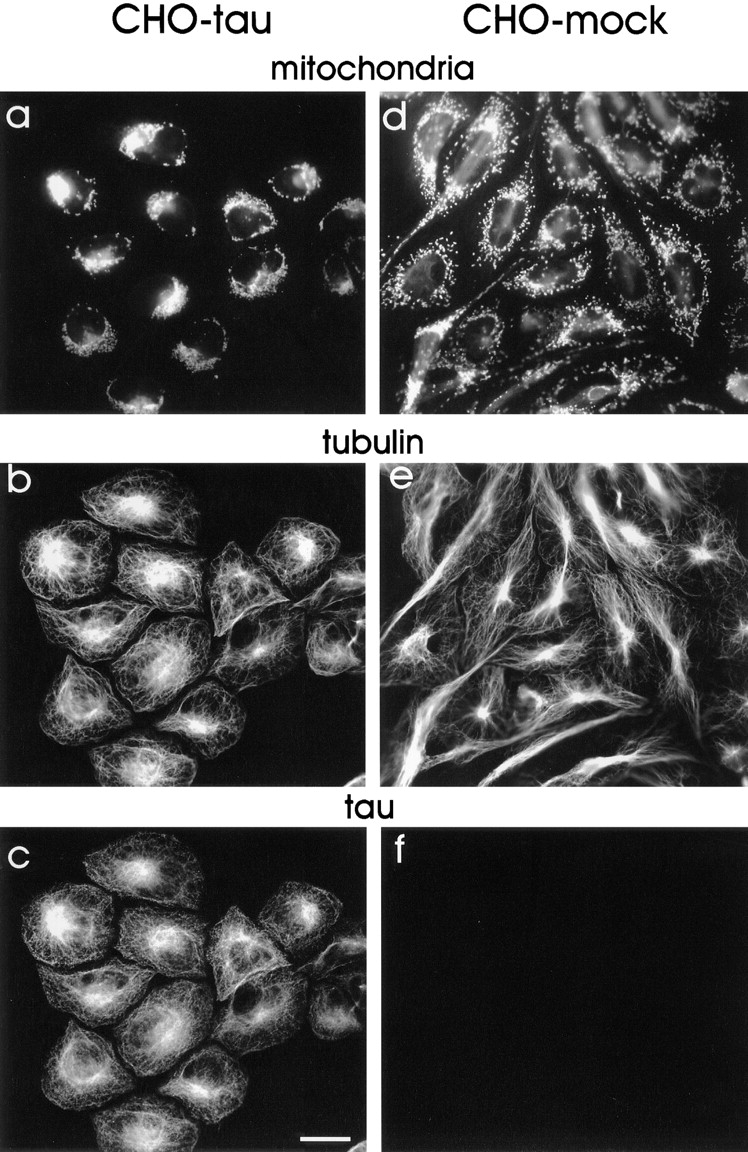
Mitochondria accumulate near the MTOC in CHO cells overexpressing tau. Cells stably transfected with tau40 (a–c) and mock-transfected CHO cells (d–f). Microtubules were stained with antibody DM1A (b and e) and tau with polyclonal anti–tau antibody K9JA (c and f). Mitochondria were visualized with MitoTracker™ Green (a and d). Cells were fixed in methanol 30 min after addition of MitoTracker™ Green. In tau-stable cells (a–c), there is a striking clustering of mitochondria near the MTOC (seen near the nucleus where the microtubule stain is most intense), whereas in mock-transfected cells (d–f) the mitochondria are dispersed throughout the cytoplasm. In addition, the tau-stable cells are more rounded up compared with control cells. Bar, 20 μm.
Figure 3.
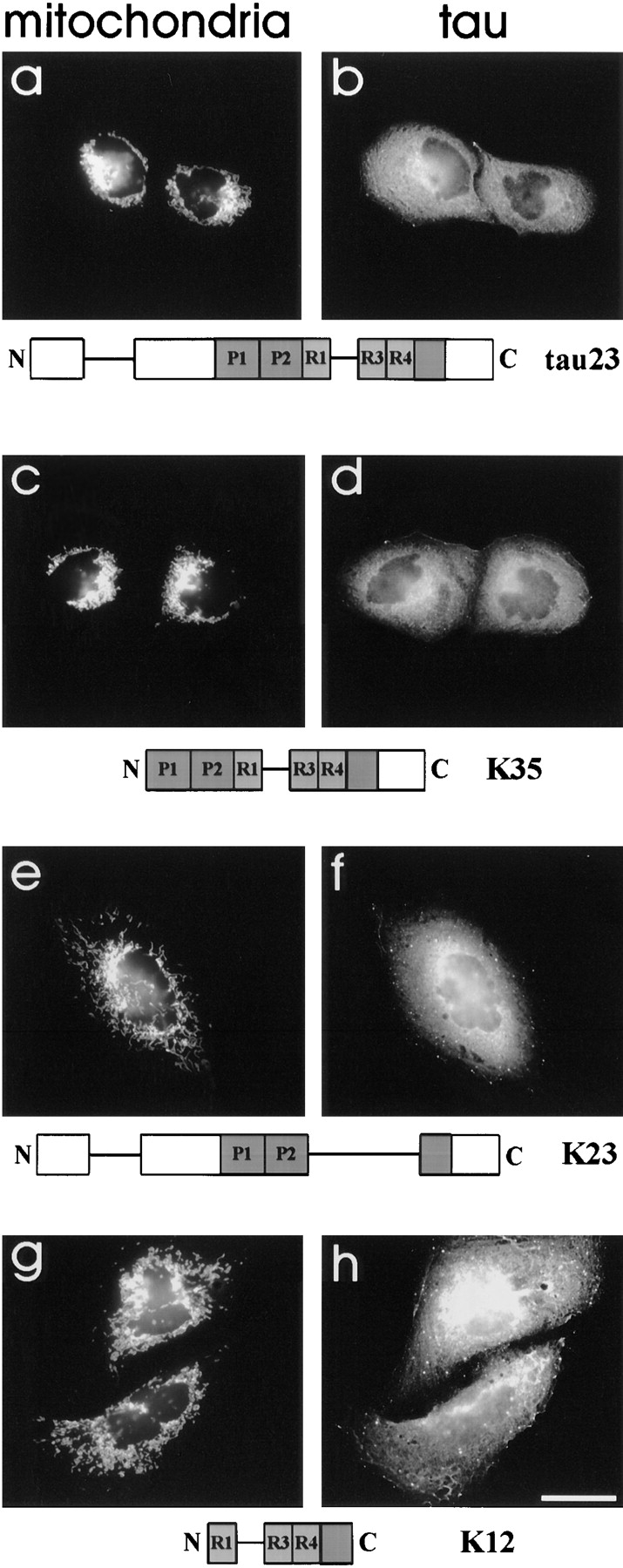
Effects of different tau constructs on mitochondrial distribution in CHO cells. Mitochondrial distribution in CHO cells stably transfected with different tau constructs was examined after paraformaldehyde fixation. This allows the visualization of total tau in the cells, but tends to dissociate tau from the microtubules (see Materials and Methods). Human tau23, the shortest tau-isoform (a and b), as well as K35 (c and d), a tau construct lacking nearly the entire projection domain (see Fig. 1) both led to the phenotype of mitochondrial clustering near the MTOC as seen in tau40-stable cells. On the other hand, K23 (e and f), a tau construct lacking the microtubule-binding region and K12 (g and h), consisting mainly of this MT-binding region (Fig. 1), did not alter the distribution of mitochondria. This argues for a tight binding of tau to microtubules as a prerequisite for impairment of plus-end–directed transport of these organelles and excludes the possibility that tau might inhibit kinesin by directly interacting with the motor protein. Mitochondria were stained with MitoTracker™ Red (a, c, e, and g) and transfected tau with polyclonal anti–tau antibody K9JA (b, d, f, and h). Bar, 20 μm.
Figure 11.
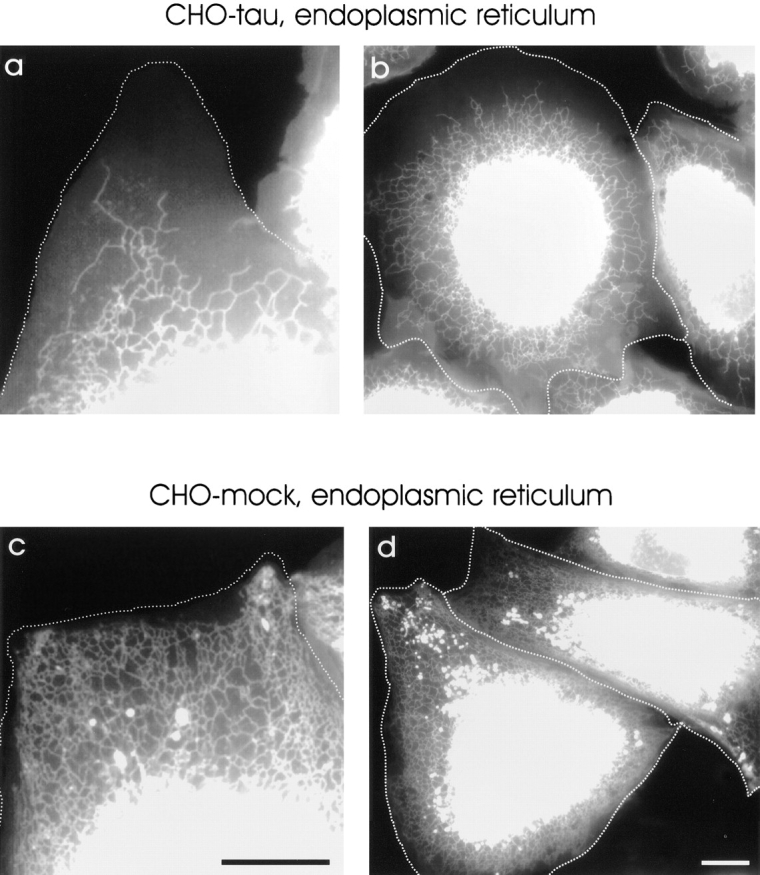
The ER organization is altered in CHO cells overexpressing tau. Tau40-stable (a and b) and mock-transfected CHO cells (c and d) were stained with DiOC6(3) to visualize the ER. In tau-stable cells, the ER is less branched and does not reach the cell periphery, while in control cells the ER extends almost to the plasma membrane and displays a more complex structure. Note that, although the extension of the ER of the smaller cell in Fig. 11 b seems to be less effected when compared with the larger cells, the branching and complexity of the endoplasmic reticulum is significantly reduced when compared with mock-transfected cells in c and d. The cell periphery is outlined for clarity. Bar, 10 μm.
Microinjection
Microinjection experiments were performed as described (Preuss et al., 1997). In brief, cells were seeded onto coverslips at a density of 2 × 104 cells in 35-mm dishes and incubated overnight. Recombinant protein, prepared as described (Biernat et al., 1993; Gustke et al., 1994) was concentrated in Microcon microconcentrators (Amicon Inc., Beverly, MA) according to the manufacturer's instructions. During this procedure, the buffer was changed to 25 mM HEPES, pH 7.2, 1 mM PMSF, and protein was then microinjected with an Eppendorf Micromanipulator 5171 at constant pressure within 15 min. MitoTracker™ Red was added 30 min before fixation at a concentration of 200 nM, and cells were finally fixed with paraformaldehyde at given time points and prepared for immunofluorescence. For quantitative analysis of the intracellular concentration of tau protein in the tau40-stable cell line, 1.9, 0.95, and 0.38 mg/ml purified recombinant tau protein, as well as buffer only, were microinjected into CHO cells. Cells were then fixed in parallel with the tau-stable cell line and stained with a polyclonal anti–tau antibody. The fluorescence of the tau stain was then quantified using the CCD camera and the MetaMorph® software package and normalized to the cell dimensions. A minimum of 80 cells were analyzed per microinjection and at least 100 tau-stable cells were scored. Regression analysis of the normalized fluorescence intensities of microinjected cells was performed with the program Sigma Plot (R = 0.9995), and the mean intracellular tau concentration of the stable cell line was calculated.
