

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride channel that is defective in cystic fibrosis, and has also been closely associated with ATP permeability in cells. Using a Xenopus oocyte cRNA expression system, we have evaluated the molecular mechanisms that control CFTR-modulated ATP release. CFTR-modulated ATP release was dependent on both cAMP activation and a gradient change in the extracellular chloride concentration. Activation of ATP release occurred within a narrow concentration range of external Cl− that was similar to that reported in airway surface fluid. Mutagenesis of CFTR demonstrated that Cl− conductance and ATP release regulatory properties could be dissociated to different regions of the CFTR protein. Despite the lack of a need for Cl− conductance through CFTR to modulate ATP release, alterations in channel pore residues R347 and R334 caused changes in the relative ability of different halides to activate ATP efflux (wtCFTR, Cl >> Br; R347P, Cl >> Br; R347E, Br >> Cl; R334W, Cl = Br). We hypothesize that residues R347 and R334 may contribute a Cl− binding site within the CFTR channel pore that is necessary for activation of ATP efflux in response to increases of extracellular Cl−. In summary, these findings suggest a novel chloride sensor mechanism by which CFTR is capable of responding to changes in the extracellular chloride concentration by modulating the activity of an unidentified ATP efflux pathway. This pathway may play an important role in maintaining fluid and electrolyte balance in the airway through purinergic regulation of epithelial cells. Insight into these molecular mechanisms enhances our understanding of pathogenesis in the cystic fibrosis lung.
Keywords: CFTR, ATP release, Cl− sensor, oocytes
CFTR is a member of the ATP-binding cassette (ABC)1 superfamily that includes over thirty proteins that share extensive sequence similarity and domain organization (Higgins et al., 1990; Ames et al., 1986; Hyde et al., 1990). Electrophysiologic analyses of the CFTR gene product has demonstrated that it is a cAMP-activated chloride channel with a conductance of 7–10 pS (Drumm et al., 1990; Gregory et al., 1990; Kartner et al., 1990; Anderson et al., 1991; Bear et al., 1992). Although altered Cl− permeability through CFTR has generally been accepted to play a role in the pathogenesis of cystic fibrosis (CF), it remains unclear whether this defect alone can explain the complex pathophysiology of associated lung disease. Recent studies have indicated that in addition to its well-defined Cl− channel activity, CFTR is also a regulator of other epithelial ion channels, including an outwardly rectifying Cl− channel (ORCC; Egan et al., 1992; Gabriel et al., 1993; Schwiebert et al., 1995; Schwiebert et al., 1998; Jovov et al., 1995), the epithelial Na+ channel (ENaC; Grubb et al., 1994; Chinet et al., 1994; Johnson et al., 1995; Hyde et al., 1993; Ismailov et al., 1996; Stutts et al., 1997; Kunzelmann et al., 1997), and a kidney K+ channel (ROMK; McNicholas et al., 1996; McNicholas et al., 1997; Ho, 1998). However, the mechanisms by which these regulatory functions contribute to the maintenance of airway electrolyte and fluid balance remain obscure.
Cantiello and colleagues first suggested that the multidrug resistance protein, a member of the ABC superfamily, might conduct ATP (Abraham et al., 1993). Several groups have provided evidence that CFTR also plays a role in ATP release (Reisin et al., 1994; Schwiebert et al., 1995; Prat et al., 1996; Pasyk and Foskett, 1997; Sugita et al., 1998, Cantiello et al., 1998). Guggino and colleagues suggested that CFTR stimulates the ORCC via a P2U purinergic receptor–dependent signal transduction pathway by modulating ATP efflux (Schwiebert et al., 1995). Similar mechanisms of ion channel regulation have been proposed for other ABC transporters that secrete ATP (Al-Awqati, 1995). However, several laboratories have failed to observe an association of CFTR with ATP permeability (Reddy et al., 1996; Li et al., 1996; Grygorczyk et al., 1996). In model systems that have been able to demonstrate CFTR-modulated ATP release, it is presently unclear whether CFTR itself, another unknown cofactor, or a CFTR-linked biologic process such as exocytosis, represents the ATP permeation/release pathway. It is also unknown if dysregulation of this process contributes to CF lung disease progression. Most recently, Foskett and coworkers demonstrated that ATP channels exhibiting slow gating kinetics were associated within a subpopulation of CFTR Cl− channels in MDCK cells (Sugita et al., 1998). They concluded that phosphorylation- and nucleotide hydrolysis–dependent CFTR gating is directly involved in gating an associated ATP channel, but that the permeation pathways for Cl− and ATP are distinct. Because the ATP conduction pathway did not appear to be obligatorily associated with CFTR expression, they suggested the existence of a cofactor that may mediate CFTR-regulated ATP conductance. However, a recent report reconstituting purified CFTR into lipid bilayers has suggested that CFTR enables permeation of both Cl− and ATP (Cantiello et al., 1998).
