

Abstract
Cdc31p is the yeast homologue of centrin, a highly conserved calcium-binding protein of the calmodulin superfamily. Previously centrins have been implicated only in microtubule-based processes. To elucidate the functions of yeast centrin, we carried out a two-hybrid screen for Cdc31p-interacting proteins and identified a novel essential protein kinase of 1,080 residues, Kic1p (kinase that interacts with Cdc31p). Kic1p is closely related to S. cerevisiae Ste20p and the p-21– activated kinases (PAKs) found in a wide variety of eukaryotic organisms. Cdc31p physically interacts with Kic1p by two criteria; Cdc31p coprecipitated with GST–Kic1p and it bound to GST–Kic1p in gel overlay assays. Furthermore, GST–Kic1p exhibited in vitro kinase activity that was CDC31-dependent. Although kic1 mutants were not defective for spindle pole body duplication, they exhibited a variety of mutant phenotypes demonstrating that Kic1p is required for cell integrity. We also found that cdc31 mutants, previously identified as defective for spindle pole body duplication, exhibited lysis and morphological defects. The cdc31 kic1 double mutants exhibited a drastic reduction in the range of permissive temperature, resulting in a severe lysis defect. We conclude that Kic1p function is dependent upon Cdc31p both in vivo and in vitro. We postulate that Cdc31p is required both for SPB duplication and for cell integrity/morphogenesis, and that the integrity/morphogenesis function is mediated through the Kic1p protein kinase.
Keywords: microtubule organizing center, spindle pole body, budding, actin, cell wall
The microtubule-based cytoskeleton in eukaryotic cells is organized and regulated by microtubule organizing centers (MTOC).1 The MTOC in animal cells is the centrosome, and the equivalent organelle in the yeast Saccharomyces cerevisiae is the spindle pole body (SPB) (for reviews see Rose et al., 1993; Winey and Byers, 1993; Kellogg et al., 1994). The centrosome consists of centrioles embedded in an amorphous pericentriolar material. In yeast, the SPB is a trilaminar structure embedded in the nuclear envelope; it contains an inner plaque that nucleates the nuclear microtubules and an outer plaque that organizes the cytoplasmic microtubules (Byers and Goetsch, 1974, 1975). Despite the structural differences between the centrosome and SPB, they have equivalent functional roles and organize the microtubules that carry out microtubule-mediated processes.
The centrosome is a complex organelle consisting of many proteins, most of which are unidentified. One of the few known conserved centrosomal proteins is centrin/caltractin, which was originally identified in the basal body of the unicellular alga Chlamydomonas reinhardtii (Huang et al., 1988a ; Salisbury et al., 1988). In Chlamydomonas, centrin is essential for accurate basal body duplication and separation (Taillon et al., 1992) as well as for microtubule severing (Sanders and Salisbury, 1989, 1994). Centrin homologues have been identified in numerous organisms (Levy et al., 1996), including yeast (Baum et al., 1986), mice (Ogawa and Shimizu, 1993), and humans (Lee and Huang, 1993; Errabolu et al., 1994; Middendorp et al., 1997). Centrin is a small calcium-binding protein that is a member of the calmodulin superfamily of proteins (Huang et al., 1988b ; Salisbury et al., 1988).
Calmodulin is also a conserved centrosomal protein (for review see Means, 1994). In yeast, there is a single essential calmodulin gene, CMD1 (Davis et al., 1986). Cmd1p localizes to the SPB (Geiser et al., 1993; Stirling et al., 1994), and some temperature-sensitive cmd1 mutants affect SPB function (Geiser et al., 1993; Ohya and Botstein, 1994). In addition to its localization to the SPB, Cmd1p localizes to regions of cell growth (Brockerhoff and Davis, 1992); in unbudded cells, it concentrates in a patch at the presumptive bud site and then, in small buds, it accumulates at cortical sites in the bud tip. As the bud enlarges, Cmd1p disperses before concentrating again at the site of cytokinesis in the neck. This localization pattern is similar to actin cortical patches, and the localization of actin and calmodulin in the bud are interdependent (Brockerhoff and Davis, 1992). At the bud tip, calmodulin binds to the unconventional myosin Myo2p (Brockerhoff et al., 1994), and its localization is largely dependent on Myo2p (Stevens and Davis, 1998). Temperature-sensitive cmd1 mutations affect actin organization, calmodulin localization, and bud emergence, in addition to SPB function (Davis, 1992; Ohya and Botstein, 1994). Therefore, calmodulin has multiple essential functions that correlate with its multiple sites of intracellular localization.
Yeast centrin, encoded by the CDC31 gene, is essential for SPB duplication (Schild et al., 1981; Baum et al., 1986). Mutations in CDC31 block the earliest steps in the duplication of the SPB and lead to enlargement of the remaining unduplicated SPB (Byers, 1981). Mutations in the KAR1 gene result in SPB phenotypes indistinguishable from those of cdc31 mutants (Rose and Fink, 1987). Kar1p is also a component of the SPB (Vallen et al., 1992; Spang et al., 1995), and is required to localize Cdc31p to the SPB (Vallen et al., 1994; Biggins and Rose, 1994; Spang et al., 1993, 1995). However, Cdc31p also shows significant localization away from the SPB (Spang et al., 1993; Biggins and Rose, 1994) and recent results show that the majority of centrin in other organisms is also found elsewhere in cells (Paoletti et al., 1996). Therefore, like calmodulin, centrin may have multiple functions.
In this paper, we report the characterization of a protein kinase that interacts with Cdc31p. Identified in a two-hybrid screen for Cdc31p-interacting proteins, Kic1p is an essential protein kinase that phosphorylates substrates in a Cdc31p-dependent manner. Characterization of kic1 mutants shows that the gene is necessary for cell integrity or morphogenesis but not for SPB duplication. In addition, re-examination of cdc31 mutants revealed that they also affect cell integrity and morphogenesis. These results indicate that Cdc31p most likely acts through Kic1p to regulate cell integrity or morphogenesis. This is the first example of a centrin acting as a regulator of a protein kinase and the first known function for centrin separate from its roles in microtubule organizing centers.
Materials and Methods
Strain Constructions and Microbiological Techniques
All yeast strains used in this study are listed in Table I. All standard microbiological techniques and media are described in Rose et al. (1990). β-Galactosidase liquid assays were performed on mid-exponential cultures of MY3366, MY3370, MY3470, MY3473, and MY3582 as described in Rose et al. (1990). For plate dilutions, single colonies were picked and resuspended in 200 μl of sterile H2O in microtiter wells in a 10-fold dilution series. Cells were replica plated to YPD with or without 1 M sorbitol at 23°, 30°, or 37°C and photographed after 4 d.
Table I.
Strains Used in This Study
| Strain | Genotype | |
|---|---|---|
| Y526 | MATα leu2 his3 trp1 gal4 gal80 gal1-lacZ::URA3 | |
| MY3278 | same as Y526 [pMR2716] | |
| MY3279 | same as Y526 [pGBT9] | |
| MY3370 | same as Y526 [pGBT9 + pGAD424] | |
| MY3366 | same as Y526 [pMR2716 + pGAD424] | |
| MY3473 | same as Y526 [pGBT9 + pMR2929] | |
| MY3470 | same as Y526 [pMR2716 + pMR2929] | |
| MY3582 | same as Y526 [pHS14 + pMR2929] | |
| MY3492 | MAT a/MATα lys2-801/+ trp1Δ63/trp1Δ63 leu2/leu2 ura3/ura3 ade2-101/ADE2 his3Δ200/his3Δ200 | |
| MY3605 | same as MY3492 but kic1Δ::URA3/KIC1 | |
| MY3606 | same as MY3492 but kic1Δ::LEU2/KIC1 | |
| MY3636 | MATα ura3-52 leu2-3,112 ade2-101 his3Δ200 trp1Δ63 kic1Δ::LEU2; [pMR3026] | |
| MY4124 | same as MY3492 [pEG(KT), URA3 2μ PGALGST] | |
| MY4125 | same as MY3492 [pMR3030, URA3 2μ PGALGST-KIC1 kinase domain] | |
| MY4126 | same as MY3492 [pMR3041, URA3 2μ PGALGST-KIC1] | |
| MY4127 | same as MY3492 [pMR3141, URA3 2μ PGALGST–kic1-1] | |
| MY3899 | MAT a ura3-52 leu2-3,112 ade2-101 his3Δ200 cdc31-1 [pMR3041] | |
| MS4048 | MAT a ura3-52 leu2-3,112 ade2-101CDC31-16 [pMR3041] | |
| MS10 | MAT a ura3-52 leu2-3,112 ade2-101 | |
| MY3779 | MAT a ura3-52 leu2-3,112 ade2-101 lys2-801 his3Δ200 kic1Δ::LEU2; [pMR3141] | |
| MY4066 | MATα ura3-52 leu2-3,112 ade2-101his3Δ200/trp1Δ63 kic1Δ::LEU2; [pMR3308] | |
| MY4065 | same as MY4066, but [pMR3310] | |
| MY4063 | same as MY4066, but {pMR3311] | |
| MY4064 | same as MY4066, but [pMR3309] | |
| MY6306 | same as MY4066, but [pMR4136] | |
| MY3873 | MAT a ura3-52 leu2-3,112 ade2-101 his3Δ200 cdc31-1 | |
| MY2260 | MATα ura3 ade2 met2 cyhr cdc31-2 | |
| MY2261 | MATα ura3 leu2 ade2 trp1 cyhr cdc31-5 | |
| MS3510 | MATα ura3-52 leu2-3,112 ade2-101CDC31-16 | |
| MY4977 | MATα ura3-52 leu2-3,112 ade2-101 trp1Δ63 kic1Δ::LEU2 cdc31-1; [pMR3141] | |
| MY4123 | same as MY3492 [pMR3071] | |
| MY5493 | same as MY3492 [pMR3071 + pMR2345] |
Y526 was from S. Fields. All other strains were constructed for this study. Strains are listed in the order that they appear in the text.
To construct a kic1Δ strain, pMR3062 was digested with XbaI and transformed into MY3492, a wild-type diploid strain. Genomic DNA from Ura+ transformants was prepared as described in Rose et al. (1990), and the presence of the deletion was confirmed by Southern analysis of the DNA. Tetrad analysis of the diploid (MY3605) determined that KIC1 is an essential gene. A LEU2-marked kic1Δ was generated by transforming a PvuII fragment containing LEU2 obtained from pRS405 (Sikorski and Hieter, 1989) into MY3605 and selecting Leu+ transformants that were Ura−. The presence of the deletion in this strain (MY3606) was confirmed by showing that tetrads segregated 2:2 for viability. In other analyses, the kic1Δ::LEU2 mutant was kept alive by a URA3 plasmid containing wild-type KIC1 (pMR3026). This strain was sensitive to 5-fluoro-orotic acid (5-FOA).
Plasmid Constructions and DNA Manipulations
All DNA manipulations were carried out as described in Sambrook et al. (1989). All enzymes were obtained from New England Biolabs Inc. (Beverly, MA) and were used according to the supplier's specifications. Plasmids are listed in Table II and/or described where appropriate below.
Table II.