Monitoring Mitochondrial Distribution and Endoplasmic Reticulum In Vivo
For the visualization of mitochondria in vivo, cells were seeded onto LabTek chambered cover glass (NUNC, Inc., Naperville IL.) at 70% confluency. 30 min before immunofluorescence examination, MitoTracker™ Green or Red was added to cells at a final concentration of 200 nM, cells were then rinsed once with medium. For monitoring mitochondrial redistribution tau-stable CHO cells were incubated for 1 h with 5 μM nocodazole and 200 nM MitoTracker™ Green. Disassembly of the microtubule network was confirmed by fixation and tubulin staining in immunofluorescence microscopy. Nocodazole was then removed and cells were examined with the cooled CCD camera on an inverted microscope (Axiovert 10; Carl Zeiss Jena GmbH) using standard filters for FITC fluorescence. Staining of the endoplasmic reticulum was achieved with DiOC6(3) at a concentration of 2.5 μg/ml, 30-min incubation at 37°C, and rinsing cells once with medium. Immunofluorescence microscopy was then performed as described above.
Transferrin Uptake and Recycling
Cells were grown in 35-mm six-well tissue culture plates for 2 d to ∼80% confluency at 37°C and 5% CO2. Supernatant was then replaced by serum-free medium, and 2 h later tetramethylrhodamine (TMR)-labeled transferrin was added to a final concentration of 380 nM. Cells were further incubated for 1 h to achieve maximal transferrin uptake. Medium containing excess or recycled transferrin was removed and new medium containing 60 μg/ml partially Fe2+-saturated transferrin (Sigma Chemical Co.) was added. Recycling was then monitored by again removing medium at given time points and quantitating fluorescence of TMR-labeled transferrin in a Fluoro Max spectrofluorometer (Spex Industries Inc., Edison, NJ) at 575 nm emission and 552 nm excitation wavelength. Cells were then trypsinized, lysed, and protein concentration was determined to normalize fluorescent signals to total protein present in the culture dishes, representing cell density. At least five independent experiments were performed with tau-stable and mock-transfected CHO cells to ensure statistical significance. To quantitate residual intracellular transferrin, cells were seeded onto coverslips, grown overnight, and subsequently saturated with TMR transferrin (see above). Cells were then fixed in methanol either immediately or 7 min after removal of TMR transferrin. Residual transferrin fluorescence was quantitated as above using standard filters for rhodamine fluorescence. Pictures were corrected by background subtraction and shadowing correction provided with the MetaMorph® software package (Visitron). At least 300 different cells per time point and cell line were quantified by thresholding.
Tau Isoforms and Constructs
Human brains contain mainly six isoforms of tau generated by alternative splicing (Goedert et al., 1989). The present work focuses on the largest (tau40, 441 residues) and smallest (tau23, 352 residues) isoform, containing four or three repeats in their microtubule-binding region (Fig. 1). Cloning, bacterial expression, and purification was done as described (Biernat et al., 1993; Gustke et al., 1994). Other tau constructs were also generated as described, such as K12 and K10 (derived from htau23, three repeats with the COOH-terminal flanking region or the whole COOH-terminal tail), K35 (which comprises the microtubule-binding domain of tau, including the proline-rich region upstream of the repeats), and K23 (a variant of tau23 lacking the three repeats). The dissociation constants of these constructs and their binding to cellular microtubules were reported previously (Gustke et al., 1994; Preuss et al., 1997): tau40 (K d = 1.1 μM), tau23 (2.5 μM), K35 (1.0 μM), K23 (6.5 μM), K10 (18.5 μM), and K12 (7.1 μM).
Figure 1.

Domain structure of tau isoforms and constructs. Tau40 is the longest tau isoform in the human CNS, occurring mostly in axons (Goedert et al., 1989). It consists of 441 amino acid residues and can be divided into an NH2-terminal projection domain and a COOH-terminal microtubule binding domain containing four imperfect repeats (R1–R4) and basic proline-rich sequences on either side that act as microtubule targeting domains (shaded dark, P1, and P2). The repeats show high homology between tau, MAP2, and MAP4. The shaded NH2-terminal inserts and repeat two can be alternatively spliced, resulting in six different tau isoforms. The predominantly embryonic isoform tau23 lacks all three inserts (352 residues). Tau constructs K12 and K10 are derived from tau23, they contain three repeats plus either a COOH-terminal flanking region (K12) or the whole COOH-terminal tail (K10). K35 contains three repeats and the basic proline-rich regions on either side, including the COOH-terminal tail. K23 contains all of tau23 except the repeats.
Inhibition of Dynein-dependent Transport by Overexpression of Dynamitin
Dynamitin (p50) expression plasmid was transfected into tau-stable CHO cells with the DOTAP-method. 6 h after transfection, cells were fixed in paraformaldehyde as described (see above). Mitochondria were stained with MitoTracker™ Red (Molecular Probes, Inc.), tubulin with rat monoclonal antitubulin antibody (YL1/2; Serotec) and transfected p50-dynamitin with mouse monoclonal antidynamitin 50-I. Immunofluorescence analysis was performed as described above. The p50 expression plasmid and antibody were generous gifts from C. Echeverri and R. Vallee (Worcester Foundation, Shrewsbury, MA) (Burkhardt et al., 1997).
Monitoring Transport of Individual Mitochondria After Nocodazole Removal
Cells were seeded onto LabTek chambered cover glass (NUNC, Inc.) at 70% confluency. Nocodazole was then added at a final concentration of 1 μM for 2 h and subsequently removed. 30 min before removal, MitoTracker™ Red was added at a concentration of 200 nM to visualize mitochondria. After an additional 30 min to allow recovery of the microtubule cytoskeleton, reaggregation of the organelles was monitored in vivo on an inverted microscope with a cooled CCD camera (see above) by recording pictures every second for 2 min with the following constraint: only mitochondria translocated with a minimum distance of 1 μm and a velocity of 0.5 μm/s were taken into consideration to exclude diffusional processes. By using the nucleus as a reference, transported mitochondria were grouped into three categories: movement towards the nucleus (centripetal; denoted as − in Fig. 7) was regarded as minus-end directed, movement towards the cell periphery (centrifugal; denoted as +) as plus-end directed and all other movements as circumferential (denoted as −/+).
Figure 7.

The ratio between plus- and minus-end–directed transport of mitochondria is altered by tau overexpression. Tau-stable and control cells were treated with nocodazole to disrupt the microtubule cytoskeleton and to achieve an even distribution of mitochondria in both cell lines. The drug was washed out after 2 h of incubation and mitochondria were stained with MitoTracker™ Red. Translocations of the organelles were then recorded for 2 min with a time interval of 1 s. The direction of movement was regarded as plus-end directed when the organelles were transported to the periphery (+), minus-end directed when transport was towards the nucleus (−), and neutral (−/+) when movement was vertical to the axis between nucleus and periphery of the cells. Note that the relative number of transported mitochondria is significantly lower for all three groups in the tau-stable cell line (P < 0.0001, two tailed Student's t test). However, in tau-stable cells, the plus-end– directed movement is significantly more affected (∼10-fold) than the reverse direction (2-fold). This leads to the phenotype of mitochondrial clustering. At least 40 cells were analyzed for quantitation, the total number of mitochondria ranged between 100 and 700 in both cell lines.
Results
Cell Shape and Mitochondrial Distribution Is Strongly Altered in Tau-overexpressing CHO Cells
To investigate the effects of human tau expression in vivo, we established CHO cells stably transfected with tau40, an isoform containing four repeats (Preuss et al., 1995, and this report). Immunofluorescent staining of microtubules showed that their network was normal and contained bound tau (Fig. 2), consistent with similar results on MAP4-transfected cells (Olson et al., 1995; Nguyen et al., 1997). In particular, no bundling of microtubules was seen, contrary to experiments with transient transfection (Kanai et al., 1989; Lee and Rook, 1992) or with microinjection (Preuss et al., 1997), where higher cellular concentrations of MAPs are obtained. To calibrate the intracellular tau concentration, we microinjected different amounts of purified, recombinant tau protein into CHO cells, and prepared them for immunofluorescence analysis in parallel with the tau-stable cells (see Materials and Methods). This yielded an average concentration of 0.04 μg/ml tau in the stably transfected cells, corresponding to ∼1 μM. Since the cellular tubulin concentration is ∼40 μM (Hiller and Weber, 1978), the ratio of tau to tubulin remained low, ∼1:40, similar to that reported for PC12 (Drubin et al., 1985) or HeLa cells (Bulinsky and Borisy, 1979). Furthermore, stable expression of tau in PC12 cells under control of the cytomegalovirus promoter (which was used in our experiments) resulted in only a twofold increase of intracellular tau-protein level (Brandt et al., 1995). Taken together, these data show that the concentration of tau in the stably transfected CHO and N2a cells is in the range of endogenous MAP levels in the control cells and increases the amount of total MAP only about two- to threefold.
In spite of the low cellular concentration, the expression of tau leads to conspicuous changes in cell shape and growth behavior. Whereas control cells were elongated, tau-transfected CHO cells rounded up (compare Fig. 2, b vs. e). This phenotype can be evoked in several ways, but one possibility is the inhibition of the motor protein kinesin as described by Rodionov et al. (1993), and a similar interpretation is appropriate in our case as well (see below). A second noticeable effect was that overexpression of tau in CHO cells slowed down proliferation approximately twofold (data not shown), similar to observations with MAP4-transfected cells (Nguyen at al., 1997).
We wanted to investigate whether the observed changes in shape and proliferation were indeed related to microtubule-dependent transport and therefore analyzed the cells with several markers for organelles. This revealed a remarkable clustering of mitochondria in tau40-expressing CHO cells near the nucleus in the vicinity of the microtubule organizing center (Fig. 2, a and c). This effect was reproduced in two independently generated tau40-stable cell lines, thus ruling out recloning artifacts. In contrast, mock-transfected CHO cells showed that mitochondria were dispersed throughout the entire cytoplasm (Fig. 2 d), frequently aligned with microtubules, suggesting a microtubule-dependent organization of mitochondria. The fraction of the cell area containing mitochondria decreased from 66.1% in mock-transfected cells to 27.7% in the tau-stable cell line (see Fig. 10). To exclude the possibility of a fixation artifact, we established CHO cells stably transfected with an enhanced green fluorescent protein (EGFP)–tau fusion protein that allowed us to monitor tau's effect on mitochondrial distribution in living cells. EGFP tau was tightly associated with microtubules (as seen by EGFP fluorescence) and led to the same effects on mitochondrial clustering (data not shown).
Figure 10.

Quantitation of mitochondrial clustering after overexpression of tau, drug treatment, and inhibition of dynein. The distribution of mitochondria was quantified as described in Materials and Methods. A minimum of 30 cells were analyzed per experiment (n). In mock-transfected cells, mitochondria occupy up to 65% of the total cell area. The same is true in cells stably transfected with K23, a tau construct lacking the microtubule-binding repeats (CHO-K23). This value drops to ∼30% in tau40-stable cells (CHO-tau). Treatment of the stably transfected cells with either nocodazole or taxotere restores the wild-type distribution of mitochondria (CHO-tau nocodazole or CHO-tau taxotere, respectively). The same effect can be seen after transfection with dynamitin (CHO-tau transfection dynamitin), but not after transfection of EGFP as a control protein (CHO-tau transfection EGFP; P < 0.0001, two-tailed Student's t test). Removal of nocodazole again leads to clustering of mitochondria at the nucleus (CHO-tau noc. removal; 4 h).