In the present study, we used a Xenopus oocyte cRNA expression system coupled with a sensitive luciferin– luciferase bioluminescence assay to explore the mechanisms that control CFTR-modulated ATP release. Our results suggest that expression of CFTR can confer a Cl−-sensitive ATP permeability to injected oocytes. Mutational analysis suggests that the interaction of extracellular Cl− at arginine residues R334 and R347 within the channel pore controls the ability of CFTR to modulate ATP release. These results suggest a novel mechanism by which changes in the extracellular Cl− concentration participate in the activation of CFTR-modulated ATP release.
Materials and Methods
Xenopus Oocyte Isolation and Maintenance
Adult female Xenopus laevis were obtained from NASCO (Fort Atkinson, WI) and Xenopus-1 (Ann Arbor, Michigan). Immediately before surgery, frogs were anaesthetized by immersion in 0.17% (wt/vol) tricaine (3-aminobenzoic acid ethyl ester methanesulphonate) for 5 min, and were then cooled by adding an equal volume of ice to the Tricaine solution for 10 min. Ovaries were surgically removed from anaesthetized frogs through a small incision in the lower abdomen. Individual oocytes were isolated and defolliculated by collagenase treatment. In brief, isolated oocytes were rinsed in Ca2+-free SOS (100 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 5 mM Hepes-NaOH, pH 7.6) and incubated with Type IV collagenase (2.0 mg/ml; Sigma Chemical Co., St. Louis, MO) in the same media for 45 min with gentle shaking. After rinsing several times with Ca2+-free SOS, defolliculated oocytes were kept in SOS (100 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM Hepes, pH 7.6) supplemented with 2.5 mM sodium pyruvate and 50 μg/ml gentamycin at 16°C until used.
During the course of this study, we found that only ∼10% of the frogs had oocytes that displayed CFTR-modulated ATP release, despite their ability to generate CFTR-dependent cAMP-inducible Cl− currents. We therefore screened all frogs to identify those whose oocytes gave ATP responses. Responder frogs were placed in a separate tank and reused at intervals of no less than 5 wk. Oocytes from responders continued to demonstrate CFTR-modulated ATP release at a similar frequency and magnitude during later harvests (up to five harvests have been performed on responder frogs without a decline in ATP responses).
Complementary RNA Synthesis and Injection into Oocytes
Synthesis of complementary RNA (cRNA) from CFTR and mutant cDNAs was performed using the Megascript cRNA synthesis kit purchased from Ambion (Austin, TX). Linearized cDNA plasmids were transcribed at 37°C for 4 h in a reaction mixture supplemented with excess ribonucleotide solution that allows an increased rate and duration of cRNA synthesis. RNA cap analogue, m7G(5′)ppp(5′)G (New England Biolabs Inc., Beverly, MA) was used to cap and protect the 5′ end of the cRNA. The final reaction mixture contained 2–4 μg of a linearized cDNA, 1× transcription buffer provided by the kit, 7.5 mM each of ATP, CTP, and UTP, 1.5 mM of GTP, 6 mM of the cap analogue, and 4 μl of the enzyme mix in a total volume of 40 μl. Xenopus oocytes were injected 24 h after isolation with 50 nl per oocyte of either DEPC water (mock-injected) or appropriate cRNA (50 ng) using a nanoliter injector (World Precision Instruments, Inc., Sarasota, FL). Typically, oocytes were analyzed for either ATP responses or electrophysiologic measurements 48 h after injection.
Measurement of the ATP Efflux in Single Xenopus Oocytes
Measurements of the ATP efflux from single oocytes were performed using a luciferin–luciferase assay in a TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA) under dim light. A single oocyte was rinsed twice in an appropriate buffer, and was then immersed in 100 μl of the same buffer containing 3.125 mg/ml luciferin–luciferase (Sigma Chemical Co.) in a 0.5-ml Eppendorf tube. Most experiments were done with either Hepes phosphate-buffered Ringers (HPBR; 10 mM Hepes, pH, 7.4, 140 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.2 mM CaGluconate, 2.4 mM K2HPO4, and 0.4 mM KH2PO4) or standard oocyte solution (SOS; 5 mM Hepes, pH 7.6, 100 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, and 1 mM MgCl2). When Cl−-free buffers were used, NaCl was replaced by equivalent amounts of sodium gluconate. The intracellular Cl− concentration of oocytes was calculated from the averaged reversal potentials of CFTR-injected oocytes in the presence of IBMX (100 μM) and forskolin (10 μM) using the Nernst equation:
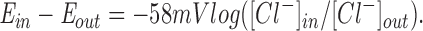 |
The optimal protocol for eliciting CFTR-modulated ATP release required the following series of sequential buffer changes that altered the external Cl− concentration: (a) Cl−-free; (b) Cl−-free + cAMP/forskolin; and (c) Cl− (100–140 mM) + cAMP/forskolin. Luminescence readings were recorded by a computer data link for 10 readings of 10 s each in the luminometer with the sensitivity level set to 89%. When indicated, 8-cpt-cAMP/forskolin cocktail was added to the experimental buffer to achieve a final concentration of 200 μM 8-cpt-cAMP and 2.5 μM forskolin. In some experiments, ionomycin (10 μM; Calbiochem-Novabiochem Corp., La Jolla, CA) replaced 8-cpt-cAMP/forskolin in the assay buffer. Buffer exchange was accomplished by rinsing the oocyte once in the new buffer; the luminescence assay was then continued for the same amount of time as described above. Care was taken never to touch the oocyte with pipette tips. The buffer rinses typically took ∼45 s, and account for the absence of data values during solution switches. To determine the absolute amount of ATP efflux from an oocyte, standard concentrations of ATP in appropriate buffer solutions were measured to establish a calibration curve. In studies that evaluated the anion dependence of the activation of CFTR-modulated ATP release, the effects of different anions (gluconate, Cl−, Br−, I−, F−, NO3, and thiocyanate) on the activity of luciferin–luciferase were evaluated in preliminary experiments. ATP calibration curves obtained in each buffer revealed that I−, F−, NO3, and thiocyanate profoundly inhibited luciferin–luciferase activity. The Cl− channel blocker diphenylamine-2-carboxylic acid (DPC) also inhibited luciferin–luciferase activity. These reagents were therefore not used in the studies. In contrast, the ATP activity calibration curves were similar in the presence of gluconate, Cl−, or Br−.