Plasmids Used in This Study
| Plasmid | Relevant yeast markers | |
|---|---|---|
| pEG(KT) | URA3 2μ PGALGST | |
| pGAD424 | LEU2 2μ PADHGAL4(768–881) | |
| pGBT9 | TRP1 2μ PADHGAL4(1–147) | |
| pHS14 | TRP1 2μ GAL4–CMD1 | |
| pMR2345 | LEU2 2μ CDC31 | |
| pMR2716 | TRP1 GAL4–CDC31 | |
| pMR2929 | LEU2 GAL4–KIC1 | |
| pMR3026 | URA3 CEN4 ARS1 KIC1 | |
| pMR3030 | URA3 2μ PGAL GST–KIC1 (kinase domain) | |
| pMR3041 | URA3 2μ PGAL GST–KIC1 | |
| pMR3062 | URA3 kic1Δ | |
| pMR3071 | URA3 CEN4 ARS1 PGAL–KIC1 | |
| pMR3141 | URA3 2μ PGAL GST–kic1-1 | |
| pMR3256 | TRP1 CEN4 ARS1 KIC1 | |
| pMR3308 | TRP1 CEN4 ARS1 kic1-4 | |
| pMR3309 | TRP1 CEN4 ARS1 kic1-2 | |
| pMR3310 | TRP1 CEN4 ARS1 kic1-3 | |
| pMR3311 | TRP1 CEN4 ARS1 kic1-5 | |
| pMR4136 | TRP1 CEN4 ARS1 kic1-101 |
pGBT9 and pGAD424 were from S. Fields; pHS14 was from H. Sundberg and T. Davis; pEG(KT) was from R. Deschenes; all other plasmids are from the Rose Laboratory.
Primers were from the Princeton University sequencing facility or from GIBCO BRL (Gaithersburg, MD). A Gal4–Cdc31p fusion plasmid, pMR2716, was constructed by isolating the entire CDC31 gene from pMR2298 (Biggins and Rose, 1994) as a NdeI–SalI fragment, with the NdeI site filled in using the Klenow fragment of DNA polymerase I. This was ligated into plasmid pGBT9 which had been digested with EcoRI, filled in using the Klenow fragment of DNA polymerase I, and then digested with SalI.
The full-length KIC1 gene was isolated from a YCp50 library (Rose et al., 1987) by plating colonies on LB ampicillin plates and then replica printing onto nitrocellulose membranes (Schleicher and Schuell, Keene, NH) as described in Scidmore (1993). Two DNA fragments from the KIC1 gene, a 2.5-kb BamHI–EagI fragment and a 600-bp BamHI–EagI fragment, were labeled by the random primer method (Feinberg and Vogelstein, 1983) and used to probe the membranes by hybridization. Plasmids from candidate positives were restriction mapped to confirm the presence of the full-length KIC1 gene. Two clones were obtained containing the entire KIC1 gene, pMR3015 and pMR3016. Since these plasmids contained >15-kb inserts, smaller subclones were constructed to facilitate further manipulations. The 3.1-kb BamHI fragment from pMR3016 containing the COOH-terminal region of KIC1 was ligated into pRS416 (Sikorski and Hieter, 1989) at the BamHI site. Plasmids were obtained with the fragment in both orientations, pMR3020 and pMR3021. To regenerate the full-length KIC1 gene, pMR3020 was digested with XbaI and SphI and ligated with the 2.9-kb XbaI–SphI fragment containing the NH2-terminal coding region of KIC1 obtained from pMR3015. The resulting plasmid, pMR3026, contains 5.9 kb of DNA containing the 3.9-kb KIC1 gene.
The glutathione-S-transferase (GST)-tagged versions of Kic1p were generated as follows. First, the catalytic domain was PCR amplified using primers 5′ (CCTCTAGACATGACGACGAAGCCAC) and 3′ (GGGAAGCTTCTCCTTTGGATCCTCATC) and plasmid pMR3015 as template. The 5′ primer had an XbaI site (underlined) engineered just upstream of the starting ATG codon and the 3′ primer had a HindIII site engineered (underlined) next to a BamHI site (italics) contained within KIC1 to allow subsequent cloning. The 790-bp PCR product was subsequently digested with XbaI and BamHI and ligated into XbaI–BamHI cut pRS416 (Sikorski and Hieter, 1989). The resulting plasmid, pMR3027, was then digested with BamHI to allow the remaining 3.1 kb of the KIC1 gene obtained from pMR3015 to be ligated in as a BamHI fragment. This yielded the final plasmid, pMR3040, which contains the full-length KIC1 gene with a XbaI site near the ATG. To generate a fusion of GST to full length Kic1p, an ∼4-kb XbaI–HindIII DNA fragment containing the entire KIC1 gene was isolated from plasmid pMR3040 and ligated into XbaI–HindIII cut pEG(KT) (Mitchell et al., 1993) to generate pMR3041. To construct a fusion between GST and the catalytic domain of Kic1p, the 790-bp catalytic domain PCR product was digested with XbaI and HindIII and ligated into the XbaI–HindIII cut vector pEG(KT) to yield plasmid pMR3030.
To generate the weak kic1-1 mutation in the catalytic domain, site-directed mutagenesis was performed on the KIC1 gene. Glutamate 198, which corresponds to the invariant residue in subdomain VIII of kinases (Hanks et al., 1988; Lindberg et al., 1992), was changed to alanine by PCR mutagenesis. An ∼570-bp DNA fragment within the KIC1 catalytic domain was PCR amplified using a 5′ primer, CTTCTAGACATGACGACGAAGCCAC, containing an engineered XbaI site (underlined), and a 3′ primer, CCTTCCATGATCACTGCTGGAGCC. The 3′ primer spanned a BclI site within the KIC1 gene (underlined). The 3′ primer was designed with a mutation, which changed TTC (encoding glutamate), to TGC (encoding alanine, bold). PCR using these primers was performed on pMR3015, the product was subsequently digested with XbaI and BclI, and then ligated into plasmid pMR3040 that had been digested with XbaI and BclI. This generated plasmid pMR3115, a centromere-based plasmid containing the full-length KIC1 gene with a mutation at amino acid 198. To construct a GST fusion containing this mutation, the ∼4-kb XbaI–HindIII fragment was isolated from pMR3115 and ligated into pEG(KT), which had been digested with XbaI and HindIII to generate pMR3141. The plasmids were transformed into yeast strains and protein expression was confirmed by Western blotting with the appropriate antibodies.
To generate the strong kic1-101 mutation in the catalytic domain, site-directed mutagenesis was performed on the KIC1 gene. The ATP binding pocket sequence GRGKFGVV was changed to GRSKFGVG using the oligonucleotide GTTATTGGGCGAAGTAAATTTGGTGTGGGTTATAAGGGCTAT and the dut-ung mutagenesis method (Bio-Rad Laboratories, Hercules, CA). Analogous mutations were sufficient to reduce the kinase activity of skeletal muscle phosphorylase kinase to negligible levels (Lee et al., 1992).
To generate temperature-sensitive mutant alleles, KIC1 was amplified using mutagenic PCR conditions (Leung et al., 1989). For these experiments, KIC1 was subcloned from pMR3026 into pRS414 to make plasmid pMR3256. PCR reactions contained 10 ng of plasmid pMR3256 linearized with EcoRI, 200 ng of forward and reverse primers, 1 mM each dGTP, dCTP, dTTP, 0.2 mM dATP, 3.0–4.0 mM MgCl2, 0.3–1.0 mM MnCl2, and 5 U TaqI DNA Polymerase. For mutagenesis, the coding sequence of the gene was divided into three regions, A, B, and C. Region A was primed with the forward primer (5′-GAGGCAACTCGTCCTACGTGAA) and the reverse primer (5′-GATCGTCTGCTGACAATCTCTC). Region B was primed with the forward primer (5′-GCGCTGGTGGTTCACTGC) and the reverse primer (5′-GCCGGAAACAGATGAGCC). Region C was primed with the forward primer (5′-GGAAGCAGCGGTAGTAACACTG) and the reverse primer (5′-CTTGCGCAGTCGAGTTCCATGT). Reactions were subjected to 30 cycles of 94°C for 1 min, 45°C for 1 min, and 72°C for 2 min. Mutant alleles were recovered according to the gap repair method of Muhlrad et al. (1992). Competent MY3636 cells (containing pMR3026; URA3 CEN KIC1) were co-transformed with PCR product from region A, B or C and pMR3256 (TRP1 CEN KIC1) gapped at the corresponding region. Region A plasmids were gapped with NarI and MscI. Region B plasmids were gapped with BsiWI and MscI. Region C plasmids were gapped with MscI and BstBI. Repair of the gapped plasmids by homologous recombination with the PCR products generated Trp+ CEN plasmids that were selected on medium lacking tryptophan. Trp+ transformants were then transferred to 5-FOA medium to select for Ura− segregants. Strains containing only the pMR3256-PCR mutants were then replica plated to YPD plates at 23°C, at 14°, 23°, 30°, and 37°C to screen for temperature-sensitive mutants.
To generate a kic1 deletion plasmid, the upstream flanking region of KIC1 was isolated as a NarI–XbaI 340-bp fragment from pMR3015 and ligated into ClaI–XbaI cut pRS406 (Sikorski and Hieter, 1989). This plasmid, pMR3029, was digested with XbaI–SacII and 1.0 kb downstream flanking region of KIC1 (containing the last 325 bps of KIC1) was isolated from pMR3021 as a XbaI–SacII fragment. These were ligated to generate pMR3062, a URA3-marked integrating vector which can be digested with XbaI for integration.
Two-Hybrid Screen
The two-hybrid screen was based on the system of Fields and Song (1989). The relevant plasmids are indicated in Table II. Strains were constructed by transformation of plasmids into reporter strain Y526, which contains a GALUAS-lacZ reporter integrated at the URA3 locus. As a preliminary control, MY3278 (Y526 containing the Gal4–Cdc31p fusion) was tested for β-galactosidase activity and none was detected (see Table III), confirming that the Gal4–Cdc31p fusion did not activate on its own. Western blotting of extracts prepared from MY3278 confirmed expression of intact Gal4–Cdc31p fusion protein. To screen for Cdc31p-interacting proteins, MY3278 was transformed with two-hybrid libraries (YL-1, YL-2, and YL-3) obtained from S. Fields (University of Washington, Seattle, WA). Transformants were plated on selective media and grown at 30°C. The colonies were replica printed to X-GAL plates (Rose et al., 1990) at 30°C and scored daily for blue color for 7 d. Colonies that turned blue were colony purified on selective media and then replica printed to X-GAL plates to confirm that they produced β-galactosidase. Colonies that re-tested twice after colony purification were subsequently plated on synthetic complete media lacking leucine, which selected for the library plasmid but not the Gal4–Cdc31p fusion plasmid. The colonies were tested for loss of the Gal4–Cdc31p plasmid, and those that lost the plasmid and maintained the library plasmid were tested on X-GAL plates for β-galactosidase expression. Plasmids that did not turn blue on X-GAL when the Gal4–Cdc31p plasmid was absent were then isolated from yeast by the method of Hoffman and Winston (1987) and amplified in bacteria. The plasmids were retransformed into strain MY3278 (containing Gal4–Cdc31p) and strain MY3279 (containing Gal4) and subsequently tested for β-galactosidase expression. Plasmids that turned strain MY3278 blue and strain MY3279 white on X-GAL plates were considered to be Cdc31p-interacting proteins. Approximately 500,000 colonies were screened and 4 plasmids were isolated. These plasmids were sequenced with a GAL4 primer encoding amino acids 856–861, 5′-TACCACTACAATGGATG. Sequencing was performed using Sequenase version 2.0 (United States Biochemical, Cleveland, OH) according to manufacturer's specifications.
Table III.