Since there are several reports (e.g., Summerhayes et al., 1983) linking the distribution of mitochondria to the integrity of the intermediate filament system, we investigated whether the vimentin network was altered through tau overexpression. In tau40-stable cells, the extension of this filamentous network is significantly reduced and becomes located in a perinuclear area (Trinczek, B., A. Ebneth, E.-M. Mandelkow, and E. Mandelkow, manuscript in preparation). Thus, one might speculate that tau's effects on the intermediate filament system may account for the aggregation of mitochondria at the MTOC. However, when treating cells with nocodazole to disrupt microtubules, mitochondria began to disperse again (see below), whereas the extension of the intermediate filament network was not altered in the tau-stable CHO cells despite disruption of microtubules. It has been shown that the disruption of microtubules leads to a collapse of the intermediate filament system (Klymkowski et al., 1989). Removal of nocodazole again did not affect the extension of the vimentin network (data not shown), but led to a reclustering of mitochondria, which indicates that the integrity of the microtubule system, not the intermediate filament system, is responsible for the observed phenotype of mitochondrial clustering in tau40-stable cells.
Interestingly, inhibition of kinesin motors by microinjection of an inhibitory antibody into fibroblasts led to a strikingly similar phenotype of a perinuclear localization of the vimentin system (Gyoeva and Gelfand, 1991).
Mitochondria are organelles that can disappear from cellular regions with lowered metabolic demand (e.g., Morris and Hollenbeck, 1993). This raises the possibility that clustering of mitochondria was not due to an impairment of the cell's transport, but to a change in the metabolic activity in the periphery due to the overexpression of tau. For this reason, we looked for other organelles known to be transported via microtubule-dependent motors and asked if they were clustered, too. This was indeed the case. When staining tau-stable cells with an antibody raised against catalase, an enzyme present in the matrix of peroxisomes, the organelles were significantly enriched in the perinuclear region, whereas in control cells they were evenly distributed (data not shown). This argues that organelle clustering near the MTOC is caused by a general influence of tau on microtubule-dependent plus-end– directed transport, rather than a selective retraction of mitochondria in response to altered metabolic demands.
To identify the domains of tau responsible for the clustering of mitochondria, we studied CHO cells stably transfected with several tau isoforms or constructs whose behavior had already been characterized in vitro (Gustke et al., 1994; Fig. 1): human tau40 (the longest isoform in the central nervous system), tau23 (the shortest isoform, found predominantly in embryonic brain), K12 (a tau construct containing only the three repeats plus the COOH-terminal flanking region), K10 (containing three repeats plus the whole COOH-terminal rest of the molecule), K35 (containing three repeats and both flanking regions), and K23 (a variant of tau23 without the repeats). The isoforms tau40, tau23, and K35 induced pronounced mitochondrial clustering around the MTOC (Figs. 2 and 3, a–d, and data not shown), whereas K10 (data not shown), K12, and K23 did not show the effect (Fig. 3, e–h). This correlates well with the strengths of binding (dissociation constants ∼1–2 μM for tau40, tau23, and K35, compared with ∼7–20 μM for the other constructs; Gustke et al., 1994). K35 binds tightly but lacks parts of the NH2-terminal projection domain so that this domain appears to be of lesser importance for mitochondrial transport. Conversely, K10 and K12 contain the repeats but lack the NH2-terminal projection and flanking domains. Thus, their binding to microtubules is weak and this explains why they have no effect on mitochondrial distribution. K23 is a special case because it contains the projection domain, the two flanking domains, but not the repeats. It binds to microtubules with reasonable affinity (K d ∼7 μM), but still does not influence microtubule-based traffic. These observations argue that a high affinity of tau to microtubules is the major determinant for observing the effects of tau overexpression and that it is highly unlikely that tau inhibits plus-end–directed transport by directly binding to kinesins.
Microinjection of Tau into CHO Cells Leads to Mitochondrial Clustering at the MTOC
Next we asked whether the elevation of tau protein was directly responsible for the phenotype of mitochondrial clustering at the MTOC, or whether it was due to other factors not related to the protein level. Recombinant tau40 was microinjected into mock-transfected CHO cells to a final concentration of ∼1.2 μM where the microtubule network is not affected (Preuss et al., 1997). Nevertheless, mitochondria began to accumulate near the nucleus within 4 h (Fig. 4). Microinjection of mouse serum at the same concentration as a control on the other hand had no effect on mitochondrial distribution (data not shown). Thus, the phenotype can be generated without affecting the microtubule cytoskeleton, suggesting that tau protein has a direct effect on the redistribution of mitochondria, presumably by retarding their plus-end directed movement (see below).
Figure 4.
Microinjection of recombinant tau protein leads to mitochondrial clustering near the MTOC. Wild-type CHO cells were microinjected with 1.3 mg/ml (∼1.2 μM) of purified recombinant tau protein and incubated for 4 h. After addition of MitoTracker™ Red (a and b), cells were fixed with paraformaldehyde and stained with antibodies against tubulin (c and d) and tau (e and f). Mitochondrial clustering is visible even within 4 h after microinjection. Note that the microtubule network retains its normal appearance and that mitochondria in microinjected cells similar as in the tau-stable CHO and N2a cells tend to cluster at one side of the nucleus in the vicinity of the MTOC. The cell periphery of microinjected cells is outlined for clarity. Bar, 10 μm.
Nocodazole and Taxotere Reverse the Effect of Tau on Mitochondrial Distribution
To investigate whether the organization and integrity of microtubules was important for the clustering of mitochondria, we applied two microtubule drugs, nocodazole and taxotere. Nocodazole causes the disassembly of microtubules, taxotere (a taxol derivative) causes their assembly and stabilization (Jordan and Wilson, 1998). In the presence of taxotere, new microtubules are nucleated randomly in the cytoplasm, independently of the MTOC, so that the defined polarity and orientation is lost (McNiven and Porter, 1986; Weisshaar et al., 1992). If microtubules mediated the relocalization of mitochondria towards the cell center, one would expect that disruption of the microtubule network in the tau-overexpressing cells might lead to a wild-type distribution. This was indeed observed. Fig. 5 shows the disruption of microtubules in tau-stable CHO cells treated with nocodazole, which resulted in an increased cytosolic background staining arising from disassembled tubulin dimers. Within 1 h after nocodazole treatment, the mitochondria were redistributed to peripheral regions of the cell. The mitochondria-containing area thereby increases from 27.7 to 63.9%, which reaches the value of control cells within the error margin (see Fig. 10). Conversely, when nocodazole was removed again, an apparently normal microtubule network reappeared within about 15 min (data not shown), originating at the MTOC, and the phenotype of mitochondrial clustering, clearly visible already within ∼1 h, was fully restored after ∼4 h, indicating that the organelles were actively transported towards the MTOC (see Figs. 6 and 10). Taken together, these data suggest that a shift in the balance between plus- and minus-end–directed motor activities through tau-overexpression is responsible for the phenotypic effects. Thus, the ratio of mitochondria actively transported towards the cell periphery (that is plus-end directed) or towards the cell center (the MTOC, minus-end directed) in tau-stable versus control cells is expected to be altered by tau overexpression. To investigate this in further detail, cells were treated with nocodazole to achieve an even distribution of mitochondria in both cell lines. The microtubule array was then allowed to recover by washing out the drug and the direction of the movement of individual mitochondria was analyzed. Although transport in general is affected by tau overexpression (Fig. 7), irrespective of the kind of motor activity, plus-end–directed transport decreased ∼10-fold, whereas minus-end–directed transport dropped only about twofold, resulting in a net inward transport of mitochondria in tau-stable cells compared with control cells.
Figure 5.
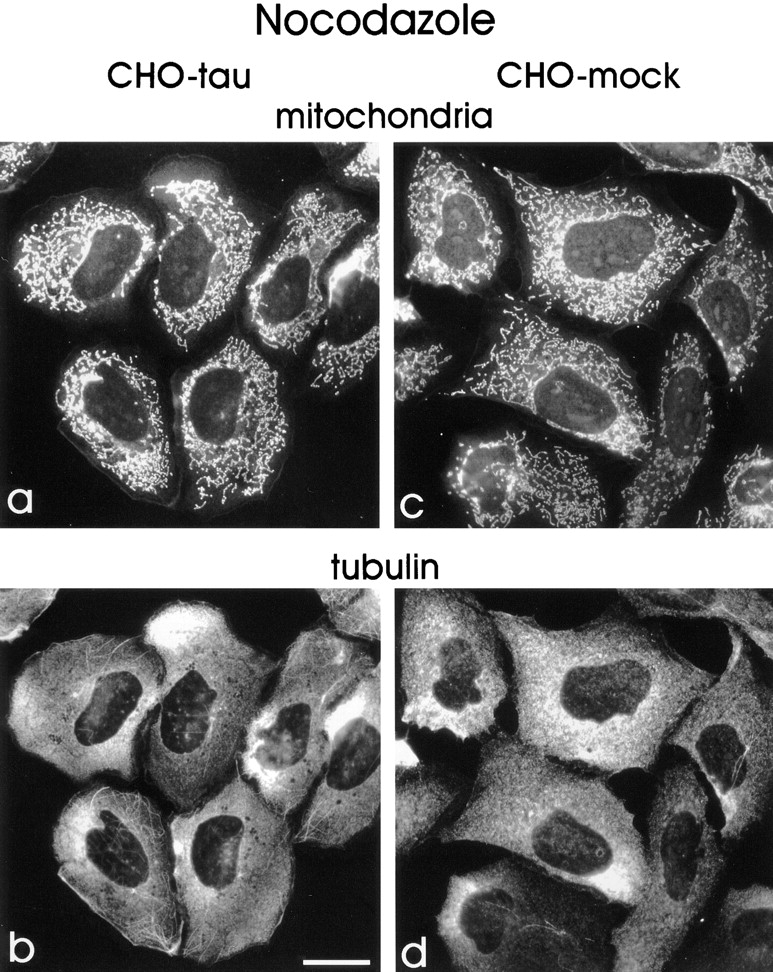
Disruption of microtubules by nocodazole leads to a random distribution of mitochondria in tau-stable CHO cells. Tau-stable cells (a and b) and mock-transfected CHO cells (c and d). Cells were treated with nocodazole to disrupt the MT cytoskeleton, fixed with methanol, and immunolabeled with antibodies against tubulin (b and d). In both cell lines, mitochondria, visualized by addition of MitoTracker™ Green before fixation (a and c), are now visible throughout the entire cytoplasm even in the cell periphery, suggesting the existence of an intact MT network as a prerequisite for mitochondrial clustering. Bar, 20 μm.
Figure 6.

Reassembly of microtubules after removal of nocodazole restores the clustering of mitochondria. The microtubule cytoskeleton of tau-transfected CHO cells was disrupted by addition of 5 μM nocodazole for 2 h. Pictures of living cells were then taken after removal of nocodazole and reassembly of microtubules at the indicated time points. Mitochondria were stained with MitoTracker™ Green. Note that, concomitant with microtubule reassembly, the phenotype of mitochondrial clustering at the MTOC near the nucleus begins to reappear after ∼1 h. The cell periphery is outlined for clarity. Bar, 10 μm.
These results demonstrate that microtubule-dependent processes are responsible for the clustering of mitochondria. It also means that mitochondria can become dispersed independently of microtubules, presumably by diffusion (in contrast to the ER, see below). The fact that reconstitution of the MT network leads to a reclustering of mitochondria near the cell center suggests that the overexpression of tau impairs predominantly the plus-end– directed transport of these organelles (see below). If this were true, one would expect that the normal distribution of mitochondria could be restored even in tau-overexpressing cells by abolishing the polar organization of microtubules. This can be achieved by treatment of cells with taxotere (at final concentrations of up to 10 μM), which leads to microtubule nucleation with random orientations throughout the cytoplasm. Fig. 8 shows that taxotere-treatment has a similar effect as nocodazole in restoring the dispersed distribution of mitochondria. Again, the mitochondria-containing area within the tau-stable cells increases to 58.1%, comparable with the 66.1% of mock-transfected cells (see Fig. 10). This result seems counterintuitive since the two drugs have opposite effects on microtubules, but it becomes understandable by considering that taxotere randomizes microtubules (at the micromolar concentrations used here), and thus destroys the basis for directed transport. In other words, a phenotype caused by impairment of plus-end–directed transport can manifest itself only in the presence of a polarized microtubule network where microtubule minus ends converge on the MTOC. Randomizing microtubule polarity by addition of taxotere allows transport of mitochondria to the cell periphery by minus-end–directed motors and thus leads to a redispersion of these organelles.