CFTR-modulated ATP release in Xenopus oocytes using this luciferin– luciferase assay was successfully reproduced at both the University of Pennsylvania and Johns Hopkins University.
Two-electrode Voltage Clamp Recording
Whole-cell (Wc) currents were measured using the two-electrode voltage clamp (TEV) method. Single oocytes were placed in a 1-ml chamber containing modified ND96 (96 mM NaCl, 1 mM KCl, 0.2 mM CaCl2, 5.8 mM MgCl2, 10 mM Hepes, pH 7.2 by NaOH) connected to a reference bath electrode by a 3 M KCl-agar bridge (Katayama and Widdicombe, 1991). Conventional TEV was performed (Stühmer, 1992) at room temperature using an OC-725C amplifier (Warner Instrument Corp., Hamden, CT) connected to a PowerMac 7100 via an ITC-16 interface (Instrutech Corp., Great Neck, NY). Pulse + PulseFit software (HEKA, Port Washington, NY) was used to ramp the applied transmembrane potential (V m) at 10-s intervals from −60 mV to 20 mV at a rate of 16 mV/s. V m was clamped at the prestimulation reversal potential between voltage ramps. Transmembrane current (I) and V m were digitized at 200 Hz during the voltage ramps, and were written directly onto hard disk. During the course of TEV recording, the major sources of equipment noise were the 60-Hz spikes induced by the power supply. Therefore, the TEV current–applied voltage data were fitted with an empirical fifth order polynomial to provide a smoothed approximation of the TEV data and eliminate the 60-Hz spikes. The membrane conductance of the oocyte was then calculated as dI/dV from this approximation. In all TEV experiments performed, a fifth order polynomial fit very well the observed current-voltage data, which had a simple monotonically increasing relations within the applied voltage range. Halide selectivity experiments were performed by replacing NaBr with NaCl in ND96 solutions. CFTR activation was accomplished by perfusing the oocyte with appropriate buffers containing 100 μM IBMX and 10 μM forskolin (Sigma Chemical Co.). In all experiments, CFTR Cl− conductance was defined as the difference in conductance measured in the presence and in the absence of 100 μM IBMX and 10 μM forskolin. The cAMP-stimulated Cl− and Br− conductances of the R347E mutant (5.3 ± 2.1 μs and 12.15 ± 3.3 μs, respectively) were much lower than that of the wild type. In control experiments, the conductance changes of mock-injected oocytes in response to IBMX/forskolin treatment were –0.61 ± 0.62 μs in the presence of Cl− and 0.11 ± 0.39 μs in the presence of Br−. These background non-CFTR conductance changes were subtracted from the measured changes of CFTR and mutants. In the case of mutant R347E, this correction caused only a small change in the calculated GBr/GCl value from 2.60 (without background correction) to 2.36 (with background correction). The Wc conductance (at −20 mV) and reversal potential (E rev) after agonist stimulation of CFTR channels (wild type or mutants) were evaluated from the slope and x intercept, respectively, of the background-corrected I–V m curve using Igor Pro software (WaveMetrics, Lake Oswego, OR). After each buffer perfusion (1 min), conductance was measured over 10 min to ensure equilibrium was reached. GBr/GCl is the ratio of the Wc conductances of stimulated CFTR channels in Br− vs. Cl− solution. The permeability ratio (PBr/PCl) was calculated from the reversal potentials E rev,Br and E rev,Cl in Br− and Cl− solutions, respectively, using
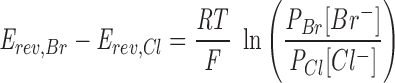 |
which is based on the Goldman–Hodgkin–Katz equation, with [Br−] and [Cl−] being the concentrations of Br− and Cl− in the solutions, and R, T, and F having their conventional meanings.