Two-Hybrid Interaction between Cdc31p and Kic1p
| Strain | Binding domain | Activation domain | Specific activity | |||
|---|---|---|---|---|---|---|
| MY3370 | Gal4 | Gal4 | 0.1 | |||
| MY3366 | Gal4–Cdc31p | Gal4 | 0.1 | |||
| MY3473 | Gal4 | Gal4–Kic1p | 0 | |||
| MY3470 | Gal4–Cdc31p | Gal4–Kic1p | 141 | |||
| MY3582 | Gal4–calmodulin | Gal4–Kic1p | 0.1 |
Liquid β-galactosidase assays were performed on the indicated strains, which contained various Gal4 DNA–binding domain fusions and activation domain fusions as indicated. Specific activity is expressed as nmoles/min/mg protein (units). The plasmids contained in Y526 are as follows: pGBT9 (Gal4p DNA–binding domain), pGAD424 (Gal4p-activation domain), pMR2716 (Gal4 DNA–binding domain Cdc31p), pMR2929 (Gal4-activation domain Kic1p) and pHS14 (Gal4–calmodulin DNA-binding domain; gift of H. Sundberg and T. Davis).
Protein Techniques
Inducible yeast GST–Kic1p fusion proteins were constructed to confirm that Kic1p interacts with Cdc31p. The NH2-terminal catalytic domain (amino acids 1–263) and the full-length Kic1p protein (amino acids 1–1,080) were both cloned into a yeast GST fusion vector under the control of the galactose-inducible GAL1 promoter (see above). To determine whether these constructs were functional we tested whether they suppressed a deletion of KIC1. Accordingly, the plasmids were transformed into a heterozygous diploid (KIC1/kic1Δ::LEU2) and the transformants were sporulated. All viable spores containing the kic1Δ also contained the full-length GST–Kic1p plasmid, indicating that this fusion can suppress the kic1Δ. In addition, the GST–Kic1p fusion could suppress on either glucose or galactose plates at 30°C and 37°C, suggesting that the fusion must be expressed at high enough levels even in the presence of glucose. In contrast, neither the catalytic domain fusion plasmid nor the GST plasmid could suppress the kic1Δ on glucose or galactose plates.
GST fusion proteins were purified by the method of Lauzé et al. (1995) with slight modifications. Strains were grown in synthetic complete media lacking uracil with 2% raffinose to early exponential phase. The production of the GST fusions was induced by the addition of 2% galactose to the media for ∼4 h. The cells were then harvested and resuspended in breaking buffer (50 mM Tris, pH 7.4, 50 mM NaCl, 1% Triton X-100) and cell extracts were prepared. The extracts were incubated either with glutathione–agarose or with overnight at 4°C. When α-GST antibodies were used, the extracts were centrifuged for 5 min in a microfuge at 4°C to precipitate particulate matter, and the supernatants were then incubated with protein A–Sepharose beads for 2 h at 4°C. The beads were washed 2× in breaking buffer, 2× in breaking buffer with 0.1% Triton X-100, 2× in breaking buffer with 0.02% Triton X-100, and 2× in breaking buffer without detergent. The washed beads were then stored at 4°C and used in subsequent kinase assays. For coprecipitation experiments, the GST–agarose beads were resuspended in sample buffer and the proteins were separated on denaturing polyacrylamide gels. The proteins were transferred to nitrocellulose membranes (Schleicher and Schuell, Inc.) and Western blotted with anti-Cdc31p antibodies to determine whether Cdc31p co-purified with Kic1p. As a control, extracts from cells expressing intact GST were analyzed in parallel. For protein microsequencing, proteins were blotted to PVDF membranes and the sequence determined at the Princeton University Protein Microsequencing Facility. For some Western blot experiments, yeast extracts were prepared by the method of Ohashi et al. (1982). Anti-GST antibodies were obtained from James Broach (Princeton University, Princeton, NJ) or GIBCO BRL and used at a 1:1,000 dilution. Anti-Cdc31p antibodies (Biggins and Rose, 1994) were used at a 1:500 dilution for Western blotting. The gel overlay assays were performed as described in Biggins and Rose (1994).
Kinase Assays
Kinase assays were performed by the method of Lauzé et al. (1995), with slight modifications, using α-GST antibodies to purify the fusion proteins. The assays were performed in a final volume of 25 μl using 5–10 μl of protein A–conjugated beads containing bound GST fusion proteins (∼50 ng of fusion protein). The reactions were preincubated in buffer without ATP (50 mM Hepes, pH 7.4, 0.1 mM MgCl2, 0.5 mM DTT, 1 mM PMSF) sometimes containing either 100 μM CaCl2 or 1 mM EGTA for 30 min at 23° or 37°C. Unlabeled ATP was added to a final concentration of 10 mM and [32P]γ-ATP was added to a final concentration of 0.4 nM. Approximately 0.5 μg of myelin basic protein was added to the reactions. The reactions were incubated at 23° or 37°C for 30 min and terminated by the addition of 10 μl of 2× sample buffer. The samples were separated on denaturing polyacrylamide gels (the unincorporated radioactivity was cut from the bottom of the gel), stained in Coomassie stain (Sambrook et al., 1989), and then dried and autoradiographed at −70°C. Chemicals were obtained from Sigma (St. Louis, MO) and isotopes were obtained from DuPont-NEN (Boston, MA). Kinase assays were quantified using a Molecular Devices PhosphorImager (Sunnyvale, CA).
Microscopic Analysis
For cell cycle analysis, strains were grown to mid-exponential phase in YPD at 23°C and then shifted to YPD with or without 1 M sorbitol. After 4 or 8 h, cells were harvested and fixed with 3:1 methanol/acetic acid. Cells were stained with 10 μg/ml DAPI (4′,6′-diamidino-2-phenylindole) for 30 min at room temperature and analyzed using DIC (differential interference contrast) or fluorescence microscopy. For actin staining, cells were fixed by adding formaldehyde directly to the culture to a final concentration of 4% and incubating at 23°C for 1 h. Fixed cells were permeabilized by incubation with 25 μg/ml Zymolyase 100,000 (ICN Pharmaceuticals Inc., Costa Mesa, CA) in 1.2 M sorbitol, 0.1 M potassium phosphate, pH 7.5, and 25 mM β-mercaptoethanol at 37°C for 30 min. Permeabilized cells were stained for immunofluorescence as described by Kilmartin and Adams (1984). Anti-actin antibodies B28 were a gift from T. Wang and A. Bretscher (Cornell University, Ithaca, NY) and used at a dilution of 1:50. FITC-conjugated goat anti–rabbit antibody was obtained from Boehringer Mannheim Biochemicals (Indianapolis, IN) and used at 1:25 dilution. DAPI was obtained from Accurate Chemicals and Scientific Corp. (Westbury, NY).
For electron microscopy cells were grown to mid-exponential phase at 23°C, shifted to 37°C for 8 h, and then fixed and stained as described in Gammie et al. (1998).
Results
A Two-Hybrid Screen to Isolate Cdc31p-interacting Proteins
A two-hybrid screen was performed to isolate potential Cdc31p-interacting proteins. The full-length CDC31 gene was fused to the DNA binding domain of the GAL4 gene to generate an in-frame fusion. A library of plasmids containing random genomic fusions to the Gal4p-activating domain was transformed into a strain expressing Gal4– Cdc31p. Four potential Cdc31p interacting clones were isolated. Three of the plasmids did not contain gene fusions but the fourth corresponded to a gene called NRK1, previously deposited in Genbank/EMBL/DDBJ by Y. Fukami (Kobe, Hyogo, Japan). No information about the function of the gene had been published; however its sequence indicates that it is an asparagine-rich kinase. We therefore named the gene KIC1 to indicate its role as a kinase that interacts with Cdc31p. KIC1 corresponds to the open reading frame YHR102W identified by the yeast Genome Sequencing Project.
Liquid β-galactosidase assays were performed to confirm the interaction between Cdc31p and Kic1p. As reported in Table III, neither the Gal4–Cdc31p fusion nor the Gal4–Kic1p fusion plasmids caused any β-galactosidase expression on their own. Expressed in a strain together, they produced 141 units of β-galactosidase activity, >1,000 times above the background. Because of the the homology between Cdc31p and calmodulin, we also tested whether the Kic1p kinase interacted with calmodulin by the two-hybrid test. A yeast Gal4–calmodulin fusion did not confer any β-galactosidase activity when coexpressed with Gal4–Kic1p (see Table III), indicating that the Kic1p interaction was specific to Cdc31p.
Kic1p Is an Essential PAK Kinase
The full-length KIC1 gene was isolated from a genomic yeast library by hybridization with a DNA probe from the two-hybrid clone. An URA3-marked deletion of the gene was constructed in a diploid by a one-step gene replacement (Rothstein, 1991). Tetrad analysis of the heterozygous diploid (MY3605) demonstrated that KIC1 is essential for viability. Microscopic examination of the inviable spores determined that they contained four or eight cells, indicating that KIC1 does not affect germination. In addition, an equivalent kic1Δ::LEU2 heterozygous diploid strain was constructed (MY3606). Leu+ haploid spores from this diploid (e.g., MY3636) were kept alive by a URA3 plasmid containing wild-type KIC1. MY3636 was inviable when the KIC1 plasmid was lost, over a wide range of temperatures (14°, 23°, and 37°C) on both rich (YPD) and synthetic defined media (see Materials and Methods). We therefore concluded that KIC1 is an essential gene.
The NH2-terminal region of Kic1p contains a 280–amino acid domain designated the “catalytic” domain (see Fig. 1) because it contains every residue conserved in protein kinases (Hanks et al., 1988; Lindberg et al., 1992). The putative ATP-binding domain consists of the GXGXXG sequence motif at position 30 and invariant lysine and aspartate residues at positions 52 and 144, respectively (Hanks et al., 1988). Kic1p is predicted to be a serine/ threonine kinase based on the presence of the sequences DIKAAN (amino acids 144–149) and GTPYWMAPE (amino acids 181–189). In addition to the NH2-terminal catalytic domain, Kic1p contains an 800–amino acid COOH-terminal tail that does not display homology to any other proteins in the database.
Figure 1.


Kic1p kinase catalytic domain. (A) The Kic1p-kinase domain is aligned with its closest homologues: SOK1/Ysk1 (Pombo et al., 1996), CELT19A5 (Wilson et al., 1994), NIK1 (Su et al., 1997), MST1 (Creasy and Chernoff, 1995), PAK1 (Brown et al., 1996), and Ste20p (Leberer et al., 1992). The length in amino acids is indicated at the end of the bars. The percentage of identical residues with the Kic1p kinase domain is shown in the far right. (B) The NH2-terminal amino acids of Kic1p are aligned with the catalytic domains of MST1 from H. sapiens, PAK1 from H. sapiens, and Ste20p from S. cerevisiae. Identical residues are indicated by the black boxes and amino acid number is indicated on the right. The 11 subdomains conserved in all kinases are indicated (Hanks et al., 1988; Lindberg et al., 1992). A short domain of additional homology is marked by asterisks above and below the text.
Homology searches indicate that the kinase domain of Kic1p shares between 43% and 52% homology with members of the extended PAK/Ste20 kinase family (Fig. 1 A). Kic1p, SOK1, CELT19A5, MST1, MST2, and Ste20p, are examples of PAKs that share a common 11–amino acid motif (AKPXSILXD/ELI) just COOH-terminal to the kinase domain (Fig. 1 B). This domain is unique to PAK kinases and is similar to a recently identified Ste20/PAK kinase sequence shown to interact with G protein β-subunits (Leeuw et al., 1998). Beyond these regions, Kic1p shows no significant homology to any other known proteins.