Figure 8.
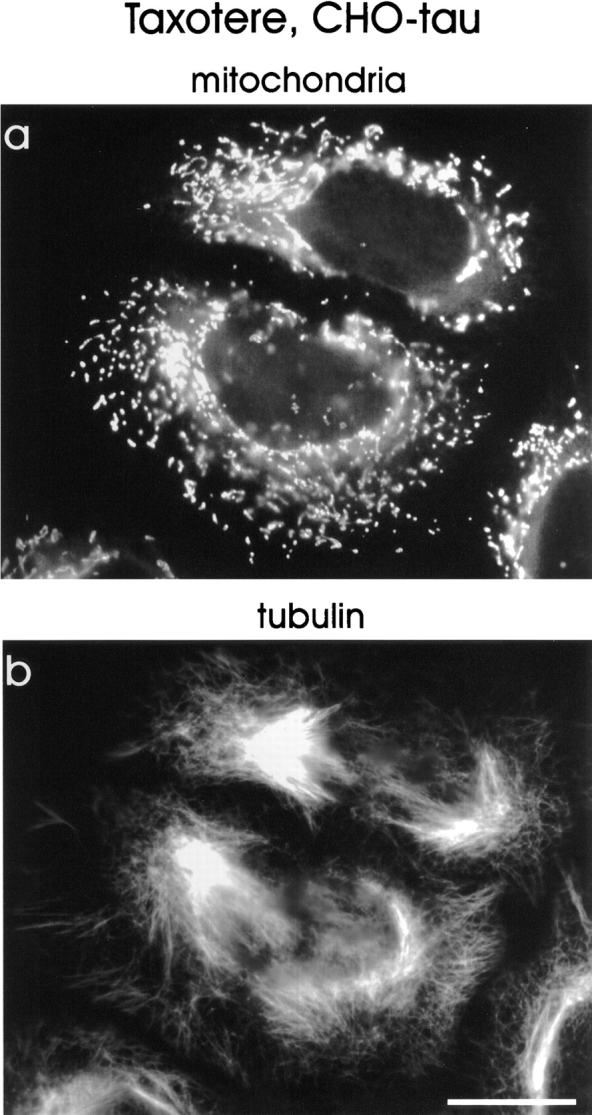
Randomization of microtubule polarity by taxotere allows clustered mitochondria to disperse again. Tau-stable cells were treated with 10 μM taxotere for 6 h, which leads to an altered microtubule organization. Note that the MTOC in taxotere-treated cells is barely visible in the tubulin stain (b), in contrast to nontreated cells (compare Fig. 2). Microtubules are nucleated randomly throughout the cytoplasm, thus abolishing the role of the MTOC as an organizing element and therefore abolishing the polarity of microtubules. Minus-end–directed motors are now able to transport mitochondria back to peripheral regions in the tau-stable cells. Cells were fixed in methanol and mitochondria were stained with MitoTracker™ Green (a), tubulin was immunostained with DM1A (b). Bar, 10 μm.
Inhibition of Dynein by Overexpression of p50-Dynamitin Reverses the Effect of Mitochondrial Clustering by Tau
The minus-end–directed transport of mitochondria is thought to depend on dynein (Hirokawa et al., 1990; Vallee et al., 1995), and therefore one would expect that inhibition of dynein would counteract the clustering of mitochondria induced by overexpression of tau. We inhibited dynein by transient transfection of the tau-overexpressing cells with dynamitin, a subunit of the dynactin complex that is crucial for the attachment between organelles and microtubules (Burkhardt et al., 1997). Overexpression of dynamitin in tau-stable cells significantly reduced the clustering of mitochondria, whereas EGFP transfection as a control had no effect on the mitochondrial distribution (Fig. 9). The area containing mitochondria within the cells after transfection increased from ∼28% in EGFP-transfected tau-stable cells to 47.9% in dynamitin-overexpressing cells (P < 0.0001, two-tailed Student's t test; Fig. 10). The fact that mitochondria were not completely redispersed (in contrast to the nocodazole-treated cells, where dispersal is complete) is probably due to fixation of cells 6 h after transfection. Since the overexpression of dynamitin affects the organization of the MTOC (Burkhardt et al., 1997), which in turn interferes with directed microtubule-dependent transport of mitochondria, we analyzed only those cells whose microtubule network was clearly unaffected. However, the experiment demonstrates that dynein motors are actively involved in the mitochondrial transport and argues against different types of mechanisms that operate in other situations (e.g., tethering to shrinking microtubules; Lombillo et al., 1995).
Figure 9.
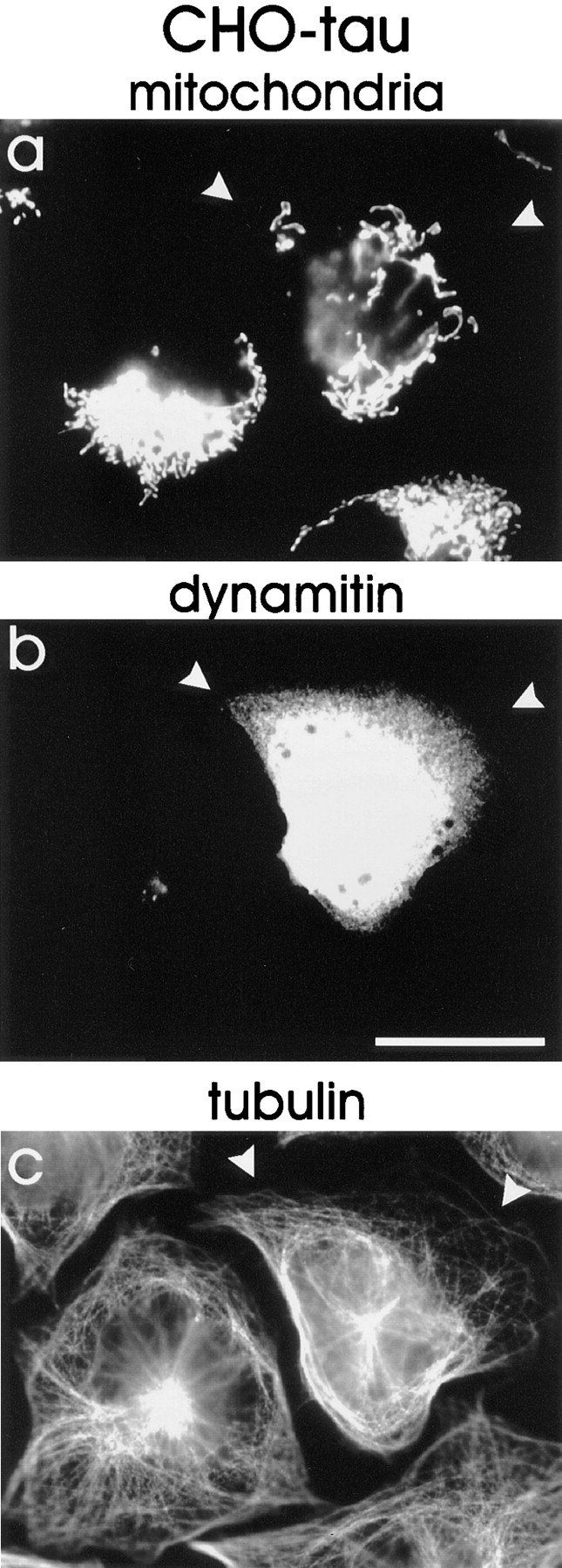
Dynamitin overexpression restores the wild-type distribution of mitochondria. Tau-stable cells were transfected with dynamitin (p50), a subunit of the dynactin complex. 6 h after transfection, the mitochondria began to disperse again, visualized with MitoTracker™ Red after paraformaldehyde fixation (a). Note that the cell transfected with tau and dynamitin (arrowheads) shows dispersed mitochondria, whereas the neighboring tau-stable cells show the typical clustering near the MTOC. Dynamitin was stained with antidynamitin 50-1 antibody (b) and tubulin with YL1/2 antibody (c). Bar, 20 μm.
Shape and Extension of the Endoplasmic Reticulum Is Altered by the Overexpression of Tau
Thus far, we concentrated on the microtubule-dependent transport of mitochondria in transfected CHO cells. However, microtubules are responsible for transporting other vesicles and organelles as well, notably the endoplasmic reticulum (Terasaki and Reese, 1994). Its distribution appears to depend both on kinesin-dependent transport and on an intact microtubule network (Vale and Hotani, 1988; Hamm-Alvarez et al., 1993; Feiguin et al., 1994). To investigate if overexpression of tau would affect the ER, we stained living cells with DiOC6(3), a fluorescent marker for the ER. Overexpression of tau caused pronounced changes in shape and extension of the ER (Fig. 11). First, in tau-stable cells, the extension of the ER was strongly reduced so that it no longer reached the cell periphery. Secondly, the density and branching of the ER was significantly decreased. As in the case of mitochondria, this result is consistent with the assumption that plus-end– directed transport along microtubules is perturbed.
On the other hand, observations with the microtubule drugs taxotere and nocodazole showed that the behavior of the ER is more complex. Taxotere partially restored the wild-type distribution, although the ER did not fully reach the cell periphery (data not shown). In contrast, after disruption of microtubules with nocodazole, the ER did not expand again. These results confirm that organized microtubules are necessary for the general constitution of the ER, in addition to kinesin (Lippincott-Schwartz, 1998).
Recycling of Transferrin in Tau-overexpressing CHO Cells Is Slowed Down
Endo- and exocytotic processes depend on an intact microtubule cytoskeleton. We therefore asked whether the overexpression of tau influences these processes as well, using rhodamine-labeled transferrin as a marker. Transferrin is endocytosed via transferrin receptors and clathrin-coated vesicles and is actively transported along microtubules by motor proteins through early and sorting endosomes to recycling compartments in the pericentriolar region of the cell. It is finally released by exocytosis, again via microtubule-dependent transport processes (Martys et al., 1995). The efflux of transferrin was measured by removal of the supernatant at different time points and by quantitating the exocytosed transferrin by fluorescence (Fig. 12 a). At early time points (a few minutes after removal of excess transferrin) the tau-stable cell lines showed a significantly lower level of exocytosed transferrin than the control cells, but this difference disappeared later on (∼20 min). Thus, the outward flow of exocytic vesicles was retarded but not interrupted.
Figure 12.

Transferrin recycling is retarded by tau overexpression. Cells were seeded on cover slips and preincubated with TMR-labeled transferrin in serum-free medium for 2 h. Excess transferrin was removed and tau-stable and mock-transfected cells were fixed in methanol. Residual transferrin fluorescence was then quantitated and plotted (a). Note that, at early time points (7 min), the transferrin fluorescence in tau-stable cells is decreased by ∼10%, whereas in control cells 50% of transferrin is exocytosed in the same time interval. 300 cells were quantitated per time point and cell line. (b) Time course of the exocytosis of transferrin after equilibration of tau-stable and mock-transfected cells with fluorescently labeled transferrin. A significant decrease of exocytosed transferrin can be seen 5–10 min after start of the experiment in tau-stable compared with control cells. Fluorescent signals were normalized to total cell numbers in each experiment. Data represent averages of four independent experiments.
This observation was confirmed by quantitating the immunofluorescence of residual TRITC-labeled transferrin in methanol-fixed tau-stable cells and mock-transfected controls. Methanol-fixed tau-stable cells yielded a significantly higher amount of transferrin 7 min after removal of transferrin-containing media compared with mock-transfected cells (Fig. 12 a; P < 0.0001, two-tailed Student's t test), mostly in the vicinity of the MTOC (the region of the recycling compartment; Martys et al., 1995). Whereas ∼50% of the initial intracellular transferrin had been exocytosed in mock-transfected cells, only 10% had been recycled in tau-stable cells during the same time period. This correlates well with the experiment shown in Fig. 12 b, where, conversely, the exocytosed transferrin was quantitated. Again, after ∼7 min, only 50% of transferrin was exocytosed in tau-stable cells compared with control cells.