Preparation of CFTR Mutants
Five CFTR mutants were analyzed for their ability to modulate ATP release using the luciferin–luciferase assay as well as their capacity to conduct Cl− and Br− using voltage clamping. For mutant R347, a cDNA segment was cut out from the PTM1-R347E CFTR construct (kindly provided by Dr. M. Welsh, University of Iowa) by restriction enzymes MroI and Bst1107I, and was subcloned into the pBQ-CFTR plasmid between the same restriction sites. Mutants R334W and R347P were constructed by replacing a SpH I–Xba I segment in the PSP-CFTR with a corresponding segment cut out from mutants PTM-R334W and PTM-R347P (provided by Dr. M.J. Welsh; Sheppard et al., 1993), respectively. Successful transfer of the mutated sequences was confirmed by sequencing. The COOH-terminal truncation mutant, TMD1 CFTR, was constructed by introducing a stop codon at K370X followed by an EcoRV restriction site using the mutagenic oligonucleotide 5′-GCAATAAACTAAATACAGGATATCTTAC-3′. The NH2-terminal truncation mutant Δ259-M265V CFTR was constructed as previously described (Piazza Carroll et al., 1995).
Immunoprecipitation of CFTR and Mutants from cRNA-injected Xenopus Oocytes
Synthesis of 35S-labeled wild-type and mutant CFTR proteins in Xenopus oocytes was achieved by coinjection of appropriate cRNAs with [35S]methionine. The injection mixtures were made by adding 50 μCi of 35S-methionine (ICN) to 2.0 μl of transcripts. Each oocyte was injected with 50 nl of a mixture containing 50 ng of cRNA and 0.5 μCi 35S. After incubating for 3 h at 18°C in MBS, oocytes were collected, quickly frozen on dry ice, and stored at −80°C until use. For immunoprecipitation, oocytes in groups of five were thawed and homogenized on ice in 30 μl of 50 mM Tris (pH 7.5), 0.25 M sucrose, 50 mM KCl, 5 mM MgCl2, and 1 mM DTT followed by solubilization with 100 μl of 1% SDS and 0.1 M Tris (pH 8.0). Samples were incubated for 30 min at 37°C, diluted in 1 ml of TX SWB buffer (0.1 M NaCl, 1% Triton X-100, 2 mM EDTA, 0.1 mM PMSF and 0.1 M Tris, pH 8.0) on ice, and further incubated at 4°C for 2 h. Before immunoprecipitation, samples were centrifuged at 16,000 g for 15 min, and supernatants were collected. Immunoprecipitation of CFTR was initiated by adding 1 μl of an epitope-specific rabbit antisera raised against a synthetic peptide corresponding to residues 45–56 in the CFTR NH2 terminus and mixing at 4°C for 1.5 h. 5 μl of Affigel protein A (Bio-Rad Laboratories, Hercules, CA) was added, and the reaction mixture was further incubated at 4°C for another 1.5 h. After washing five times in TX SWB, protein A beads were pelleted by brief centrifugation in a microcentrifuge and resuspended in 1X sample loading buffer. Samples were denatured at 37°C for 30 min and analyzed on 7.5% SDS PAGE. After electrophoresis, gels were fixed in 35% methanol and 10% HAc, dried, and exposed to film for autoradiography.
Results
ATP Release from CFTR-expressing Xenopus Oocytes
ATP release from Xenopus oocytes was studied using a single oocyte luciferin–luciferase luminometric assay. Initial experiments compared water and CFTR cRNA– injected oocytes under conditions that promote Cl− flux through CFTR. In the presence of Cl−-free Ringers containing 200 μM 8-(4-chlorophenylthio)-adenosine 3′:5′- cyclicmonophosphate (8-cpt-cAMP)/2.5 μM forskolin, no activation of ATP efflux was observed for up to 10 min (Fig. 1 and data not shown). Similarly, no ATP efflux was observed when either CFTR- or water-injected oocytes were incubated in the presence of high (140 mM) extracellular Cl− and cAMP/forskolin for 10 min (data not shown). However, prestimulation of oocytes with cAMP/forskolin in Cl−-free Ringers, followed by an acute exposure to 140 mM Cl− Ringers, was associated with a significant and immediate ATP efflux in a subset (∼50%, N = 41) of CFTR-injected oocytes (Fig. 1, A and B). Activation of ATP efflux in oocytes was dependent on CFTR expression, and was never observed in water-injected oocytes (N = 41). CFTR activation was required to elicit ATP efflux since it was not seen when similar protocols were carried out in the absence of cAMP agonists (data not shown). The latency as well as the initial rate of ATP efflux during the first 45 s could not be assessed because of the time required for buffer washes. However, steady-state rates of ATP efflux were quantitated by calculating the slope of relative light units vs. time for each individual oocyte, and correlating the per minute light unit change with standards of known ATP concentration. In a select number of experiments, steady-state rates of ATP efflux in CFTR-injected oocytes remained constant for periods extending up to 3 min, which was the longest time course analyzed. However, in CFTR-injected oocytes that gave very high levels of ATP efflux, slight time-dependent declines in the steady-state rates could be seen, and may represent transient depletions of available intracellular ATP pools. The cumulative data depicted in Fig. 1 C demonstrate a mean ATP efflux rate of 1.99 ± 0.55 pmoles ATP/min (N = 41) for CFTR cRNA-injected oocytes, compared with −0.005 ± 0.004 pmoles ATP/min (N = 41) for water-injected oocytes. The various treatment conditions and their effects on CFTR-modulated ATP release are summarized in Table I.