Despite the abundance of PAK kinases, relatively little is known about their specific functions. SOK1/YSK1 is a human kinase that is activated three- to sevenfold by oxidant stress (Pombo et al., 1996; Osada et al., 1997). CELT19A5 is a putative open reading frame (ORF) identified in the C. elegans sequencing project (Wilson et al., 1994). NIK1 is a mouse kinase that interacts with the SH3 domains of Nck, an adapter protein that is the common target for a number of cell surface receptors (Su et al., 1997). MST1 and MST2 are human kinases that are not involved in the regulation of any known mammalian MAPK pathway, but MST1 activity is apparently stimulated by protein phosphatase 2A (Creasy and Chernoff, 1995). Perhaps more relevant to Kic1p function, PAK1, MUK2, mPAK-3, and β-PAK (Manser et al., 1994, 1995; Bagrodia et al., 1995; Brown et al., 1996; Osada et al., 1997) are all thought to be activated by p21/CDC42/Rac and are implicated in rearrangements of the actin cytoskeleton. Interestingly, hPAK1 and mPAK-3 can both substitute for Ste20p in yeast (Bagrodia et al., 1995; Brown et al., 1996). Ste20p is involved in signal transduction during the yeast pheromone activated mating response (Ramer and Davis, 1993; Herskowitz, 1995). Although Ste20p and the PAKs contain p21/CDC42/Rac-binding sites, there are no predicted binding sites in Kic1p and the other Ste20-like kinases. In S. cerevisiae there are at three additional Ste20p-related protein kinases (for review see Hunter and Plowman, 1997): Sps1p, which is induced during meiosis and required for spore wall formation; Cla4p, which is required for cytokinesis; and Skm1p, whose function is unclear but which may be involved in morphogenesis. Kic1p and Sps1p are more closely related to each other than to Ste20p (45% identity over the catalytic domain) and share a similar NH2-terminal location for the kinase domain. The two other protein kinases are more distantly related to Kic1p and, like Ste20p, have Cdc42-binding sites and COOH-terminal catalytic domains.
Kic1p Interacts Directly with Cdc31p
The two-hybrid analysis suggested that Cdc31p and Kic1p formed a complex in vivo. We wanted to confirm that these proteins interacted by other methods. Inducible GST–Kic1p fusion plasmids were transformed into wild-type strains and protein extracts were prepared using glutathione–agarose. We found that Cdc31p copurified with both the Kic1p catalytic domain fusion (amino acids 1–263) and the full-length Kic1p fusion (Fig. 2 A). The interaction was specific because Cdc31p did not coprecipitate with GST alone (Fig. 2 A). In an attempt to determine the relative amounts of each protein in the complex, extracts from wild-type cells (not expressing GST–Kic1p) were immunoprecipitated with α-Cdc31p antibodies. The majority of the endogenous Kic1p protein coprecipitated with Cdc31p, as determined by Western blotting with α-Kic1p antibodies (data not shown). These results indicate that a significant fraction of Kic1p is associated with Cdc31p in vivo and therefore the interaction is likely to be functionally relevant.
Figure 2.

Cdc31p binds Kic1p directly and is part of a larger complex. (A) Cdc31p coprecipitates with Kic1p. GST, GST catalytic domain, and GST full-length Kic1p fusion proteins were purified from yeast (strains MY4124, MY4125, and MY4126, respectively) using glutathione–agarose under native conditions, separated on denaturing gels, and then Western blotted with anti-Cdc31p antibodies. Cdc31p band is indicated for part A and B. (B) Cdc31p interaction with Kic1p is calcium independent. The full-length Kic1p fusion protein was purified from MY4126 in the presence of 10 mM EGTA or 10 mM calcium. Equal amounts of purified Kic1p were run on a gel and Western blotted with anti-Cdc31p antibodies. (C) Coomassie staining of the pure GST–Kic1p fusion proteins as well as copurifying proteins. Purified GST (lane 1, strain MY4124), GST–catalytic domain (lane 2, strain MY4125), and GST full-length fusion proteins (lane 3, MY4126) were separated on an 8% denaturing gel. Lane 4 contains the total yeast extract from MY4126 expressing GST fused to full-length Kic1p. Copurifying peptides of 200, 80, 70, and 40 kD are indicated with asterisks. The GST protein is not observed because it is <30 kD and therefore ran off the gel. Higher percentage gels were run to monitor GST purification (not shown). M, protein molecular weight markers. (D) Anti-GST Western blotting showing full-length GST–Kic1p and several degradation products. Lanes marked 1 and 3 are as in C. (E) Cdc31p binds to full-length GST– Kic1p and its degradation products. Fusion proteins were transferred to membrane and a gel overlay was performed with radiolabeled Cdc31p (Biggins and Rose, 1994). Lanes are marked as in C.
Since Cdc31p is a calcium-binding protein (Spang et al., 1993; Biggins and Rose, 1994), we tested whether the interaction with Kic1p depended on the presence of calcium. Full-length GST–Kic1p fusion was precipitated using glutathione–agarose in the presence of either 10 mM calcium or 10 mM EGTA. We found that equivalent amounts of Cdc31p copurified with Kic1p in the presence of EGTA or calcium (Fig. 2 B), indicating that the Kic1p–Cdc31p interaction is calcium independent.
To determine whether Kic1p and Cdc31p were part of a larger complex in vivo, we examined the precipitates by Coommassie blue staining (Fig. 2 C). Several additional proteins coprecipitated with the GST–Kic1p fusions but not with GST alone (Fig. 2 C). Three major bands coprecipitated with both the full-length and the catalytic domain fusion, migrating at 40, 70, and 80 kD (Fig. 2 C). In addition, a minor protein of 200 kD also coprecipitated with the full-length fusion protein. We performed peptide microsequencing on the coprecipitating bands. The 70- and 200-kD proteins were NH2-terminally blocked. The 80-kD protein was determined to be the heat shock protein Hsc82p (Borkovich et al., 1989), a member of the Hsp90 class. It is of interest that an Hsp90p also coprecipitates with centrin purified from Xenopus extracts (Uzawa et al., 1995), suggesting that centrins may interact with Hsp90s in all eukaryotes. By Western blotting we determined that the additional bands present in the full-length GST–Kic1p fusion lanes were proteolytic fragments (see Fig. 2 D, lane 3).
Although Kic1p and Cdc31p interact by the two-hybrid test and coimmunoprecipitation, these assays do not distinguish whether the interaction is via a direct or indirect interaction. Therefore, a gel blot overlay assay was performed. This technique was successful in detecting Cdc31p binding to the Kar1 fusion proteins expressed in yeast and in bacteria (Biggins and Rose, 1994; Spang et al., 1995). Both the catalytic domain and full-length GST–Kic1p fusions were purified from yeast under native conditions (Fig. 2 E) and binding to radiolabeled Cdc31p purified from bacteria was assayed. We found that Cdc31p bound directly to the full-length GST–Kic1 protein (and proteolytic fragments), but not to the Kic1p catalytic domain (Fig. 2 E). Cdc31p binding to GST–Kic1p was also detected in a total yeast protein extract expressing the GST– Kic1p fusion (Fig. 2 E, lane 4). In addition, Cdc31p bound to a 116-kD protein band, which might correspond to wild-type Kic1p (Fig. 2 E, lane 4). In the control experiment, Cdc31p did not bind to purified GST alone (data not shown). Because Cdc31p did not bind directly to the catalytic domain fusion in the gel blot experiment, the Cdc31p-binding site is most likely COOH-terminal to residue 263. This is consistent with the Gal4p fusion protein, which starts at amino acid 260 of the kinase. Interestingly, adjacent to the kinase domain is a stretch of 14 amino acids, LKELISRYLLFRDK, that is 64% similar (50% identical) to the Cdc31p-binding domain described for Kar1p, LIESKWHRLLFHDK (Vallen et al., 1994; Spang et al., 1995). This sequence is just COOH-terminal to, and partially overlaps, the potential G protein β-subunit consensus (Leeuw et al., 1998) described above (Fig. 1 B).
Kic1p Has Kinase Activity In Vitro, Which Depends upon Cdc31p
To confirm that Kic1p was a kinase, we tested whether Kic1p had kinase activity in vitro. The GST–Kic1p fusion proteins were purified from yeast using α-GST antibodies and tested for kinase activity using myelin basic protein (MBP) as substrate. We found that GST–Kic1p was associated with a high level of protein kinase activity (Fig. 3 A). The level of activity was the same in either 1 mM EGTA or 100 μM calcium, indicating that the Kic1p kinase activity is not regulated by calcium levels. The purified GST protein control exhibited only a slight background kinase activity (Fig. 3 A).
Figure 3.

GST–Kic1p kinase activity is Cdc31p-dependent, but calcium-independent. (A) GST–Kic1p and GST alone were expressed in yeast (strains MY4126 and MY4124, respectively) and purified under native conditions by immunoprecipitation with anti-GST antibodies. The kinase activity of GST–Kic1p and GST preparations was assayed at 23°C using γ-labeled ATP. The conditions used are shown above the gel with the addition (+) or omission (−) of the following: the test substrate MBP (myelin basic protein), Mg2+, Ca2+ or EGTA. (B) The kinase activity in the reactions is Kic1p specific. GST, GST-Kic1p, and GST-Kic1-1p were expressed in yeast (strains MY4124, MY4126, and MY4127, respectively) and purified under native conditions by immunoprecipitation with anti-GST antibodies. Kinase assays were performed in standard kinase buffer (see Materials and Methods) using equal amounts of GST–Kic1p or GST–Kic1-1p (proteins amounts were normalized by Coomassie staining and anti-GST Westerns). Protein preparations were assayed for kinase activity at 23°C and 37°C. Both autophosphorylation (auto-P) of GST-Kic1p and phosphorylation of MBP are shown. (C) Cdc31p function is required for GST–Kic1p kinase activity. GST was purified from a CDC31 strain (MY4124) and GST-Kic1p was purified from CDC31, cdc31-1, and CDC31-16 strains (MY4126, MY3899, and MS4048, respectively) under native conditions by immunoprecipitation with α-GST antibodies. Strains containing either cdc31-1 or CDC31-16 are temperature sensitive for growth at 37°C. Equivalent amounts of GST–Kic1p kinase were used in each assay. Phosphorylation of MBP is shown. GST–Kic1p kinase activity is reduced in the presence of mutant Cdc31-1p or Cdc31-16p at 23°C and is at background levels at 37°C. Kinase activities were quantified by PhosphorImager analysis (see Materials and Methods).
A GST–Kic1 mutant protein was constructed to determine whether the kinase activity was due to Kic1p or another copurifying kinase. Site-directed mutagenesis was performed on the KIC1 gene to change the conserved glutamate in subdomain VIII of the catalytic region (amino acid 189) to an alanine. The corresponding mutation in cAMP-dependent protein kinase reduced but did not eliminate kinase activity (Gibbs and Zoller, 1991). This G189A mutation was made in the GST–Kic1p plasmid and the mutant protein was designated GST–Kic1-1p. Equal amounts of wild-type GST–Kic1p and GST–Kic1-1p were purified from strains grown at 23°C and tested for autophosphorylation and MBP phosphorylation at 23° and 37°C (Fig. 3 B). The mutant GST–Kic1-1p fusion protein showed reduced kinase and autophosphorylation activity at 23°C and kinase activity that was further reduced at 37°C (Fig. 3 B). Therefore, the kinase activity could be attributed to GST–Kic1p and not to a coprecipitating kinase.
Since Kic1p interacts with Cdc31p we wished to determine if the kinase activity required functional Cdc31p. To test this we purified wild-type GST–Kic1p from CDC31, cdc31-1, and CDC31-16 strains. Both cdc31-1 and CDC31-16 strains are temperature sensitive for growth at 37°C. GST–Kic1p fusions were purified from each of these strains at the permissive temperature and then GST– Kic1p kinase activity was assayed as above (Fig. 3 C). GST–Kic1p purified from both cdc31-1 and CDC31-16 strains showed reduced kinase activity at 23°C (∼20% and 40%, respectively compared with the wild type) and activity was further reduced to background levels at 37°C (Fig. 3 C). Therefore we concluded that Kic1p kinase activity is dependent upon Cdc31p.