Transport of Mitochondria and Endoplasmic Reticulum into the Neurites of Differentiated N2a-Neuroblastoma Cells Is Impaired by Overexpression of Tau
In the experiments described so far, we focussed on CHO cells to investigate tau's effects on transport processes. The chief reasons were that CHO cells are well suited for studying transport due to their flattened cell form and their size, which facilitates observation and microinjection. Secondly, CHO cells do not normally express tau so that the discrimination between endogenous MAPs (e.g., MAP4) and stably introduced tau is much easier than in neuroblastoma cells. On the other hand, tau is expressed mainly in neuronal tissue, raising the question whether the effects of tau overexpression manifest themselves in neuronal cells as well. For this reason, we stably transfected human tau40 into the mouse neuroblastoma cell line N2a. Although the cell body of N2a cells is very compact and small (∼10–20 μm, compared with ∼40 μm for CHO cells) so that it is difficult to detect differences in mitochondrial localization, the organelles appear to be clustered in the vicinity of the nucleus in tau-stable compared with control cells. Transport processes in the neurites of differentiated N2a cells on the other hand can be monitored much more easily because transport deficits in these structures accumulate to a more significant extent. As shown by several reports, tau protein is important for the generation of neurites, and transport of material into the neurites depends on kinesin (Baas et al., 1991; Feiguin et al., 1994).
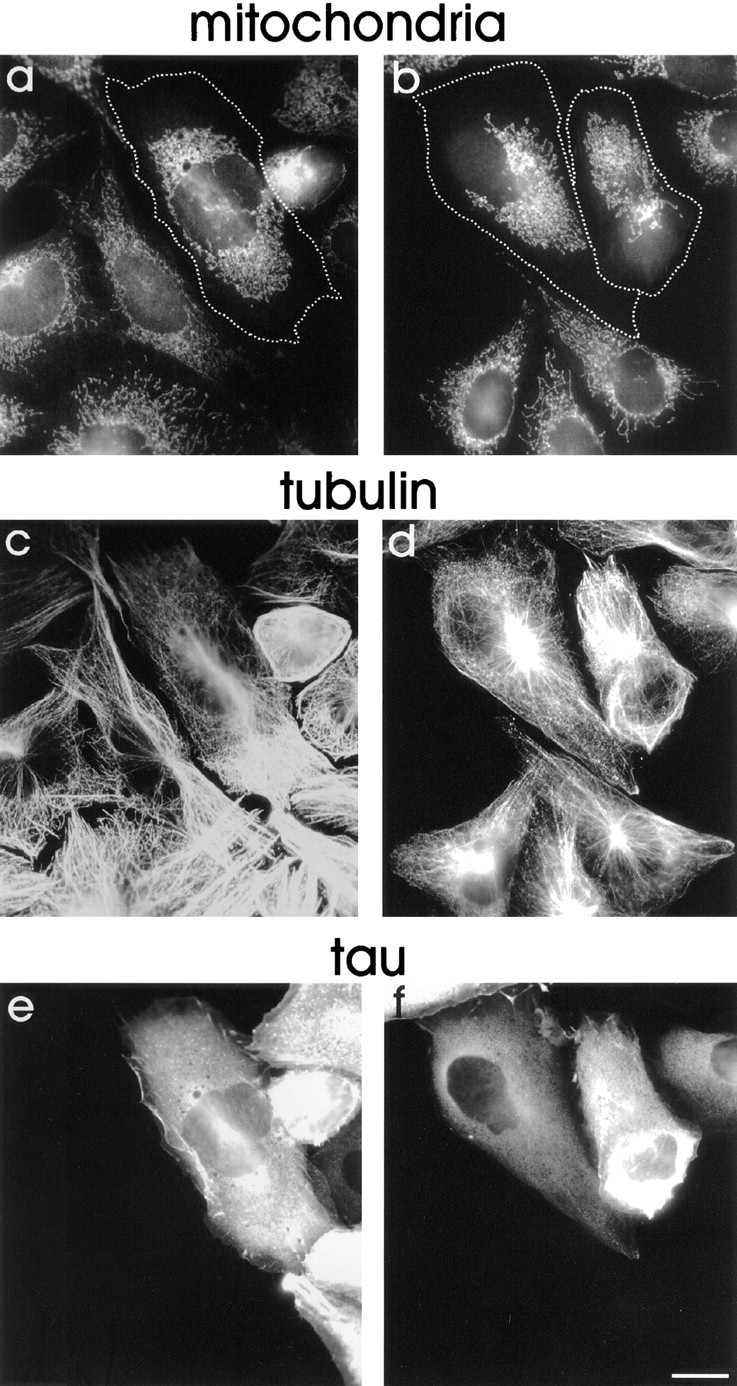
Differentiated wild-type N2a cells contain abundant mitochondria in their neurites, extending up to the tip (Fig. 13 d). In striking contrast, the neurites of differentiated N2a cells transfected with tau showed very few mitochondria (Fig. 13 a and Table I). The remarkable depletion of mitochondria from neuritic extensions in these cells indicates that tau overexpression in neuronal cells leads to the same phenotype as seen in CHO cells, and again can be explained by an impairment of plus-end–directed microtubule-dependent transport of mitochondria. The absence of these organelles would severely limit the energy supply of neurites and could cause their degeneration. These observations have implications for the cellular causes of Alzheimer's disease where elevated and mislocalized tau occurs concomitant with neuronal degeneration (as discussed later).
Figure 13.

Transport of mitochondria into neurites of differentiated neuroblastoma cells is impaired by overexpression of tau. Tau40-stable (a–c) and mock-transfected N2a cells (d–f) were differentiated for 2 d by addition of 1 μM retinoic acid into medium containing 0.1% fetal calf serum and fixed in methanol. Mitochondria were visualized with MitoTracker™ Green (a and d), immunostaining was done with antibodies against tubulin (b and e) and tau (c and f). It can be clearly seen that significantly fewer mitochondria are present in the neurites of tau-stable cells in contrast to control cells (compare a and d, and Table I). In addition, mitochondria in tau-stable cells appear to be more clustered in the vicinity of the MTOC (a), similar to tau-stable CHO cells. The comparable staining intensity of the cell bodies indicates that the clustering effect is not due to a lower number of mitochondria in the tau-transfected cell. Bar, 10 μm.
Table I.
Quantitation of the Phenotypic Effects of Tau Overexpression in N2a-Neuroblastoma Cells
| N2a tau40 | N2a mock | |||
|---|---|---|---|---|
| Neurites | 5.4% | 15% | ||
| (length >30 μm) | ||||
| Mitochondria in neurites | 17% | 94% | ||
| (>6 per neurite) | ||||
| ER distribution | 82% | 23% | ||
| (clustered) |
A minimum of 150 cells were analyzed in four (neurite length and mitochondria) or three (ER distribution) independent experiments. In differentiated, mock-transfected cells, 15% of the cells developed neurites longer than 30 μm, whereas, in tau-stable cells, this value drops to only 5.4%. The effect of tau-overexpression becomes even more impressive when analyzing the mitochondria present in the neuritic structures: only 17% of tau-stable cells transported more than six mitochondria into their neurites, compared with 94% of control cells. The average number of mitochondria in the control cells was ∼10–20 per neurite (data not shown). The ER was regarded as clustered when it did not surround the nucleus of the cells. In tau-stable cells, up to 82% displayed this phenotype. In contrast, in mock-transfected cells, only 23% of cells had a similar appearance.
Since the distribution of mitochondria was impaired in differentiated N2a neuroblastoma cells overexpressing tau, we asked whether the ER was affected as well (judging by immunofluorescence with an ER-specific antibody; Louvard et al., 1982). Control cells showed ER elements throughout the cell body mostly surrounding the nucleus, but visible in the neuritic structures. By contrast, the majority of tau-overexpressing cells (∼82%, Table I) showed that the ER was collapsed towards the cell center, without extending around the nucleus (Fig. 14 a). The overall intensity of ER-staining remained the same in the two cell types. In particular, the ER extending into the neurites was strongly reduced in the tau-stable cells (compare Fig. 14, a and d). Thus, an elevated level of tau in differentiated N2a cells causes a retraction of the ER structure in the cell body similar to the effect seen in CHO cells, leading to a decrease of the ER in neurites, presumably by affecting intracellular transport processes. Consistent with this, the neurites of tau-overexpressing cells are significantly shorter than those of control cells (e.g., the number of neurites extending ≥30 μm decreases approximately threefold, Table I).
Figure 14.
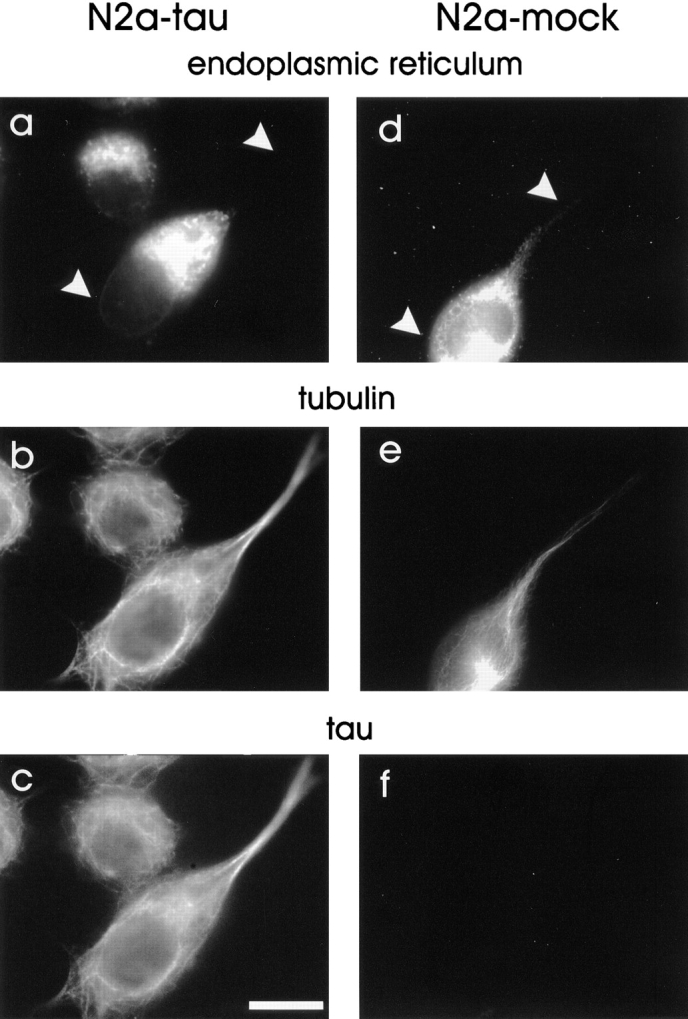
The expansion of the endoplasmic reticulum in differentiated neuroblastoma cells is significantly reduced by overexpression of tau. Tau-stable N2a cells (a–c) or mock-transfected cells (d–f) were induced to differentiate, and then fixed in paraformaldehyde and triple-labeled with antibodies against the ER (a and d), tubulin (b and e), and tau (c and f). In contrast to control cells, ∼82% of tau-stable neuroblastoma cells show the ER clustered at one side of the nucleus (a), whereas in mock-transfected cells the ER surrounds the entire nucleus (d; compare Table I). Furthermore, the expansion of the ER into the neurites in control cells (d) can clearly be seen. In tau-stable cells, only a faint signal of ER-immunoreactivity is visible in neuritic processes. The total intensity of the ER stain is the same in both cells. Arrowheads in a and d are indicating the position of the neurite relative to the cell body. Bar, 10 μm.