Figure 1.

Luciferin–luciferase detection of CFTR-modulated ATP release in cRNA-injected Xenopus oocytes. Isolated Xenopus oocytes were injected 24 h after harvest with either 50 ng of CFTR cRNA or an equivalent volume of water, and allowed to incubate at 15°C for 2 d. Single oocytes were then assessed for ATP efflux after serial incubation in: (1) 140 mM Na-gluconate/ HPBR; (2) 140 mM Na-gluconate/HPBR, 200 μM 8-cpt-cAMP, 2.5 μM forskolin; and (3) 140 mM NaCl/HPBR, 200 μM 8-cpt-cAMP, 2.5 μM forskolin. All buffers contained 3.125 mg/ml luciferin–luciferase. A depicts the kinetics of ATP efflux as the mean raw relative light units (± SEM) vs. time for all oocytes (N = 41) analyzed from five independent responder frogs. Arrows denote the time of buffer washes with a duration of ∼45 s. The rates of ATP efflux during each buffer condition (pmoles/min) were calculated for individual oocytes from the slope of relative light units vs. time (for the 100-s reading) using ATP concentration curve standards (1 pmole ATP equaled 90 light units at the sensitivity used in this study). B depicts the raw individual rates of ATP efflux for all oocytes analyzed in each treatment group (N = 41). These cumulative results demonstrate an average of 50% responsiveness in CFTR cRNA-injected and 0% in water-injected oocytes for ATP efflux after elevating extracellular Cl− in the presence of cAMP agonists. The mean rates (± SEM) of ATP efflux for each set of oocytes in B are shown in the inset (C). All experiments represent cumulative data from five independent frogs for which equal numbers of water- and CFTR cRNA-injected oocytes were assessed for ATP efflux within each batch of oocytes. All oocytes analyzed were included in these results. D correlates the magnitude of CFTR-modulated ATP release and forskolin/IBMX-inducible Cl− conductances in oocytes from two independent frogs. These studies were performed by sequentially measuring ATP efflux followed by Wc conductance on the same CFTR cRNA-injected oocyte. ATP efflux experiments were performed as in A, and rates indicated are in the presence of buffer 3 (140 mM NaCl/cAMP/Forskolin). Solid points represent oocytes that gave positive ATP efflux rates, while open boxes are oocytes with no ATP efflux. The dashed line depicts the correlation (r = 0.74) in the magnitude of ATP efflux rates in those oocytes that secreted ATP, with cAMP-inducible Cl− conductances from the same oocytes.
Table I.
Buffer Conditions and Their Effects on ATP Efflux
| Buffer sequence | ATP release | |
|---|---|---|
| 140 mM Na-gluconate | No | |
| 140 mM Na-gluconate, 140 mM Na-gluconate + cAMP/Forsk | No | |
| 140 mM NaCl | No | |
| 140 mM NaCl, 140 mM NaCl + cAMP/Forsk | No | |
| 140 mM Na-gluconate, 140 mM NaCl | No | |
| 140 mM Na-gluconate, 140 mM Na-gluconate + cAMP/Forsk, 140 mM NaCl + cAMP/Forsk | Yes | |
| 140 mM Na-gluconate, 140 mM Na-gluconate + cAMP/Forsk, 140 mM NaCl | Yes | |
| 140 mM NaCl, 140 mM NaCl + cAMP/Forsk, 140 mM Na-gluconate + cAMP/Forsk | No |
Buffer sequences analyzed for CFTR modulated ATP release are represented in order. Various lengths of exposure ranged from 2–10 min as described in Results. Buffer conditions are based on an HPBR solution as described in Materials and Methods. ATP release was indicated as any observable increase in the overall magnitude of relative fluorescence.
Because these studies used mammalian Ringer solutions, additional experiments were performed to compare responses directly using SOS. In these studies, the ATP efflux rate (2.0 ± 1.1 pmoles ATP/min) in SOS buffer (100 mM NaCl) was threefold lower than that seen in the mammalian buffer (140 mM NaCl; 6.3 ± 2.2 pmoles ATP/min). However, activation of CFTR-modulated ATP release was still dependent on cAMP activation and the switch from low to high extracellular Cl−. These findings suggest that buffer osmolarity does not significantly alter the profile of CFTR-modulated ATP release. There were no significant changes in buffer osmolarity after anion replacement for either the HPBR or the SOS buffer series (Table II). Therefore, it is unlikely that the pattern of ATP release seen after changes in extracellular chloride is due to osmolarity changes in assay buffers. As addressed later, the lower ATP efflux rates seen in SOS are likely due to the dependence of CFTR-modulated ATP release on extracellular Cl− concentrations.
Table II.