KIC1 Is Required for Cell Integrity and Morphogenesis
To understand the functional role of KIC1 and its interactions with CDC31, we generated additional temperature-sensitive alleles. Random PCR mutagenesis of KIC1 produced 58 different temperature-sensitive alleles. The mutants were divided into three classes. Class A mutants grew poorly at 37°C. Class B mutants showed no growth at 37°C, but the growth defect was suppressed by 1 M sorbitol (an osmotic stabilizer). Class C mutants failed to grow at 37°C but were not suppressed by 1 M sorbitol. Two examples of each class are shown in Fig. 4 in order of their severity, class A being the least severe and class C the worst.
Figure 4.

kic1 mutants fall into three classes. Wild-type (MS10), kic1-1 (MY3779), kic1-4 (MY4066), kic1-3 (MY4065), kic1-5 (MY4063), kic1-2 (MY4064), and kic1-101 (MY6306) strains were serially diluted and aliquots were spotted onto YPD or YPD containing 1 M sorbitol media (+ sorbitol) and incubated at the indicated temperatures. At 37°C, class A mutants grew poorly compared with the wild type. Class B mutants fail to grow at 37°C but the growth defect was suppressed by the addition of 1 M sorbitol to the media. Class C mutants failed to grow at 37°C in both the presence and absence of 1 M sorbitol.
Surprisingly, the kinase-down mutant kic1-1 (Fig. 3 B) was found to be a member of the least severe mutant class (class A), growing poorly at 23° and 37°C, but like wild type at 30°C (data not shown). To determine the phenotype of a mutant with a more severe defect in kinase activity, we constructed a mutation in the ATP-binding pocket that is expected to completely block kinase activity (based on Lee et al., 1992). This mutation, designated kic1-101, was expressed from a CEN plasmid and found to partially complement a complete deletion of kic1. Although viable, the kic1-101 mutant exhibits the most severe defect among our collection of mutants, apart from the deletion (Fig. 4). The Gal4–Kic1p fusion plasmid isolated from the two-hybrid screen also complemented a complete deletion of kic1 and resulted in a mutant phenotype indistinguishable from kic1-101. This construct contained only the last 20 amino acids of the kinase domain and the entire COOH-terminal portion of the Kic1p protein. These results demonstrate that the kinase activity of Kic1p is critically important, although not essential, for normal cell function. In addition, they suggest that the non-kinase domain may have a separate important role in cell growth.
Cell cycle analysis demonstrated that the kic1 mutations resulted in multiple severe morphological defects. Mutants from each class were found to have similar phenotypes. For simplicity, only one mutant from each class is described. The kic1-1 (class A), kic1-3 (class B), and kic1-2 (class C) strains were shifted to 37°C for 8 h, and then analyzed microscopically (Table IV). Wild-type strains were found to have the same cell cycle distributions at 23° and 37°C in both the presence and absence of 1 M sorbitol. Accordingly, only the data for wild type at 23°C in the absence of 1 M sorbitol is shown (Table IV, line 1). In contrast to the wild type, the class A kic1-1 mutant grew in chains and clusters of cells, each of which contained a nucleus (Table IV, lines 2–4; Fig. 5). Many of the cells had abnormally wide bud necks (Fig. 5, arrows), and three percent of the cells had lysed (Table IV, line 3). These results suggested that the kic1 class A mutations resulted in defects in cell wall assembly and cell separation. The more severe class B kic1-3 mutant had a strong lysis phenotype at 37°C as evidenced by the frequent presence of cell “ghosts” and cell debris (Fig. 5, asterisks; Table IV, lines 5–7). The inviability and lysis defect of the kic1-3 mutant was suppressed by the presence of 1 M sorbitol. Similar to the class A mutants, unlysed kic1-3 cells exhibited wide bud necks at 37°C. When the lysis defect was osmotically suppressed with sorbitol, the kic1-3 mutant took on the appearance of a class A mutant, growing in clusters and chains (Fig. 5). The kic1-3 mutant cultures also contained cells with small buds that had completed anaphase (divided nuclei, Table IV, line 6). The observation that there was no accumulation of binucleate cells with large buds suggested that the mutants might be defective for bud growth. The class C kic1-2 mutant behaved very much like kic1-3 at 37°C except that its growth defect was not suppressed by the addition of 1 M sorbitol (Fig. 5; Table IV, lines 8–10). In the presence of sorbitol, the kic1-2 mutants were larger and round in appearance but most cells were lysed (Fig. 5, asterisks).
Table IV.
Cell Cycle Analysis of WT, cdc31, kic1, and cdc31-1 kic2-2 Strains
| Strain | temp |

|
lysed | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| °C | ||||||||||||||||||
| (1) | WT | 23 | − | 33 | 35 | 0 | 7 | 25 | 0 | |||||||||
| (2) | kic1-1 | 23 | − | 40 | 29 | 0 | 9 | 22 | 0 | |||||||||
| (3) | 37 | − | Clusters & chains | 3 | ||||||||||||||
| (4) | 37 | + | Clusters & chains | 0 | ||||||||||||||
| (5) | kic1-3 | 23 | − | 21 | 39 | 1 | 4 | 27 | 0 | |||||||||
| (6) | 37 | − | 13 | 8 | 12 | 2 | 3 | 62 | ||||||||||
| (7) | 37 | + | Clusters & chains | 0 | ||||||||||||||
| (8) | kic1-2 | 23 | − | 28 | 36 | 0 | 8 | 28 | 0 | |||||||||
| (9) | 37 | − | 29 | 4 | 8 | 1 | 2 | 57 | ||||||||||
| (10) | 37 | + | 22 | 2 | 2 | 8 | 2 | 54 | ||||||||||
| (11) | cdc31-1 | 23 | − | 43 | 31 | 0 | 9 | 17 | 0 | |||||||||
| (12) | 37 | − | 9 | 2 | 0 | 89 | 0 | 0 | ||||||||||
| (13) | 37 | + | 22 | 9 | 0 | 67 | 2 | 0 | ||||||||||
| (14) | cdc31-2 | 23 | − | 33 | 41 | 0 | 6 | 20 | 0 | |||||||||
| (15) | 37 | − | 15 | 4 | 0 | 72 | 0 | 9 | ||||||||||
| (16) | 37 | + | 22 | 21 | 0 | 54 | 3 | 0 | ||||||||||
| (17) | cdc31-5 | 23 | − | 40 | 35 | 0 | 5 | 20 | 0 | |||||||||
| (18) | 37 | − | 4 | 1 | 0 | 39 | 1 | 55 | ||||||||||
| (19) | 37 | + | 30 | 6 | 0 | 30 | 2 | 32 | ||||||||||
| (20) | CDC31-16 | 23 | − | 32 | 34 | 0 | 9 | 25 | 0 | |||||||||
| (21) | 37 | − | 30 | 7 | 0 | 61 | 2 | 3 | ||||||||||
| (22) | 37 | + | 29 | 16 | 0 | 53 | 2 | 0 | ||||||||||
| (23) | cdc31-1 kic1-1 | 23 | − | 25 | 27 | 0 | 20 | 28 | 0 | |||||||||
| (24) | 37 | − | 25 | 14 | 0 | 3 | 5 | 53 | ||||||||||
| (25) | 37 | + | 21 | 20 | 0 | 12 | 7 | 40 | ||||||||||
Wild-type (MS10), kic1-1 (MY3779), kic1-3 (MY4065), kic1-2 (MY4064), cdc31-1 (MY3873), cdc31-2 (MS2260), cdc31-5 (MS2261), CDC31-16 (MS3510), and cdc31-1, kic1-1 (MY4977) strains were grown to early exponential phase and shifted to the indicated media and temperature for the times shown. Cells were then fixed in 3:1 methanol/acetic acid and stained with the DNA-specific dye DAPI. In each case 200 cells were examined. Where “Clusters & chains” is indicated, cell cycle analysis could not be performed.
Figure 5.

Images of wild-type and kic1 strains. Wild-type (MS10), kic1-1 (MY3779), kic1-2 (MY4064), and kic1-3 (MY4065) strains were incubated in YPD at 37°C for 8 h, with (+) or without (−) 1 M sorbitol and analyzed microscopically. The asterisks indicate lysed cells or lysed cell contents. Single arrows indicate wide bud necks.
Taken together, the cell cycle analysis suggested that KIC1 is required for some aspect of morphogenesis and to maintain cell integrity. To characterize this further we examined the mutants by electron microscopy. The cell walls in wild type are trilaminate in appearance (for review see Cid et al., 1995). The plasma membrane–cell wall interface stains darkly, whereas the β1-3 glucan–rich middle layer is lighter in appearance. The outer protein-rich mannan layer also stains darkly (Fig. 6). In cross section, the primary septum (PS), composed of chitin, can be seen at the bud neck. Under these conditions, chitin is very electron translucent and typically appears as a thin white line. Surrounding the primary septum at the outer periphery of the bud neck is a ring of chitin, which appears as two white crescents in cross section (Fig. 6, B and D).
Figure 6.
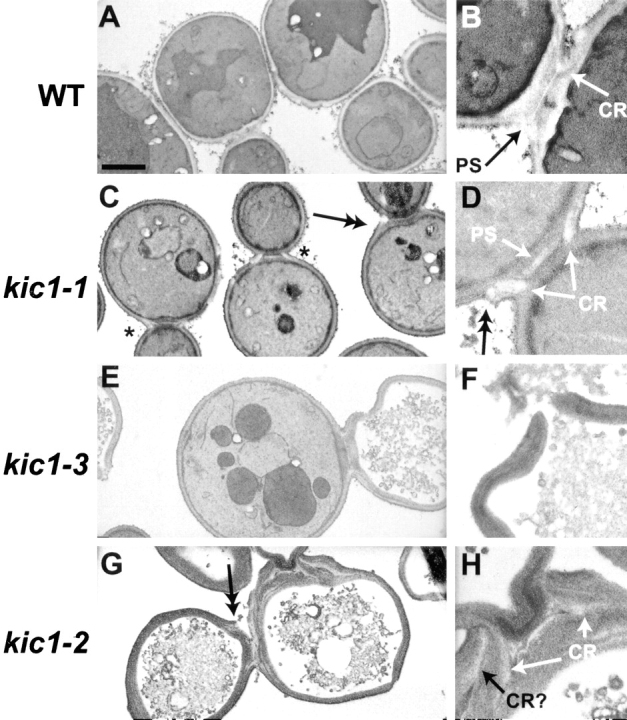
Electron microscopic images of wild-type and kic1 strains. Wild-type (MS10), kic1-1 (MY3779), kic1-2 (MY4064), and kic1-3 (MY4065) strains were grown to exponential phase at 23°C and then shifted to 37°C for 8 h before being fixed and stained for electron microscopy as described in Gammie et al. (1998). Cells in the center and right columns are magnified 3.2× compared with the left column. Chitin rings (CR) and primary septa are indicated (PS). “Frayed” or discontinuous cell walls are indicated with double arrows. Bar in left column, 2 μm.
Electron microscopic analysis of the kic1 mutants revealed a series of distinct cell wall assembly defects. The class A kic1-1 mutant had a noticeable increase in electron translucent material, presumably chitin, at the bud neck (Fig. 6 C, asterisks). The extra material was often associated with a “frayed” appearance of the cell wall at or near the bud neck (Fig. 6, C and D, double arrows). The primary septa in these cells were thicker than wild type (Fig. 6 D, PS). In some cases the primary septum appeared as two white lines instead of one.