Discussion
Tau Retards Plus-End–directed Transport of Mitochondria, ER, and Transport Vesicles Along Microtubules
In this study, we have investigated how an increased level of tau protein (generated by overexpression or microinjection) affects intracellular transport in CHO and neuroblastoma cells. The main results can be summarized as follows: (a) increased tau leads to a clustering of mitochondria and peroxisomes near the MTOC (Fig. 2), to a contraction of the intermediate filament system and the ER (Fig. 11), and to a reduced rate of exocytosis (e.g., of transferrin receptor, Fig. 12). The cells lose their elongated shape and become rounded (Fig. 2), and their growth rate slows down. Mitochondria and ER are decreased in neuritic processes (Figs. 13 and 14; Table I). (b) All of the above effects are observed at relatively low levels of tau overexpression where the microtubule network is not impaired (no bundling, no detachment from MTOC); in fact, they depend on the integrity of the microtubule network. The effects also depend on a strong binding of tau to microtubules (K d ∼1 μM, provided by the repeats and proline-rich flanking domain). (c) If the microtubule network is disrupted by drugs (i.e., disassembly by nocodazole, Fig. 5), or detachment of microtubules from MTOC and randomization by taxotere (Fig. 8), the tau-induced changes in the distribution of mitochondria are reversed so that the organelles become dispersed throughout the cell again (Fig. 10). (d) Inhibition of dynein (via overexpression of dynamitin) reverses the effects of tau overexpression and allows the dispersion of mitochondria (Figs. 9 and 10).
These effects can be explained by the assumption that tau interferes preferentially with the plus-end–directed transport along microtubules involving kinesin, while the minus-end transport involving dynein is less affected. The directionality of organelle transport observed with the tau-transfected cells is consistent with a number of reports showing that plus-end–directed transport is achieved mainly by kinesin-like motors, and the reverse transport by dynein (Vallee and Sheetz, 1996; Rickard and Kreis, 1996; Hirokawa, 1998; Lippincott-Schwartz, 1998). For example, mitochondria are transported by the kinesin motors KIF1B (Nangaku et al., 1994) and KIF5B (Tanaka et al., 1998), or in the opposite direction by dynein (Hirokawa et al., 1990), and, therefore, inhibition of kinesin leads to a net retrograde transport of mitochondria. Rodionov et al. (1993) have shown that microinjection of an inhibitory kinesin antibody, which cross-reacts with several members of the kinesin superfamily, leads to a strikingly similar phenotype of mitochondrial clustering. Furthermore, the targeted disruption of mouse conventional kinesin heavy chain KIF5B results in the perinuclear clustering of mitochondria (Tanaka et al., 1998). Similarly, the extension of the ER and the intermediate filament system or the trafficking of vesicles is determined by a balance between kinesin- and dynein-dependent movements so that the ER and the vimentin-network collapses when kinesin is suppressed (Feiguin et al., 1994; Gyoeva and Gelfand, 1991). (The term “anterograde” is used for the plus-end–directed axonal transport along microtubules, but also for the minus-end–directed transport from ER to Golgi, so that we prefer the term “plus-end–directed” for the present discussion.)
The function of tau or MAPs is usually discussed in terms of their role in microtubule stabilization (Matus, 1994), the generation of cell processes (Kosik and McConlogue, 1994), in their spacer function between microtubules (Chen et al., 1992), or as anchoring sites for other proteins (Obar et al., 1989; Ookata et al., 1995). The data presented here point to a new function of tau; that is, regulation of transport within cells by the variation of the intracellular concentration of tau on the surface of microtubules. Since related effects have been observed with MAP4 (Bulinski et al., 1997), this may be a general function of MAPs. In this context, it is of interest to note that, although MAP4 efficiently stabilizes microtubules in vitro and in vivo, the mitochondrial distribution in MAP4-overexpressing cells has been reported to not be altered (Nguyen et al., 1997). This argues against the possibility that a change in lifetime of microtubules accounts for the effect of mitochondrial clustering, because the lifetime would be increased both by MAP4 and tau. More importantly, the comparison perhaps points to different functions of tau and MAP4 in vivo.
Inhibition of Motor Activity by MAPs
The question of whether and how MAPs and motors could interfere with each other on a microtubule has been the subject of extended discussions, mainly because experimental evidence obtained in vitro remained ambiguous (e.g., Paschal et al., 1989; Von Massow et al., 1989; Rodionov et al., 1990; Lopez and Sheetz, 1993; Hagiwara et al., 1994). The results depended not only on the type of MAPs and motors used, their effective concentrations (in solution or bound to a surface), but also on the experimental design (microtubule gliding or bead movement assay). The majority of the in vitro results suggested that tight binding of MAPs to microtubules via the microtubule-binding domain and a large projection domain was a prerequisite for observing any inhibition in motility. Thus, MAP2 could retard kinesin or dynein, while smaller MAPs (e.g., tau, MAP2c) had a much smaller effect or none at all (Von Massow et al., 1989; Lopez and Sheetz, 1993).
Conceptually, one can analyze the interference between MAPs and motors in terms of competition for common binding sites on a microtubule and/or in terms of steric hindrance (Lopez and Sheetz, 1993). A purely competitive mechanism would require only the microtubule-binding domain of MAPs (repeats plus flanking regions), while steric hindrance would depend on both microtubule binding plus the NH2-terminal projection domain. Since several of the earlier studies had emphasized that large projection domains of MAPs were necessary for motor inhibition, we were surprised to find that a small MAP such as tau, overexpressed only moderately in cells, could be inhibitory for plus-end–directed transport. This could be achieved even with constructs having no projection domain (K35). Constructs with lower affinity generally had no effect (K10, K12), even when the projection domain was present, as in K23 (Fig. 3). Obviously the cellular environment reacts more sensitively to MAPs than the experimental systems used in vitro. This could be explained by several factors: in the crowded environment of a cell, even shorter MAPs could have noticeable effects on transport, which would be most apparent for small motors (note that KIF1B, responsible for mitochondrial transport, is only 35-nm long, compared with 75 nm for conventional kinesin; Nangaku et al., 1994; Scholey et al., 1989). Secondly, the periods of observation are long, compared with the in vitro experiments, so that small differences in transport velocities can accumulate to a measurable extent. Thirdly, the cellular effects result from an interplay between plus- and minus-end motors, which are differently affected by MAPs, while in vitro experiments were done with one type of motor only. Finally, the in vitro studies are usually done with an excess of motors that tend to overwhelm the resistance that MAPs might offer. A comparison between the different isoforms and constructs of tau shows that the inhibition of vesicle movement is dominated by the competition between tau and motors for binding sites on the microtubules, while steric hindrance is of lesser importance.
The direction of microtubule-based transport is thought to be determined by particular motor proteins anchored at the organelle by means of adaptor complexes (e.g., kinectin, dynactin; Kumar et al., 1995; Burkhardt et al., 1997). The variety of motor proteins in cells makes it possible, in principle, for each type of vesicle or organelle to select one or more type of motor, depending on the required destination (Hirokawa, 1998; Hamm-Alvarez et al., 1993). In spite of attached motors, the movement of vesicles is not simply unidirectional, but rather saltatory, with frequent detachments and even reversals (Wacker et al., 1997). The net movement is therefore the result of many trials, biased in a particular direction. Inside a cell, MAPs could inhibit the activity of a motor in several ways; e.g., by preventing the initial attachment, by reducing the velocity, or by reducing the run length. In the present context, the important point is that tau most likely affects the balance between different directions, impairing the plus-end–directed movements of mitochondria more than the minus-end–directed ones, and this results in the clustering of these organelles near the MTOC, whereas the phenotype of ER retraction or the slow down in the export of transferrin might be achieved by simply reducing the overall transport processes or influencing the run length of motor proteins on microtubules.
Besides saltatory particle movement with directional bias, one should consider other mechanisms as well. For example, extension of the ER network can be driven by microtubule polymerization where the tips of microtubules are attached to the membrane by special attachment complexes (Allan and Vale, 1994; Waterman-Storer et al., 1995), and anaphase transport of chromosomes can be driven by microtubule depolymerization (Lombillo et al., 1995; Merdes et al., 1997). The attachment complexes can contain motor-like proteins that, in this case, operate as microtubule anchors rather than as independent motors. In practice, motor- and assembly-driven mechanisms may coexist within the cell (Rodionov and Borisy, 1997). However, in our case, the direct motor-dependent processes appear to be the dominant ones, as shown by the dynamitin-induced inhibition of dynein (Figs. 9 and 10; Burkhardt et al., 1997). Moreover, since tau stabilizes microtubules, it would be expected to enhance plus-end– directed transport if this were assembly driven, rather than inhibiting it as observed. Finally, we note that the distribution of organelles in the cytoplasm can be influenced by two further mechanisms, diffusion and actomyosin-based movement. The latter appears to be restricted to specialized regions (e.g., near the cell cortex or the axon; Morris and Hollenbeck, 1995; Mermall et al., 1998), but a diffusional component is likely to operate at all times. It explains the dispersion of clustered mitochondria upon microtubule disassembly by nocodazole, but this is not likely to be affected by tau.
Having determined the effects of tau overexpression in CHO cells, an obvious question was whether analogous effects would be observable in cells of neuronal origin. In this case, the experimental approaches are more limited because of the smaller size of the cells. For example, a redistribution of organelles in the cell body is difficult to observe and the level of endogenous tau is too low for many biochemical assays. On the other hand, vesicles and organelles are easily detectable in the long neuritic processes of differentiated cells. Thus, the neurites of control N2a cells show staining with the markers of mitochondria and the ER, but N2a cells overexpressing tau only at a significantly lower level. In agreement with the previous interpretation, this means that plus-end–directed transport, at least in the case of mitochondria, is impaired, leading to an accumulation of these elements in the cell body.
The effect of tau on vesicle trafficking described here represents a new function that has attracted little attention thus far. This was due in part to the fact that the influence of MAPs on the activity of motor proteins were mainly investigated in vitro, showed only weak effects, visible only at high MAP concentrations, and were apparently restricted to high molecular weight MAPs (Paschal et al., 1989; Von Massow et al., 1989; Lopez and Sheetz, 1993; Hagiwara et al., 1994). Nevertheless, in the context of cells, the subtle effects become magnified and visible as redistribution of cellular components. The most important prerequisite for tau's influence on intracellular traffic is the tight binding to microtubules due to the repeats and the flanking domains (∼1–2 μM) while the projection domain has only a minor effect. This implies that the phosphorylation of tau should play an important role since this regulates tau's affinity to microtubules. One can broadly distinguish two types of phosphorylation of tau, proline directed (affecting Ser-Pro or Thr-Pro motifs and induced by proline-directed kinases such as MAP kinase, GSK3, cdk5) and non–proline directed (induced by second-messenger–dependent kinases such as PKA, PKC, CaMK, MARK; for review see Mandelkow et al., 1995). Proline-directed phosphorylation has only a moderate influence on tau-microtubule interactions (Biernat et al., 1993), whereas two other sites potently disrupt the interaction, Ser262 (phosphorylated by MARK; Drewes et al., 1997) and Ser214 (phosphorylated by PKA; Zheng-Fischhöfer et al., 1998; both these sites are enhanced in Alzheimer's tau). Thus far, such sites were mainly seen as modulators of microtubule stability. However, in the light of our present results, one should also view the MAP-microtubule affinity-regulating phosphorylation reactions as modulators of intracellular traffic. In this scenario, tau bound to microtubules would present resistance to plus-end–directed movement, which would be relieved by tau's phosphorylation and detachment from microtubules. Since the outward flow of material is a prerequisite for cell polarity and differentiation (Feiguin et al., 1994; Bradke and Dotti, 1997), the activity of tau kinases would become important at this point. It is interesting to note that vesicle transport can be inhibited by MAP2, but relieved by a kinase that is probably related to MARK (Drewes et al., 1997; Lopez and Sheetz, 1995), a kinase that phosphorylates MAP2c and MAP4 in vitro (Illenberger et al., 1996), and therefore can be expected to regulate the affinity of MAPs to microtubules in general. This points to another interesting observation: it has been shown that MAP1A can compensate for the lack of tau in knock-out mice (Harada et al., 1994), suggesting that the function of MAPs is partially redundant. Whereas axonal elongation was not affected in these animals, microtubule stability was decreased in some axons. It is conceivable that different MAPs regulate the transport of different organelles and in different cell compartments. Thus, it would be interesting to correlate the distribution of organelles and MAPs in mouse models lacking or overexpressing MAPs to get further clues for the functions of MAPs in intracellular transport.