Osmolarity of Buffers Used in ATP Efflux Assay
| Buffer base | Predominant anion composition | Osmolarity | ||
|---|---|---|---|---|
| mOsmol | ||||
| HPBR | 140 mM NaCl | 303 ± 5 | ||
| 140 mM Na-gluconate | 295 ± 3 | |||
| 140 mM NaBr | 293 ± 4 | |||
| SOS | 100 mM NaCl | 205 ± 2 | ||
| 100 mM Na-gluconate | 204 ± 2 | |||
| 100 mM NaBr | 204 ± 3 |
The osmolarity of these solutions was determined on a Multi-Osmette Model 2430 Automatic Osmometer (Precision Systems, Natick, MA).
ATP Release Cannot be Explained by CFTR-facilitated Changes in Membrane Potential or Cell Volume
Not all CFTR cRNA-injected oocytes that expressed CFTR, as demonstrated by high whole cell (Wc) cAMP-activated Cl− conductances, released ATP after a shift from low to high Cl− extracellular medium in the presence of cAMP agonists (Fig. 1 D). Thus, CFTR expression was necessary, but not sufficient, to confer the ATP permeability. Nevertheless, we considered that elevation of extracellular Cl− might hyperpolarize the cell membrane and drive ATP anions from the cell through unspecified pathways. To determine whether changes in membrane potential during the experimental protocol contributed to the CFTR-associated ATP effluxes, we evaluated the effects of stimulating CFTR-independent Cl− channels in our ATP release assay. Ionomycin (10 μM), which activates endogenous Cl− channels in oocytes by releasing Ca2+ from intracellular stores (Yoshida and Plant, 1992; Liu and Harmann, 1978; Boton et al., 1990), failed to evoke ATP efflux from mock-injected oocytes (Fig. 2, A and B). In contrast, CFTR-injected oocytes from the same batches of oocytes demonstrated substantial ATP effluxes in response to CFTR activation (Fig. 2, A and B). The lack of ATP efflux in ionomycin-treated oocytes was not due to inhibitory effects of ionomycin on luciferin–luciferase activity, as determined by performing the standard ATP concentration calibration in the presence of ionomycin (Fig. 2 C). Treatment of uninjected oocytes with 10 μM ionomycin elicited Cl− conductances (104 ± 28 μs) of comparable magnitude to those in CFTR cRNA-injected oocytes (121 ± 35 μs) stimulated with IBMX/forskolin. Preincubation of oocytes for 5 min in a Cl−-free solution before TEV did not significantly lower the intracellular Cl− concentration (42.54 ± 1.65 mM), as assessed by reversal potential determination. These data therefore demonstrate that activation of a Cl− permeability per se is incapable of stimulating the ATP efflux, and suggest that the efflux observed during activation of CFTR cannot be accounted for by changes in electrical driving forces.
Figure 2.

Absence of ATP efflux after activation of Ca+-activated Cl− conductance in oocytes. To assess whether membrane potential changes due to opening of Cl− channels in oocytes could nonspecifically facilitate ATP efflux, the rates of ATP efflux in cAMP/forskolin stimulated CFTR cRNA- injected oocytes, and ionomycin-treated water-injected oocytes were compared. A demonstrates the mean (± SEM) relative light units produced from oocytes derived from two frogs after exposure to the following sequence of buffers: (1) 140 mM Na-gluconate/HPBR; (2) 140 mM Na-gluconate/HPBR + (200 μM 8-cpt-cAMP, 2.5 μM forskolin, or 10 μM ionomycin); and (3) 140 mM NaCl/HPBR + (200 μM 8-cpt-cAMP, 2.5 μM forskolin, or 10 μM ionomycin). Buffer changes are denoted by arrows, and the treatment conditions are indicated in the legend. B depicts the mean (± SEM) rate of ATP efflux for each buffer condition as calculated from the slope of relative light units vs. time (for the 100-s reading) for each individual oocyte. ATP concentration curve standards were used to calibrate rates in terms of pmoles/min (1 pmole ATP = 90 light units). All oocytes analyzed were included in these results. To demonstrate that ionomycin has no effect on the activity of luciferin–luciferase, we compared standard curves for ATP in the presence of buffers and agonists used for the above study (C). Ionomycin standard curves were measured in Na-gluconate buffers.
Although osmolarity was held constant in the protocols, we evaluated the potential involvement of cell volume perturbations that might arise from Cl− fluxes during the changes in extracellular Cl− concentration. The volumes of oocytes injected with CFTR cRNA were assessed with digital imaging as previously described (Takahashi et al., 1996; Jentsch, 1996). Batches of CFTR cRNA–injected oocytes that demonstrated CFTR-modulated ATP release exhibited undetectable changes (<2%) in cell volume (9.0 × 10−4 ± 0.11 × 10−4 cm3) after exposure to the series of solutions used to activate CFTR-modulated ATP release. The data suggest that substantial changes in oocyte volume do not account for the observed ATP efflux.