The cell wall defects of the class B and C mutants, kic1-3 and kic1-2, were more severe than that of kic1-1. Many cells were lysed and the cell walls were grossly abnormal (Fig. 6, E–H). Instead of the normal trilaminar appearance, the mutant cell walls were thicker and uniformly electron dense (most apparent in Fig. 6, F and H). They lacked the distinct electron translucent β-glucan layer seen in wild-type cells. The bud necks of class B and C mutants were also wider and thicker than in wild type. Many had multiple layers of electron-translucent material, presumably chitin, at their necks (Fig. 6, G and H). The cell walls also had a frayed appearance near the bud necks (Fig. 6 G, double arrows). These observations are in contrast with the phenotype of PKC1 mutants. PKC1 mutants lyse at a point of visible thinning of the cell wall, usually at the tips of small buds. In contrast, the kic1 mutants showed no visible thinning of the cell wall at the lysis point (Fig. 6 F) and lysis typically occurred near the bud necks (data not shown).
CDC31 Is Also Required for Morphogenesis and Cell Integrity
We were surprised to find that the kic1 mutants exhibited phenotypes that were not like the published phenotypes of cdc31 mutants (Schild et al., 1981; Baum et al., 1986; Vallen et al., 1994). The cdc31 mutants arrest as large-budded cells with unduplicated spindle poles. In contrast, after shift to 37°C, the kic1 mutants grew as chains of cells, each containing a nucleus, indicating they had neither a SPB duplication defect nor underwent cell cycle arrest. Since CDC31 is required for Kic1p kinase activity (Fig. 3) and the kinase mutations exhibit morphological defects, we re-examined the morphology of four cdc31 mutants at the non-permissive temperature. The cdc31-1, cdc31-2, cdc31-5, and CDC31-16 strains were shifted to 37°C for 8 h and analyzed microscopically. As previously published, the cdc31 mutants were found to be defective in SPB duplication, as evidenced by the accumulation of mononucleate large-budded cells (Table IV, lines 11–22). However, the cdc31 mutants also exhibited distinct allele-specific morphological defects at the nonpermissive temperature (Fig. 7).
Figure 7.

Morphological defects of cdc31 strains. The cdc31-1 (MY3873), cdc31-2 (MS2260), cdc31-5 (MS2261), and CDC31-16 (MS3510) strains were incubated in YPD 37°C for 8 h and analyzed microscopically. Single asterisks, lysed cells; double asterisks indicate cell debris; double arrows, long narrow bud.
Most strikingly, the cdc31-5 mutant showed a severe lysis defect similar to that of the class B and C kic1 mutants (Table IV, lines 17–19; Fig. 7). Lysis was weakly suppressed by the addition of 1 M sorbitol (Fig. 7; Table IV, line 19). The cdc31-2 and CDC31-16 mutants also exhibited weak lysis phenotypes, which were suppressed by 1 M sorbitol (Table IV, lines 14–16, 20–22). In Fig. 7, asterisks mark examples of lysed cells, and double asterisks mark cell debris. In addition to the lysis defect, both the cdc31-1 and cdc31-2 mutants arrested with an unusual bud morphology; ≤50% of the arrested cells had long narrow buds (Fig. 7, double arrows; and Table IV, lines 11–16). This phenotype was also partially suppressed by the presence of 1 M sorbitol.
Neither the long bud morphology nor lysis has been reported for mutants with defects in spindle assembly that cause a G2/M arrest. We therefore re-examined the large bud arrest phenotype of several yeast β-tubulin mutants (Huffaker et al., 1988) and found no evidence of either elongated buds or cell lysis (data not shown). Furthermore, the long bud phenotype did not lead to lysis in cdc31-1 (Table IV, lines 11–13; Fig. 7), nor did lysis of cdc31-5 and CDC31-16 require the presence of long buds (Table IV, lines 17-22; Fig. 7). From these data, we concluded that CDC31 is also required for cell morphology and integrity.
CDC31 and KIC1 Interact In Vivo
We generated a kic1-1 cdc31-1 double mutant to look for genetic interactions between KIC1 and CDC31. Neither kic1-1 nor cdc31-1 exhibited lysis at 37°C. We reasoned that if the two proteins interact in vivo, then by combining two mild mutations we should be able to generate the lysis defect of the most severe single mutants. When the cdc31-1 kic1-1 strain was shifted to 37°C it showed a strong lysis defect that was only weakly suppressed by the addition of 1 M sorbitol (Table IV, lines 23–25; Fig. 8). Thus the double mutant phenotype was equivalent to the severe class C kic1 mutant. These results strongly suggest that Kic1p and Cdc31p interact in vivo, and that their combined functions are required for normal cell integrity.
Figure 8.

CDC31 and KIC1 interact in vivo. (A) cdc31-1 kic1-1 double mutants have a synthetic lysis defect. The cdc31-1 kic1-1 strain (MY4977) was incubated in YPD at 37°C for 8 h in the presence (+) or absence (−) of 1 M sorbitol and analyzed microscopically. Asterisks, lysed cells. (B) The elongated bud defect of Kic2p overexpression is suppressed by increasing the expression of Cdc31p. Overexpression of the Kic1p (Kic1p O/E) protein induces elongated bud growth (−CDC31 2μ, strain MY4123) that is suppressed when CDC31 is expressed off a high copy vector (+CDC31 2μ, strain MY5493).
The interaction between Kic1p and Cdc31p was further examined by co-overexpression in wild-type cells. When Kic1p was overexpressed, the cultures were viable but grew with elongated cell morphology (Fig. 8) very reminiscent of cdc31-1 and cdc31-2 mutants at 37°C (Fig. 7). When Cdc31p was co-overexpressed with Kic1p the cells regained the normal rounded cell morphology (Fig. 8). These results support the view that Kic1p and Cdc31p interact to affect cell morphology.
kic1 Mutants Are Defective for Actin Localization
The cell lysis and abnormal bud phenotypes of kic1 mutants prompted us to look at their actin distribution. During vegetative growth, actin function and localization is required for normal cell polarity and bud growth (for review see Ayscough and Drubin, 1996). Wild-type cells show a distinct actin localization pattern during the cell cycle (Fig. 9). Actin patches are directed to the bud site and become highly concentrated in the emerging small bud. In large budded cells before cytokinesis, actin redistributes to the bud neck.
Figure 9.

kic1 and cdc31 strains have actin localization defects. Wild-type (MS10), kic1-1 (MY3779), kic1-3 (MY4065), kic1-2 (MY4064), cdc31-1(MY3873), and cdc31-1 kic1-1 (MY4977) strains were grown to exponential phase at 23°C, shifted to 37°C for 4 h, and then prepared for immunofluorescence as described in Material and Methods. A representative gallery of cells (unbudded, budded, and large budded) is shown for each strain. Wild-type (WT) cells show a characteristic actin localization pattern during the cell cycle. Unbudded WT cells show no actin concentration. Actin becomes highly concentrated in the emerging small bud of WT cells. Medium-budded WT cells show staining primarily in the bud tip. In WT large-budded cells before cytokinesis, actin redistributes to the septum between the two daughter cells. In the kic1 mutants actin was often prematurely localized to the necks of emerging buds (*) and buds appeared to be wider than normal (**). In addition, kic1 mutants also accumulated actin bars (ab), which are thought to be agglomerations of improperly folded actin. The kic1 mutants also accumulated cells with elongated bud necks containing two constrictions (arrows).
All the kic1 mutants exhibited actin defects (Fig. 9). Small-budded kic1 cells often had actin at their bud necks (Fig. 9, asterisks) instead of being restricted to the bud. This aberrant actin continued to localize to the bud neck during bud growth (see budded panel). Often kic1 mutant cells had abnormally wide bud-necks (Fig. 9, double asterisks). The kic1 mutant cells also often contained actin bars (Fig. 9, ab), that are thought to be agglomerations of improperly folded actin. Actin bars were observed in 7% of kic1-3 cells (n = 82), and 17% of kic1-2 cells (n = 62). Furthermore, many kic1 cells showed an elongated bud neck when the cell wall was removed (Fig. 9, arrows). Finally, in contrast to wild type, actin was absent from the bud necks of large-budded kic1 mutant cells.
Given the actin phenotype in the large-budded kic1 mutants, it was of interest to examine actin in a cdc31 mutant. In the cdc31 arrested large-budded cells actin was not localized to the mother-bud junction (Fig. 9). This may reflect the failure in cell cycle progression in the cdc31 mutant. In the unlysed kic1-1 cdc31-1 double-mutant cells, actin showed an aberrant localization quite similar to the kic1-1 mutant. Taken together, these data suggest that Cdc31p and Kic1p may function in cell integrity by effecting the localization of the underlying actin cytoskeleton.
Discussion
The identification of a Cdc31p-interacting kinase, Kic1p, marks the first report of a centrin associated with a protein kinase. In addition, this is the first report of a non-SPB (centrosome) function for a centrin protein. We demonstrate that Kic1p's kinase activity is dependent upon Cdc31p and that the kinase domain is required for proper cell wall assembly and cell integrity. Examination of four temperature-sensitive cdc31 alleles, previously characterized as defective for SPB duplication, revealed that these mutants exhibited allele-specific cell integrity and morphology defects. We conclude that that Cdc31p is critically involved in Kic1p's function in cell integrity and morphogenesis, but that Kic1p is not required for Cdc31p's SPB duplication function. Consistent with this, the combination of cdc31-1 and kic1-1, two alleles that do not generate lysis on their own, resulted in a severe synthetic lysis defect, similar to the most severe kic1 mutation. The simplest interpretation of these results is that Kic1p and Cdc31p interact, in vivo, to regulate some aspect of cell wall assembly.
The identification of allele-specific cell integrity and morphology defects for different cdc31 alleles was surprising. The cdc31-2, cdc31-5, and CDC31-16 mutants had lysis defects of varying severity, and the cdc31-1 and cdc31-2 mutants had elongated bud growth. The variation in cdc31 phenotypes suggests that CDC31 might be required for more than one morphological process. One possibility is that, like calmodulin, Cdc31p may interact with several different proteins with distinct functions. Different mutations may have more or less severe defects depending upon the specific interactions that are most affected. It is intriguing that cdc31-1 maps between the two NH2-terminal EF-hands (A48T), cdc31-5 maps to the central α-helical domain (P94S), and both cdc31-2 and cdc31-16 map between the two COOH-terminal EF-hands (D131N and E133K, respectively; Vallen et al., 1994). Given the location of the mutations, it is tempting to speculate that the central domain is important for cell integrity, whereas the NH2-terminal half of the protein preferentially effects bud morphology. Whereas cdc31-1 does not exhibit a lysis defect on its own, the synthetic lysis defect observed for the cdc31-1 kic1-1 double mutant suggests that this mutation may partially compromise the activity of Kic1p or the interaction between Kic1p and Cdc31p.
In contrast to the effects of loss of function mutations, when wild-type Kic1p was overexpressed it caused an elongated cell phenotype (Fig. 8 B), similar to that observed for cdc31-1 and cdc31-2. Interestingly, co-overexpression of Cdc31p caused cells to regain their normal rounded morphology. If the elongated cell phenotype were simply due to overexpression of Kic1p kinase activity, which is Cdc31p dependent, we would expect that co-overexpression of Cdc31p would increase kinase activity and further exacerbate the elongated cell phenotype. However, we found the opposite to be true. We observed suppression of the elongated cell phenotype, and therefore the phenotype appears to be independent of Kic1p kinase activity. Indeed the elongated cell phenotype was also observed when kic1-1 (a kinase down mutant) was overexpressed (data not shown). We propose that overexpression of Kic1p titrates Cdc31p away from another protein(s) whose interaction with Cdc31p is required for normal cell morphology. However, we cannot rule out the possibility that overexpression of the non-kinase domain of Kic1p generates the elongated cell phenotype.