Implications for Neuronal Degeneration in Alzheimer's Disease
The effects of tau overexpression on mitochondrial distribution, vesicle-trafficking, and the ER have important implications for Alzheimer's disease. In AD, tau is abnormally phosphorylated, aggregated into paired helical filaments, and mislocalized from the axon to the somatodendritic compartment (Binder et al., 1985; Wischik et al., 1988; Braak et al., 1994). In addition, it has been reported that intracellular tau-protein concentration in AD brain is elevated (Khatoon et al., 1992). In neurons, this mislocalization and elevated intracellular concentration of tau would affect the transport of mitochondria or ER to the most peripheral regions of the axons and would lead to a decrease in glucose metabolism and ATP synthesis in these compartments, causing energy deficits, loss of Ca2+ homeostasis, or lipid synthesis (Futerman and Banker, 1996; Mattson and Guo, 1997). Finally, this may lead to degeneration of the neuron, presumably beginning from the most distal regions, leading to the so called “dying back” of axons (Braak et al., 1994; Trojanowski and Lee, 1995). In this context, it is of special interest that compromised mitochondrial function has indeed been shown to associate with late onset Alzheimer's disease (Davis et al., 1997). In addition, a slowing down of transport processes could affect the proper distribution of proteins, particularly of amyloid precursor protein (APP), another key player in the progression of Alzheimer's disease that is rerouted by transcytosis (Simons et al., 1995), and whose proteolytic processing to the amyloidogenic peptide Aβ1-42 takes place in the endoplasmic reticulum (Cook et al., 1997; Hartmann et al., 1997). Thus, a transport inhibition due to the overexpression and mislocalization of tau in neurons might increase production of Aβ1-42. Conversely, overproduction of amyloidogenic variants of APP correlates with an increased level of tau protein and tau mRNA (Lee et al., 1996), and, in the Alzheimer's-related Down syndrome, APP and tau are both upregulated (Oyama et al., 1994). Finally, the mutant presenilins PS1 and PS2 that are responsible for familial Alzheimer's disease have been shown to interact with APP in the endoplasmic reticulum, thereby enhancing the production of Aβ (Xia et al., 1997). Thus, future work might give new clues to an understanding of Alzheimer's disease since it might experimentally link the function of AD-specific gene products, such as APP and the presenilins, with mitochondrial energy supply and tau protein.
Acknowledgments
We thank I. Thielke and K. Alm for excellent technical assistance, and J. Biernat for generously providing the tau constructs. We gratefully acknowledge the gift of ER antibody from D. Louvard (Institut Curie, Paris, France), the gift of dynamitin (p50) expression plasmid, and the antidynamitin 50-1 antibody from Dr. R.B. Vallee (Worcester Foundation, Shrewsbury, MA), and the gift of the anticatalase antibody from Dr. W. Just. This article is based in part on the doctoral thesis by R. Godemann and K. Stamer in the Faculty of Biology, University of Hamburg (Hamburg, Germany).
Abbreviations used in this paper
- AD
Alzheimer's disease
- EGFP
enhanced green fluorescent protein
- MAP
microtubule-associated protein
- MT
microtubule
- MTOC
microtubule organizing center
- TMR
tetramethylrhodamine
Footnotes
This project was supported by grants from the Deutsche Forschungsgemeinschaft.
Address all correspondence to Eckhard Mandelkow or Andreas Ebneth, MPG-ASMB, Notkestrasse 85, D-22607 Hamburg, Germany. Tel.: 49-40-8998-2810. Fax: 49-40-8971-6822. E-mail: mand@mpasmb.desy.de or ebneth @mpasmb.desy.de
References
- Allan VJ, Vale RD. Movement of membrane tubules along microtubules in vitro: evidence for specialized sites of motor attachment. J Cell Sci. 1994;107:1885–1897. doi: 10.1242/jcs.107.7.1885. [DOI] [PubMed] [Google Scholar]
- Baas PW, Slaughter T, Brown A, Black MM. Microtubule dynamics in axons and dendrites. J Neurosci Res. 1991;30:134–153. doi: 10.1002/jnr.490300115. [DOI] [PubMed] [Google Scholar]
- Barlow S, Gonzalezgaray ML, West RR, Olmsted JB, Cabral F. Stable expression of heterologous microtubule-associated proteins (MAPs) in chinese-hamster ovary cells—evidence for differing roles of MAPs in microtubule organization. J Cell Biol. 1994;126:1017–1029. doi: 10.1083/jcb.126.4.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow E-M, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, Mandelkow E-M. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti C. Neuronal polarity: vectorial cytoplasmic flow precedes axon formation. Neuron. 1997;19:1175–1186. doi: 10.1016/s0896-6273(00)80410-9. [DOI] [PubMed] [Google Scholar]
- Brady ST. Biochemical and functional diversity of microtubule motors in the nervous system. Curr Opin Neurobiol. 1995;5:551–558. doi: 10.1016/0959-4388(95)80058-1. [DOI] [PubMed] [Google Scholar]
- Brandt R, Leger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol. 1995;131:1327–1340. doi: 10.1083/jcb.131.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulinski JC, Borisy GG. Self-assembly of microtubules in extracts of cultured HeLa cells and the identification of HeLa microtubule-associated proteins. Proc Natl Acad Sci USA. 1979;76:293–297. doi: 10.1073/pnas.76.1.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulinski JC, McGraw TE, Gruber D, Nguyen H-L, Sheetz MP. Overexpression of MAP4 inhibits organelle motility and trafficking in vivo. J Cell Sci. 1997;110:3055–3064. doi: 10.1242/jcs.110.24.3055. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J Cell Biol. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapin SJ, Bulinski JC. Non-neuronal 210×103 Mr microtubule associated protein (MAP4) contains a domain homologous to the microtubule-binding domains of neuronal MAP2 and tau. J Cell Sci. 1991;98:27–36. doi: 10.1242/jcs.98.1.27. [DOI] [PubMed] [Google Scholar]
- Chapin SJ, Bulinski JC. Microtubule stabilization by assembly-promoting microtubule-associated proteins: a repeat performance. Cell Motil Cytoskelet. 1992;23:236–243. doi: 10.1002/cm.970230403. [DOI] [PubMed] [Google Scholar]
- Chen J, Kanai Y, Hirokawa N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature. 1992;360:674–677. doi: 10.1038/360674a0. [DOI] [PubMed] [Google Scholar]
- Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM-Y, Doms RW. Alzheimer's Abeta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- Davis ED, Miller S, Herrnstadt C, Ghosh SS, Fahy E, Shinobu LA, Galasko D, Thal LJ, Beal MF, Howell N, Parker WD., Jr Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer's disease. Proc Natl Acad Sci USA. 1997;94:4526–4531. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Cell Biol. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Preuss U, Mandelkow E-M, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Drubin DG, Feinstein SC, Shooter EM, Kirschner MW. Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J Cell Biol. 1985;101:1799–1807. doi: 10.1083/jcb.101.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Feiguin F, Ferreira A, Kosik KS, Caceres A. Kinesin-mediated organelle translocation revealed by specific cellular manipulations. J Cell Biol. 1994;127:1021–1039. doi: 10.1083/jcb.127.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, Banker GA. The economics of neurite outgrowth—the addition of new membrane to growing axons. Trends Neurosci. 1996;19:144–149. doi: 10.1016/s0166-2236(96)80025-7. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO (Eur Mol Biol Organ) J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson HV, Valetti C, Kreis TE. Motors and membrane traffic. Curr Opin Cell Biol. 1997;9:18–28. doi: 10.1016/s0955-0674(97)80147-0. [DOI] [PubMed] [Google Scholar]
- Gustke N, Trinczek B, Biernat J, Mandelkow E-M, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- Gyoeva FK, Gelfand VI. Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature. 1991;353:445–448. doi: 10.1038/353445a0. [DOI] [PubMed] [Google Scholar]
- Hagiwara H, Yorifuji H, Sato-Yoshitake R, Hirokawa N. Competition between motor molecules (kinesin and cytoplasmic dynein) and fibrous microtubule-associated proteins in binding to microtubules. J Biol Chem. 1994;269:3581–3589. [PubMed] [Google Scholar]
- Hamm-Alvarez SF, Kim PY, Sheetz MP. Regulation of vesicle transport in CV-1 cells and extracts. J Cell Sci. 1993;106:955–966. doi: 10.1242/jcs.106.3.955. [DOI] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Hartmann T, Bieger SC, Bruhl B, Tienari JP, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer's disease Abeta40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- Hiller G, Weber K. Radioimmunoassay for tubulin: a quantitative comparison of the tubulin content of different established tissue culture cells and tissues. Cell. 1978;14:795–804. doi: 10.1016/0092-8674(78)90335-5. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Yoshida T, Kawashima T. Brain dynein (MAP1c) localizes on both anterogradely and retrogradely transported membranous organelles in vivo. J Cell Biol. 1990;111:1027–1037. doi: 10.1083/jcb.111.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N. Microtubule-organization and dynamics dependent on microtubule-associated proteins. Curr Opin Cell Biol. 1994;6:74–81. doi: 10.1016/0955-0674(94)90119-8. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Hollenbeck P, Swanson J. Radial extension of macrophage tubular lysosomes supported by kinesin. Nature. 1990;346:864–866. doi: 10.1038/346864a0. [DOI] [PubMed] [Google Scholar]
- Illenberger S, Drewes G, Trinczek B, Biernat J, Meyer H, Olmsted J, Mandelkow E-M, Mandelkow E. Phosphorylation of microtubule-associated proteins MAP2 and MAP4 by the protein kinase p110mark. J Biol Chem. 1996;271:10834–10843. doi: 10.1074/jbc.271.18.10834. [DOI] [PubMed] [Google Scholar]
- Illenberger S, Zheng-Fischhöfer Q, Preuss U, Stamer K, Baumann K, Trinczek B, Biernat J, Godemann R, Mandelkow E-M, Mandelkow E. The endogenous and cell-cycle dependent phosphorylation of tau protein in living cells: implications for Alzheimer's disease. Mol Biol Cell. 1998;9:1495–1512. doi: 10.1091/mbc.9.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol. 1998;10:123–130. doi: 10.1016/s0955-0674(98)80095-1. [DOI] [PubMed] [Google Scholar]
- Kaech S, Ludin B, Matus A. Cytoskeletal plasticity in cells expressing neuronal microtubule-associated proteins. Neuron. 1996;17:1189–1199. doi: 10.1016/s0896-6273(00)80249-4. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Takemura R, Oshima T, Mori H, Ihara Y, Yanagisawa M, Masaki T, Hirokawa N. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. J Cell Biol. 1989;109:1173–1184. doi: 10.1083/jcb.109.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatoon S, Grundke-Iqbal I, Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem. 1992;59:750–753. doi: 10.1111/j.1471-4159.1992.tb09432.x. [DOI] [PubMed] [Google Scholar]
- Klymkowski M, Bachant J, Domingo A. Functions of intermediate filaments. Cell Motil Cytoskelet. 1989;14:309–331. doi: 10.1002/cm.970140302. [DOI] [PubMed] [Google Scholar]
- Kosik KS, McConlogue L. Microtubule-associated protein function: lessons from expression in spodoptera frugiperda cells. Cell Motil Cytoskelet. 1994;28:195–198. doi: 10.1002/cm.970280302. [DOI] [PubMed] [Google Scholar]