Dissociation of Cl− Conductance and ATP Efflux Functions Associated with CFTR
Because the results indicated that CFTR was necessary but not sufficient for ATP permeability, we attempted to identify the important domains in CFTR by comparing wild-type CFTR with two deletion mutants, TMD1 and Δ259-M265V. The TMD1 mutant encompassing the NH2-terminal portion of CFTR encoded the first 369 amino acids (first six transmembrane helices), while in the Δ259-M265V mutant the first 259 amino acids were deleted and the methionine 265 was mutated to a valine. Oocytes expressing the TMD1 CFTR mutant generated ∼30% of the wild-type CFTR Cl− conductance. However, ATP efflux was not associated with this mutant (Fig. 3). In contrast, expression of the Δ259-M265V mutant was associated with near wild-type rates of ATP efflux in the absence of detectable Cl− conductances (Fig. 3). These results demonstrate that the ability of the CFTR to modulate ATP release can be dissociated from its Cl− conducting activity.
Figure 3.

Comparison of CFTR-modulated ATP release between wild-type, TMD1, and Δ259-M265V CFTR cRNA– injected Xenopus oocytes. Results in A demonstrate the mean (± SEM) relative light units produced by CFTR cRNA–injected oocytes exposed to the following order of buffers: (1) 140 mM Na-gluconate/HPBR; (2) 140 mM Na-gluconate/HPBR, 200 μM 8-cpt-cAMP, 2.5 μM forskolin; and (3) 140 mM NaCl/HPBR, 200 μM 8-cpt-cAMP, 2.5 μM forskolin. Buffer changes are denoted by arrows, and cRNA injected is indicated in the legend. B depicts the mean (± SEM) rate of ATP efflux for each buffer condition as calculated from the slope of relative light units vs. time (for the 100-s reading) for each individual oocyte. C depicts the mean (± SEM) Wc chloride conductance values representing the total and delta conductance after adding forskolin/IBMX to the bathing solution (corrected for water-injected background). D gives the mean I/V relationships for each construct in the presence and absence of the cAMP agonists forskolin/IBMX. An equal number (N) of wt CFTR, TMD1, and Δ259-M265V cRNA-injected oocytes were assessed for ATP efflux within each of two batches of oocytes. All oocytes analyzed were included in these results derived from two independent frogs.
Activation of CFTR-modulated ATP Release Demonstrates Selectivity for Extracellular Halides
In addition to Cl−, CFTR is permeable to other halides and small anions, including Br−, I−, F−, and NO3 − (Anderson et al., 1991; Tabcharani et al., 1993). Because changing the extracellular Cl− concentration appeared to be critical for activating CFTR-modulated ATP release in oocytes, we explored the anion dependence of this activation. As indicated in Materials and Methods, all anions tested, with the exception of Br− and Cl−, significantly inhibited the luciferin–luciferase reaction (Fig. 4 B). The anion dependence experiments were therefore limited to Cl− and Br−. Replacement of 140 mM extracellular Cl− with 140 mM Br− caused a fourfold decrease in the rate of CFTR- dependent ATP release (Fig. 4 A), which was reversed when Br− was replaced with Cl− (Fig. 4 A). This halide specificity was also seen in SOS buffers. No effect of changing the extracellular halide was observed in water-injected oocytes (data not shown). Although the magnitude of CFTR-modulated ATP release was threefold lower in the presence of 100 mM Cl− when compared with 140 mM Cl−, the Cl−:Br− halide dependence of ATP release was significantly greater in SOS buffers (Fig. 4 C). The sensitivity of CFTR-modulated ATP release to the external Cl− concentration and the apparent halide selectivity of this phenomenon suggested that a Cl− sensor may exist within CFTR that responds to Cl− concentration changes in the external environment by activating ATP efflux. To test this hypothesis, we examined the dependence of ATP release on the concentration of extracellular Cl− in CFTR cRNA–injected oocytes.
Figure 4.

CFTR-modulated ATP release is influenced by extracellular halides. A depicts the mean (± SEM) relative light units produced by CFTR cRNA-injected oocytes (N = 25) from five frogs. Oocytes were exposed to the following order of buffers: (1) 140 mM Na-gluconate/HPBR; (2) 140 mM Na-gluconate/HPBR, 200 μM 8-cpt-cAMP, 2.5 μM forskolin; (3) 140 mM NaCl/HPBR, 200 μM 8-cpt-cAMP, 2.5 μM forskolin; (4) 140 mM NaBr/HPBR, 200 μM 8-cpt-cAMP, 2.5 μM forskolin; and (5) 140 mM NaCl/HPBR, 200 μM 8-cpt-cAMP, 2.5 μM forskolin. Buffer changes are denoted by arrows. Only oocytes that demonstrated release in the presence of extracellular Cl− (arrow 3) were pooled for this analysis. The mean (± SEM) rates of ATP efflux calculated from the rates for each individual oocyte were 3.3 ± 1.5 pmoles ATP/min (buffer 3), 1.0 ± 0.5 pmoles ATP/min (buffer 4), and 4.0 ± 1.7 pmoles ATP/min (buffer 5). B depicts the affects of various halides and anions on the activity of luciferin–luciferase using standard curves for ATP (no differences were seen in Cl and Br-containing buffers). The halide selectivity of CFTR-modulated ATP release in SOS and HPBR was compared in C. These studies evaluated ATP efflux in wtCFTR cRNA-injected oocytes using the specified order of buffers as shown in the figure (details of buffer composition are described in Materials and Methods). Results depict the mean (± SEM, N = 6) relative light units produced in each buffer with mean (± SEM) rates of ATP efflux shown above each line. Calculated rates of ATP efflux are the mean of rates from individual oocytes in pmoles ATP/min. In this analysis, oocytes that demonstrated no ATP efflux under all of the buffer conditions were not included in these calculations that attempted to compare Cl to Br dependence of ATP release.