Our results complement recent unpublished data from the laboratory of F. Klis (Vossen, J., and F. Klis, personal communication). These researchers independently identified KIC1 on the basis of the sensitivity of the mutant to calcofluor white. They have also found that kic1 mutants are highly fragile and defective in cell separation. The mutants were resistant to K1 killer toxin, because of reduced β1-6 glucan in their cell walls. They also found that the cell walls were resistant to Zymolyase, a β1-3 glucanase, indicting that the outer protein or mannan layer was less permeable than in wild-type cells. These results are consistent with our electron microscopy observations that show that kic1 cell walls are disorganized, lack a wild-type glucan layer, and have increased levels of electron-dense mannans. In addition, these researchers have shown that loss of Kic1p function upregulates the HOG pathway (high osmolarity/glycerol). When the HOG pathway is upregulated, cells produce more of the EXG1 exoglucanase with reduces β1-6 glucan levels. In addition, upregulation of the HOG pathway triggers accumulation of glycerol in cell increasing their osmotic potentials. This could explain part of the lysis defect of kic1 mutants. Increased osmotic potential would draw more water into the cells creating additional pressure on the cell walls leading to lysis.
The kic1 mutants lyse near the bud neck suggesting that they are lysing either at the neck, or at the bud or birth scars. Mutations in chitin deposition in the cell wall often lead to lysis at bud or birth scars which are largely composed of chitin (for review see Cid et al., 1995). Our electron microscopic analysis of the kic1 mutants identified aberrant electron transparent structures, presumably comprised of chitin, at the bud neck. Mutants sometimes had what appeared to be either multiple primary septa or chitin rings. These structures often appeared frayed and looked like weak spots in the wall. It is important to note that the kic1 defect is not the same as that caused by mutations in PKC1, the gene for protein kinase C (Levin and Bartlett-Heubusch, 1992; for review see Cid et al., 1995). Like kic1 mutants, pkc1 mutants exhibit lysis that is suppressed by the addition of 1 M sorbitol to the media. However, unlike kic1-1 the pkc1 mutants lyse at the tips of small buds, where there is a noticeable thinning of the cell wall. Furthermore, pkc1 mutants arrest at the G2–M transition with duplicated DNA content and small buds, whereas kic1 mutants do not arrest at the G2–M transition and complete multiple rounds of the cell cycle. Accordingly, the kic1 mutants accumulate fewer small-budded cells and the lysis defect is not always suppressed by sorbitol. The kic1 mutants also exhibit an abnormal bud neck phenotype that is not seen in pkc1 mutants. Therefore we propose that Kic1p and Pkc1p act in separate morphogenic pathways.
Kic1p is a member of a sub-group of PAK1/Ste20p-like kinases that have NH2-terminal kinase domains and COOH- terminal regulatory regions. Unlike other members of the PAK1/Ste20p family, this subfamily lacks Cdc42- and Rac-binding domains. The demonstration that the kinase activity of Kic1p is Cdc31p dependent raises the possibility that centrin may regulate the activity of NH2-terminal Ste20p-like kinases in other organisms just as Cdc42p/Rac regulates the function of the COOH-terminal kinases. Interestingly, Kic1p and a subset of PAK kinases contain an eleven amino acid motif (AKPXSILXD/ELI) that is found just COOH-terminal to the kinase domain (Fig. 1 B). It has recently been reported that this sequence regulates the binding of G protein β-subunits to PAK/Ste20 kinases (Leeuw et al., 1998). This domain partially overlaps the putative Cdc31p-binding domain of Kic1p (Vallen et al., 1994; Spang et al., 1995). The G protein β-subunit that binds to Ste20p was Ste4p, a protein of 46 kD. It is tempting to speculate that the 40-kD protein we see associated with Kic1p might be a β-subunit that co-operatively or competitively regulates Kic1p function with Cdc31p.
The class B and C mutants (Table IV) also exhibited mild spindle migration defects, which resulted in small-budded binucleate cells. These may be due to the mislocalization of actin during the stages of bud morphogenesis. Previous work has documented that spindle migration and orientation in yeast requires both functional astral microtubules (Palmer et al., 1992; Sullivan and Huffaker, 1992) and a polarized actin cytoskeleton (Palmer et al., 1992; Wang and Bretscher, 1995). In the absence of any obvious defects in microtubule structure (data not shown), it is reasonable to suggest that the kic1 binucleate phenotype is due primarily to actin defects. The wide bud neck phenotype could also be caused by the observed actin defects. In many cases actin was found at the bud site and neck after bud emergence had progressed significantly. This is not seen in wild-type cells. Since secretion is actin directed this might lead to increased deposition of cell wall materials. In this respect it is important to note that some PAK kinases have been implicated in regulating actin organization.
Cdc31p is involved in both SPB duplication and cell integrity/morphogenesis. Is this a coincidence or are they coordinated in some way? The earliest step in SPB duplication, the appearance of the SPB satellite body, occurs after telophase but before START. Bud growth is initiated at START and must be completed for proper completion of both the nuclear and cell division pathways. The involvement of Cdc31p in two disparate processes raises the possibility that Cdc31p plays an explicit role in the coordination of these two major cell cycle processes.
Acknowledgments
This work was supported by a National Institutes of Health grant to M.D. Rose (GM52526). D.S. Sullivan was supported by a National Research Service Award (GM17998).
Abbreviations used in this paper
- 5-FOA
5-fluoro-orotic acid
- DAPI
4′,6′-diamidino-2-phenylindole
- DIC
differential interference contrast
- GST
glutathione-S-transferase
- MAP kinase
mitogen-activated protein kinase
- MTOC
microtubule organizing centers
- PAK
p-21–activated kinases
- SPB
spindle pole body
- YPD
yeast extract with peptone and dextrose
Footnotes
We are very grateful to J. Vossen and F. Klis for communication of results before publication. We thank J. Nickels, E. Lauzé, E. Weiss, and M. Winey for helpful discussions. We thank J. Goodhouse for assistance with the electron microscopy. We thank all members of the Rose lab for helpful discussions, especially C. Beh, A. Gammie, L. Satterwhite, and S. Clark. We thank I. Ivanovska, W. Khalfan, and A. Gammie for editorial comments on the manuscript. We are grateful to S. Fields, B. Deschenes, H. Sundberg, and T. Davis for strains and plasmids. We thank J. Kilmartin, T. Bretscher, and T. Wang for antibodies.
D. Sullivan's current address is Mitotix, Inc., One Kendall Square, Building 600, Cambridge, MA 02139.
S. Biggins's current address is Department of Physiology, University of California, San Francisco, CA 94143-0444.
Address all correspondence to Mark D. Rose, Department of Molecular Biology, Princeton University, Princeton, NJ 08544-1014. Tel.: (609) 258-2804. Fax: (609) 258-6175. E-mail: mrose@molecular.princeton.edu
References
- Ayscough KR, Drubin DG. Actin: general principles from studies in yeast. Annu Rev Cell Dev Biol. 1996;12:129–160. doi: 10.1146/annurev.cellbio.12.1.129. [DOI] [PubMed] [Google Scholar]
- Bagrodia S, Taylor SJ, Creasy CL, Chernoff J, Cerione RA. Identification of a mouse p21Cdc42/Rac activated kinase. J Biol Chem. 1995;270:22731–22737. doi: 10.1074/jbc.270.39.22731. [DOI] [PubMed] [Google Scholar]
- Baum P, Furlong C, Byers B. Yeast gene required for spindle pole body duplication: homology of its product with Ca2+-binding proteins. Proc Natl Acad Sci USA. 1986;83:5512–5516. doi: 10.1073/pnas.83.15.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Rose MD. Direct interaction between yeast spindle pole body components: Kar1p is required for Cdc31p localization to the spindle pole body. J Cell Biol. 1994;125:843–852. doi: 10.1083/jcb.125.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkovich KA, Farrelly FW, Finkelstein DB, Taulien J, Lindquist S. Hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol Cell Biol. 1989;9:3919–3930. doi: 10.1128/mcb.9.9.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockerhoff SE, Davis TN. Calmodulin concentrates at regions of cell growth in Saccharomyces cerevisiae. . J Cell Biol. 1992;118:619–629. doi: 10.1083/jcb.118.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockerhoff SE, Stevens RC, Davis TN. The unconventional myosin, Myo2p, is a calmodulin target at sites of cell growth in Saccharomyces cerevisiae. . J Cell Biol. 1994;124:315–323. doi: 10.1083/jcb.124.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Stowers L, Baer M, Trejo J, Coughlin S, Chant J. Human Ste20 homologue hPAK1 links GTPases to the JNK MAP kinase pathway. Curr Biol. 1996;6:598–605. doi: 10.1016/s0960-9822(02)00546-8. [DOI] [PubMed] [Google Scholar]
- Byers, B. 1981. Multiple roles of the spindle pole bodies in the life cycle of Saccharomyces cerevisiae. In Molecular Genetics in Yeast. D. von Wettstein, A. Stenderup, M. Kielland-Brandt, and J. Friis, editors. Alfred Benzon Symposia 16. Munksgaard, Copenhagen. pp. 119–133.