- Kumar J, Yu H, Sheetz MP. Kinectin, an essential anchor for kinesin-driven vesicle motility. Science. 1995;267:1834–1837. doi: 10.1126/science.7892610. [DOI] [PubMed] [Google Scholar]
- Lee G, Rook S. Expression of tau protein in non-neuronal cells: microtubule binding and stabilization. J Cell Sci. 1992;102:227–237. doi: 10.1242/jcs.102.2.227. [DOI] [PubMed] [Google Scholar]
- Lee RKK, Wurtman RJ, Yang BPS, Neve R. Increased tau protein in primary cortical astrocytes expressing human Aβ(1-42) and APP751. Soc Neurosci Abstr. 1996;22:1419. [Google Scholar]
- Li Y, Black MM. Microtubule assembly and turnover in growing axons. J Neurosci. 1996;16:531–544. doi: 10.1523/JNEUROSCI.16-02-00531.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J. Cytoskeletal proteins and Golgi dynamics. Curr Opin Cell Biol. 1998;10:52–59. doi: 10.1016/s0955-0674(98)80086-0. [DOI] [PubMed] [Google Scholar]
- Lombillo VA, Stewart RJ, McIntosh JR. Minus-end-directed motion of kinesin-coated microspheres driven by microtubule depolymerization. Nature. 1995;373:161–164. doi: 10.1038/373161a0. [DOI] [PubMed] [Google Scholar]
- Lopez L, Sheetz M. Steric inhibition of cytoplasmic dynein and kinesin motility by MAP2. Cell Motil Cytoskelet. 1993;24:1–16. doi: 10.1002/cm.970240102. [DOI] [PubMed] [Google Scholar]
- Lopez LA, Sheetz MP. A microtubule-associated protein (MAP2) kinase restores microtubule motility in embryonic brain. J Biol Chem. 1995;270:12511–12517. doi: 10.1074/jbc.270.21.12511. [DOI] [PubMed] [Google Scholar]
- Louvard D, Reggio H, Warren G. Antibodies to the Golgi complex and the rough endoplasmic reticulum. J Cell Biol. 1982;92:92–107. doi: 10.1083/jcb.92.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E, Mandelkow E-M. Microtubules and microtubule-associated proteins. Curr Opin Cell Biol. 1995;7:72–81. doi: 10.1016/0955-0674(95)80047-6. [DOI] [PubMed] [Google Scholar]
- Mandelkow E-M, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging. 1995;16:355–362. doi: 10.1016/0197-4580(95)00025-a. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Banker G. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. 1996;16:5727–5740. doi: 10.1523/JNEUROSCI.16-18-05727.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martys JL, Shevell T, McGraw TE. Studies of transferrin recycling reconstituted in streptolysin O permeabilized Chinese hamster ovary cells. J Biol Chem. 1995;270:25976–25984. doi: 10.1074/jbc.270.43.25976. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Guo Q. Cell and molecular neurobiology of presenilins: a role for the endoplasmic reticulum in the pathogenesis of Alzheimer's disease? . J Neurosci Res. 1997;50:505–513. doi: 10.1002/(SICI)1097-4547(19971115)50:4<505::AID-JNR1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Matus A. Stiff microtubules and neuronal morphology. Trends Neurosci. 1994;17:19–22. doi: 10.1016/0166-2236(94)90030-2. [DOI] [PubMed] [Google Scholar]
- McNiven MA, Porter KR. Microtubule polarity confers direction to pigment transport in chromatophores. J Cell Biol. 1986;103:1547–1555. doi: 10.1083/jcb.103.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merdes A, Cleveland DW. Pathways of spindle pole formation: different mechanisms; conserved components. J Cell Biol. 1997;138:953–956. doi: 10.1083/jcb.138.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermall V, Post PL, Mooseker MS. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science. 1998;279:527–533. doi: 10.1126/science.279.5350.527. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131:1315–1326. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Nguyen H-L, Chari S, Gruber D, Lue C-M, Chapin SJ, Bulinski JC. Overexpression of full- or partial-length MAP4 stabilizes microtubules and alters cell-growth. J Cell Sci. 1997;110:281–294. doi: 10.1242/jcs.110.2.281. [DOI] [PubMed] [Google Scholar]
- Obar RA, Dingus J, Bayley H, Vallee RB. The RII subunit of cAMP-dependent protein kinase binds to a common amino-terminal domain in microtubule-associated proteins 2A, 2B, and 2C. Neuron. 1989;3:639–645. doi: 10.1016/0896-6273(89)90274-2. [DOI] [PubMed] [Google Scholar]
- Olson KR, McIntosh JR, Olmsted JB. Analysis of MAP 4 function in living cells using green fluorescent protein (GFP) chimeras. J Cell Biol. 1995;130:639–650. doi: 10.1083/jcb.130.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ookata K, Hisanaga S, Bulinski JC, Murofushi H, Aizawa H, Itoh TJ, Hotani H, Okumura E, Tachibana K, Kishimoto T. Cyclin B interaction with microtubule-associated protein 4 (MAP4) targets p34cdc2 kinase to microtubules and is a potential regulator of M-phase microtubule dynamics. J Cell Biol. 1995;128:849–862. doi: 10.1083/jcb.128.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama F, Cairns NJ, Shimada H, Oyama R, Titani K, Ihara Y. Down's syndrome: up-regulation of beta-amyloid protein precursor and tau mRNAs and their defective coordination. J Neurochem. 1994;62:1062–1066. doi: 10.1046/j.1471-4159.1994.62031062.x. [DOI] [PubMed] [Google Scholar]
- Paschal BM, Obar RA, Vallee RB. Interaction of brain cytoplasmic dynein and MAP2 with a common sequence at the C terminus of tubulin. Nature. 1989;342:569–572. doi: 10.1038/342569a0. [DOI] [PubMed] [Google Scholar]
- Preuss U, Doring F, Illenberger S, Mandelkow E-M. Cell-cycle dependent phosphorylation and microtubule-binding of tau-protein stably transfected into Chinese-hamster ovary cells. Mol Biol Cell. 1995;6:1397–1410. doi: 10.1091/mbc.6.10.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss U, Biernat J, Mandelkow E-M, Mandelkow E. The ‘jaws' model of tau-microtubule interaction examined in CHO cells. J Cell Sci. 1997;110:789–800. doi: 10.1242/jcs.110.6.789. [DOI] [PubMed] [Google Scholar]
- Rickard JE, Kreis TE. Clips for organelle-microtubule interactions. Trends Cell Biol. 1996;6:178–183. doi: 10.1016/0962-8924(96)10017-9. [DOI] [PubMed] [Google Scholar]
- Rodionov V, Gyoeva F, Kashina A, Kuznetsov S, Gelfand VI. Microtubule-associated proteins and microtubule-based translocator have different binding-sites on tubulin molecule. J Biol Chem. 1990;265:5702–5707. [PubMed] [Google Scholar]
- Rodionov VI, Gyoeva FK, Tanaka E, Bershadsky AD, Vasiliev JM, Gelfand VI. Microtubule-dependent control of cell-shape and pseudopodial activity is inhibited by the antibody to kinesin motor domain. J Cell Biol. 1993;123:1811–1820. doi: 10.1083/jcb.123.6.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov VI, Borisy GG. Microtubule treadmilling in vivo. Science. 1997;275:215–218. doi: 10.1126/science.275.5297.215. [DOI] [PubMed] [Google Scholar]
- Sato-Harada R, Okabe S, Umeyama T, Kanai Y, Hirokawa N. Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct. 1996;21:283–295. doi: 10.1247/csf.21.283. [DOI] [PubMed] [Google Scholar]
- Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 1997;90:1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Scholey JM, Heuser J, Yang JT, Goldstein LS. Identification of globular mechanochemical heads of kinesin. Nature. 1989;338:355–357. doi: 10.1038/338355a0. [DOI] [PubMed] [Google Scholar]
- Simons M, Ikonen E, Tienari PJ, Cid-Arregui A, Monning U, Beyreuther K, Dotti C. Intracellular routing of human amyloid protein precursor: axonal delivery followed by transport to the dendrites. J Neurosci Res. 1995;41:121–128. doi: 10.1002/jnr.490410114. [DOI] [PubMed] [Google Scholar]
- Summerhayes IC, Wong D, Chen LB. Effect of microtubules and intermediate filaments on mitochondrial distribution. J Cell Sci. 1983;61:87–105. doi: 10.1242/jcs.61.1.87. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Terasaki M, Reese TS. Interactions among endoplasmic reticulum, microtubules, and retrograde movements of the cell surface. Cell Motil Cytoskelet. 1994;29:291–300. doi: 10.1002/cm.970290402. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM-Y. Phosphorylation of paired helical filament tau in Alzheimer's disease neurofibrillary lesions: focusing on phosphatases. FASEB J. 1995;9:1570–1576. doi: 10.1096/fasebj.9.15.8529836. [DOI] [PubMed] [Google Scholar]
- Vale RD, Hotani H. Formation of membrane networks in vitro by kinesin-driven microtubule movement. J Cell Biol. 1988;107:2233–2241. doi: 10.1083/jcb.107.6.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee RB, Vaughan KT, Echeverri CJ. Targeting of cytoplasmic dynein to membranous organelles and kinetochores via dynactin. Cold Spring Harbor Symp Quant Biol. 1995;60:803–811. doi: 10.1101/sqb.1995.060.01.086. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Sheetz MP. Targeting of motor proteins. Science. 1996;271:1539–1544. doi: 10.1126/science.271.5255.1539. [DOI] [PubMed] [Google Scholar]
- Von Massow A, Mandelkow E-M, Mandelkow E. Interaction between kinesin, microtubules, and microtubule-associated protein 2. Cell Motil Cytoskelet. 1989;14:562–571. doi: 10.1002/cm.970140413. [DOI] [PubMed] [Google Scholar]
- Wacker I, Kaether C, Kromer A, Migala A, Almers W, Gerdes HH. Microtubule-dependent transport of secretory vesicles visualized in real time with a GFP-tagged secretory protein. J Cell Sci. 1997;110:1453–1463. doi: 10.1242/jcs.110.13.1453. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Gregory J, Parsons SF, Salmon ED. Membrane/microtubule tip attachment complexes (TACs) allow the assembly dynamics of plus ends to push and pull membranes into tubulovesicular networks in interphase Xenopusegg extracts. J Cell Biol. 1995;130:1161–1169. doi: 10.1083/jcb.130.5.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer C, Salmon ED. Microtubule dynamics: treadmilling comes around again. Curr Biol. 1997;7:R369–R372. doi: 10.1016/s0960-9822(06)00177-1. [DOI] [PubMed] [Google Scholar]
- Weisshaar B, Doll T, Matus A. Reorganization of the microtubular cytoskeleton by embryonic microtubule-associated protein 2 (MAP2c) Development (Camb) 1992;116:1151–1161. doi: 10.1242/dev.116.4.1151. [DOI] [PubMed] [Google Scholar]
- West RR, Tenbarge KM, Olmsted JB. A model for microtubule-associated protein 4 structure. Domains defined by comparisons of human, mouse, and bovine sequences. J Biol Chem. 1991;266:21886–21896. [PubMed] [Google Scholar]
- Wiemer EAC, Wenzel T, Deerinck TJ, Ellisman MH, Subramani S. Visualization of the peroxisomal compartment in living mammalian cells: dynamic behavior and association with microtubules. J Cell Biol. 1997;136:71–80. doi: 10.1083/jcb.136.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, Klug A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer's disease. Proc Natl Acad Sci USA. 1988;85:4506–4510. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Zhang J, Perez R, Koo E, Selkoe DJ. Interaction between amyloid precursor protein and presenilins in mammalian cells: implications for the pathogenesis of Alzheimer's disease. Proc Natl Acad Sci USA. 1997;94:8208–8213. doi: 10.1073/pnas.94.15.8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng-Fischhöfer Q, Biernat J, Mandelkow E-M, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of tau-protein by GSK-3b and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired helical filament-like conformation. Eur J Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]