- Byers B, Goetsch L. Duplication of spindle plaques and integration of the yeast cell cycle. Cold Spring Harbor Symp Quant Biol. 1974;38:123–131. doi: 10.1101/sqb.1974.038.01.016. [DOI] [PubMed] [Google Scholar]
- Byers B, Goetsch L. Behavior of spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. . J Bacteriol. 1975;124:511–523. doi: 10.1128/jb.124.1.511-523.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid VJ, Durán A, del Rey F, Snyder MP, Nombela C, Sánchez M. Molecular basis of cell integrity and morphogenesis in Saccharomyces cerevisiae. . Microbiol Rev. 1995;59:345–386. doi: 10.1128/mr.59.3.345-386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creasy CL, Chernoff J. Cloning and characterization of a human protein kinase with homology to Ste20. J Biol Chem. 1995;270:21695–21700. doi: 10.1074/jbc.270.37.21695. [DOI] [PubMed] [Google Scholar]
- Davis TN. Mutational analysis of calmodulin in Saccharomyces cerevisiae. . Cell Calcium. 1992;13:435–444. doi: 10.1016/0143-4160(92)90056-x. [DOI] [PubMed] [Google Scholar]
- Davis TN, Urdea MS, Masiarz FR, Thorner J. Isolation of the yeast calmodulin gene: calmodulin is an essential protein. Cell. 1986;47:423–431. doi: 10.1016/0092-8674(86)90599-4. [DOI] [PubMed] [Google Scholar]
- Errabolu R, Sanders MA, Salisbury JL. Cloning of a cDNA encoding human centrin, an EF-hand protein of centrosomes and mitotic spindle poles. J Cell Sci. 1994;107:9–16. doi: 10.1242/jcs.107.1.9. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–256. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Gammie AE, Brizzio V, Rose MD. Distinct morphological phenotypes of cell fusion mutants. Mol Biol Cell. 1998;9:1395–1410. doi: 10.1091/mbc.9.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser JR, Sundberg HA, Chang BH, Muller EG, Davis TN. The essential mitotic target of calmodulin is the 110-kilodalton component of the spindle pole body in Saccharomyces cerevisiae. . Mol Cell Biol. 1993;13:7913–7924. doi: 10.1128/mcb.13.12.7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs CS, Zoller MJ. Rational scanning mutagenesis of a protein kinase identifies functional regions involved in catalysis and substrate interactions. J Biol Chem. 1991;266:8923–8931. [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Herskowitz I. MAP kinase pathways in yeast: for mating and more. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- Hoffman CS, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. . Gene (Amst) 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- Huang B, Watterson DM, Lee VD, Schibler MJ. Purification and characterization of a basal-body associated Ca2+-binding protein. J Cell Biol. 1988a;107:121–131. doi: 10.1083/jcb.107.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Mengersen A, Lee VD. Molecular cloning of a cDNA for caltractin, a basal body-associated Ca2+-binding protein: Homology in its protein sequence with calmodulin and the yeast CDC31gene product. J Cell Biol. 1988b;107:133–140. doi: 10.1083/jcb.107.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffaker TC, Thomas JH, Botstein D. Diverse effects of β-tubulin mutations on microtubule formation and function. J Cell Biol. 1988;106:1997–2010. doi: 10.1083/jcb.106.6.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Plowman GD. The protein kinases of budding yeast: six score and more. Trends Biochem Sci. 1997;22:18–22. doi: 10.1016/s0968-0004(96)10068-2. [DOI] [PubMed] [Google Scholar]
- Kellogg DR, Moritz M, Alberts BM. The centrosome and cellular organization. Annu Rev Biochem. 1994;63:639–674. doi: 10.1146/annurev.bi.63.070194.003231. [DOI] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AE. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. . J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauzé E, Stoelcker B, Luca F, Weiss E, Schutz AR, Winey M. Yeast spindle pole body duplication gene MPS1encodes an essential dual specificity protein kinase. EMBO (Eur Mol Biol Organ) J. 1995;14:1655–1663. doi: 10.1002/j.1460-2075.1995.tb07154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberer E, Dignard D, Harcus D, Thomas DY, Whiteway M. The protein kinase homologue Ste20p is required to link the yeast pheromone response G-protein βγ subunits to downstream signalling components. EMBO (Eur Mol Biol Organ) J. 1992;11:4815–4824. doi: 10.1002/j.1460-2075.1992.tb05587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Maeda S, Angelos KL, Kamita SG, Ramachandran C, Walsh DA. Analysis by mutagenesis of the ATP binding site of the γ subunit of skeletal muscle phosphorylase kinase expressed using a baculovirus system. Biochemistry. 1992;31:10610–10625. doi: 10.1021/bi00158a026. [DOI] [PubMed] [Google Scholar]
- Lee VD, Huang B. Molecular cloning and centrosomal localization of human caltractin. Proc Natl Acad Sci USA. 1993;90:11039–11043. doi: 10.1073/pnas.90.23.11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuw T, Wu C, Schrag JD, Whiteway M, Thomas DY, Leberer E. Interaction of a G-protein β-subunit with a conserved sequence in Ste20/PAK family protein kinases. Science. 1998;391:191–195. doi: 10.1038/34448. [DOI] [PubMed] [Google Scholar]
- Leung DW, Chen E, Goeddel DV. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique. 1989;1:11–15. [Google Scholar]
- Levin DE, Bartlett-Heubusch E. Mutants in the S. cerevisiae PKC1gene display a cell cycle-specific osmotic stability defect. J Cell Biol. 1992;116:1221–1229. doi: 10.1083/jcb.116.5.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy YY, Lai EY, Remillard SP, Heintzelman MB, Fulton C. Centrin is a conserved protein that forms diverse associations with centrioles and MTOCs in Naegleria and other organisms. Cell Motil Cytoskeleton. 1996;33:298–323. doi: 10.1002/(SICI)1097-0169(1996)33:4<298::AID-CM6>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Lindberg RA, Quinn AM, Hunter T. Dual-specificity protein kinases: will any hydroxyl do? . Trends Biochem Sci. 1992;17:114–119. doi: 10.1016/0968-0004(92)90248-8. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao Z, Lim L. A brain serine/ threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- Manser E, Chong C, Zhao ZS, Leung T, Michael G, Hall C, Lim L. Molecular cloning of a new member of the p21-Cdc42/Rac-activated kinase (PAK) family. J Biol Chem. 1995;270:25070–25078. doi: 10.1074/jbc.270.42.25070. [DOI] [PubMed] [Google Scholar]
- Means AR. Calcium, calmodulin and cell cycle regulation. FEBS (Fed Eur Biochem Soc) Lett. 1994;347:1–4. doi: 10.1016/0014-5793(94)00492-7. [DOI] [PubMed] [Google Scholar]
- Middendorp S, Paoletti A, Schiebel E, Bornens M. Identification of a new mammalian centrin gene, more closely related to Saccharomyces cerevisiae CDC31gene. Proc Natl Acad Sci USA. 1997;94:9141–9146. doi: 10.1073/pnas.94.17.9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Marshall TK, Deschenes RJ. Vectors for the inducible overexpression of glutathione-S-transferase fusion proteins in yeast. Yeast. 1993;9:715–723. doi: 10.1002/yea.320090705. [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Hunter R, Parker R. A rapid method for localized mutagenesis of yeast genes. Yeast. 1992;8:79–82. doi: 10.1002/yea.320080202. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Shimizu T. cDNA sequence for mouse caltractin. Biochim Biophys Acta. 1993;1216:126–128. doi: 10.1016/0167-4781(93)90048-i. [DOI] [PubMed] [Google Scholar]
- Ohashi A, Gibson J, Gregor I, Schatz G. Import of proteins into mitochondria. The precursor of cytochrome c1 is processed in two steps, one of them heme-dependent. J Biol Chem. 1982;257:13042–13047. [PubMed] [Google Scholar]
- Ohya Y, Botstein D. Diverse essential functions revealed by complementing yeast calmodulin mutants. Science. 1994;263:963–966. doi: 10.1126/science.8310294. [DOI] [PubMed] [Google Scholar]
- Osada S, Izawa M, Saito R, Mizuno K, Suzuki A, Hirai S, Ohno S. YSK1, a novel mammalian protein kinase structurally related to Ste20 and SPS1, but is not involved in the known MAPK pathways. Oncogene. 1997;14:2047–2057. doi: 10.1038/sj.onc.1201043. [DOI] [PubMed] [Google Scholar]
- Palmer RE, Sullivan DS, Huffaker T, Koshland D. Role of astral microtubules and actin in spindle orientation and migration in the budding yeast, Saccharomyces cerevisiae. . J Cell Biol. 1992;119:583–593. doi: 10.1083/jcb.119.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti A, Moudjou M, Paintrand M, Salisbury JL, Bornens M. Most of centrin in animal cells is not centrosome-associated and centrosomal centrin is confined to the distal lumen of centrioles. J Cell Sci. 1996;109:3089–3102. doi: 10.1242/jcs.109.13.3089. [DOI] [PubMed] [Google Scholar]
- Pombo CM, Bonventre JV, Molnar A, Kyriakis J, Force T. Activation of a human Ste20-like kinase by oxidant stress defines a novel stress response pathway. EMBO (Eur Mol Biol Organ) J. 1996;15:4537–4546. [PMC free article] [PubMed] [Google Scholar]
- Ramer SW, Davis RW. A dominant truncation allele identifies a new gene, STE20, that encodes a putative protein kinase necessary for mating in Saccharomyces cerevisiae. . Proc Natl Acad Sci USA. 1993;90:452–456. doi: 10.1073/pnas.90.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MD, Fink GR. KAR1, a gene required for function of both intranuclear and extranuclear microtubules in yeast. Cell. 1987;48:1047–1060. doi: 10.1016/0092-8674(87)90712-4. [DOI] [PubMed] [Google Scholar]
- Rose MD, Novick P, Thomas JH, Botstein D, Fink GR. A Saccharomyces cerevisiaegenomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–247. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Heiter. 1990. Methods in yeast genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 198 pp.
- Rose MD, Biggins S, Satterwhite LL. Unraveling the tangled web at the microtubule organizing center. Curr Opin Cell Biol. 1993;5:105–115. doi: 10.1016/s0955-0674(05)80015-8. [DOI] [PubMed] [Google Scholar]
- Rothstein R. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Salisbury JL, Baron AT, Sanders M. The centrin-based cytoskeleton of Chlamydomonas reinhardtii: Distribution in interphase and mitotic cells. J Cell Biol. 1988;107:635–641. doi: 10.1083/jcb.107.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: a Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Sanders MA, Salisbury JL. Centrin-mediated microtubule severing during flagellar excision in Chlamydomonas reinhardtii. . J Cell Biol. 1989;108:1751–1760. doi: 10.1083/jcb.108.5.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders MA, Salisbury JL. Centrin plays an essential role in microtubule severing during flagellar excision in Chlamydomonas reinhardtii. . J Cell Biol. 1994;124:795–805. doi: 10.1083/jcb.124.5.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild D, Anathaswamy HN, Mortimer RK. An endomitotic effect of a cell cycle mutant of Saccharomyces cerevisiae. . Genetics. 1981;97:551–562. doi: 10.1093/genetics/97.3-4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore, M.A. 1993. A genetic analysis of KAR2, the yeast homologue of mammalian BiP. Ph.D. thesis. Princeton University, Princeton, NJ. 255 pp.
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, Courtney I, Fackler U, Matzner M, Schiebel E. The calcium-binding protein cell division cycle 31 of Saccharomyces cerevisiaeis a component of the half bridge of the spindle pole body. J Cell Biol. 1993;123:405–416. doi: 10.1083/jcb.123.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, Courtney I, Grein I, Matzner K, Schiebel E. The Cdc31p binding protein Kar1p is a component of the half bridge of the yeast spindle pole body. J Cell Biol. 1995;128:863–877. doi: 10.1083/jcb.128.5.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens RC, Davis TN. Mlc1p is a light chain for the unconventional myosin Myo2p in Saccharomyces cerevisiae. . J Cell Biol. 1998;142:711–722. doi: 10.1083/jcb.142.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DA, Welch KA, Stark MJ. Interaction with calmodulin is required for the function of Spc110p, an essential component of the yeast spindle pole body. EMBO (Eur Mol Biol Organ) J. 1994;13:4329–4342. doi: 10.1002/j.1460-2075.1994.tb06753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YC, Han J, Xu S, Cobb M, Skolnik EY. NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO (Eur Mol Biol Organ) J. 1997;16:1279–1290. doi: 10.1093/emboj/16.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DS, Huffaker TC. Astral microtubules are not required for anaphase B in Saccharomyces cerevisiae. . J Cell Biol. 1992;119:379–388. doi: 10.1083/jcb.119.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taillon BE, Adler SA, Suhan JP, Jarvik JW. Mutational analysis of centrin: An EF-hand protein associated with three distinct contractile fibers in the basal body of Chlamydomonas. . J Cell Biol. 1992;119:1613–1624. doi: 10.1083/jcb.119.6.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzawa M, Grams J, Madden B, Toft D, Salisbury JL. Identification of a complex between centrin and heat shock proteins in CSF–arrested Xenopus oocytes and dissociation of the complex following oocyte activation. Dev Biol. 1995;171:51–59. doi: 10.1006/dbio.1995.1259. [DOI] [PubMed] [Google Scholar]
- Vallen EA, Scherson TY, Roberts T, van Zee K, Rose MD. Asymmetric mitotic segregation of the yeast spindle pole body. Cell. 1992;69:505–515. doi: 10.1016/0092-8674(92)90451-h. [DOI] [PubMed] [Google Scholar]
- Vallen EA, Ho W, Winey M, Rose MD. Genetic interactions between CDC31 and KAR1, two genes required for duplication of the microtubule organizing center in Saccharomyces cerevisiae. . Genetics. 1994;137:407–422. doi: 10.1093/genetics/137.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Bretscher A. The rho-GAP encoded by BEM2regulates cytoskeletal structure in budding yeast. Mol Biol Cell. 1995;6:1011–1024. doi: 10.1091/mbc.6.8.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, et al. 2.2 Mb of contiguous nucleotide sequence from chromosome III of C. elegans. . Nature. 1994;368:32–38. doi: 10.1038/368032a0. [DOI] [PubMed] [Google Scholar]
- Winey M, Byers B. Assembly and functions of the spindle pole body in budding yeast. Trends Genet. 1993;9:300–304. doi: 10.1016/0168-9525(93)90247-f. [DOI] [PubMed] [Google Scholar]