Dependence of CFTR-modulated ATP on the External Cl− Concentration
The effect of extracellular Cl− on the rate of ATP efflux in CFTR-expressing oocytes was determined by increasing in a step-wise fashion the extracellular concentration of Cl− from 0 to 140 mM in the presence of cAMP/forskolin. Osmolarity was kept constant throughout the experiments by gluconate replacement. The cumulative results from two independent batches of oocytes demonstrated a sharp threshold for activation of CFTR-modulated ATP release at extracellular concentrations of Cl− >100 mM (Fig. 5 A). Results from two additional batches of oocytes narrowed the range of Cl− necessary for activation of ATP release. The cumulative results from four frogs demonstrated that ATP efflux rates were dramatically increased between 110 and 120 mM extracellular Cl− (Fig. 5 B). In these studies, maximum levels of ATP efflux were fivefold greater in the presence of extracellular Cl− when compared with Br−. Interestingly, when the external Cl− concentration was decreased step-wise after a strong ATP response was induced with 140 mM NaCl, the rate of ATP efflux remained elevated at maximum levels at external Cl− concentrations >100 mM, and only returned to baseline once more significant reductions in extracellular Cl− (<80 mM) were imposed (data not shown). These results suggest that CFTR-modulated ATP release may have differential requirements for extracellular Cl− depending on the direction of the gradient change and/or the state of prior activation. Together, these findings suggest that a chloride sensor within CFTR may be capable of regulating ATP efflux near physiologic concentrations (85–130 mM) of Cl− seen in the airway (Joris et al., 1993; Gilljam et al., 1989; Smith et al., 1996).
Figure 5.

Extracellular Cl− concentration determines the relative activity of CFTR-modulated ATP release. Two independent batches of wtCFTR cRNA- injected Xenopus oocytes were analyzed for the effects of extracellular Cl− concentration on the activation of ATP efflux. An HPBR solution was used throughout these experiments with alterations in the final NaCl concentration facilitated by exchange of an equal molar amount of Na-gluconate with NaCl. The total concentration of NaCl and Na-gluconate was equal to 140 mM throughout these experiments with a constant osmolarity. A demonstrates the mean (± SEM) relative light units produced by CFTR-expressing oocytes for which the extracellular Cl− concentration was incrementally increased by adding NaCl-containing buffers with a constant concentration of luciferin–luciferase enzymes. Between seven and eleven oocytes were evaluated at each Cl− concentration from two independent frogs. At the end of the experiment, Cl− was replaced with Br− to confirm the halide selectivity of the oocytes analyzed. Oocytes that did not demonstrate ATP efflux above water-injected controls (data not shown) were not included in this data set. B depicts the mean (± SEM) rates of ATP efflux from cumulative data of four independent batches of CFTR cRNA-injected oocytes as a function of Cl− concentration. The rate of ATP efflux in the presence of extracellular Br− is also included for reference. The concentration of extracellular Cl− necessary for activation of CFTR-modulated ATP efflux was ∼115 mM (arrow).
Mutations in the CFTR Channel Pore Alter the Halide Dependence of ATP Release
To explore the mechanisms by which changes in the extracellular Cl− concentration affect activation of ATP release, we examined the CFTR mutants R334W, R347P, and R347E. We hypothesized that positively charged residues within the channel pore might bind Cl− and elicit structural changes in CFTR necessary for modulating ATP release. Immunoprecipitation studies using a rabbit antisera raised against a synthetic peptide corresponding to residues 45–65 in the CFTR NH2 terminus demonstrated similar levels of protein expression in wtCFTR, R334W, R347P, and R347E cRNA–injected oocytes (Fig. 6 A). No CFTR protein was detected in water-injected oocytes.
Figure 6.

Electrophysiological properties of CFTR mutants using Wc two-electrode voltage clamp measurements. Mutants of CFTR that alter charged arginine residues within the channel pore including R334W, R347E, and R347P were used. A demonstrates the amount of [35S]methionine-labeled CFTR immunoprecipitated with anti-hCFTR antibodies from oocytes injected with mutant and wild-type CFTR cRNAs. The arrow depicts unglycosylated CFTR protein produced 3 h after injection. The Wc I/V relationships for wtCFTR, R347P, R347E, R334W cRNA, and water-injected oocytes are given in B. Arrows denote the I/V relations before forskolin/IBMX activation in the presence of extracellular Cl− (marked baseline) and after adding forskolin/ IBMX in the presence of extracellular Cl− (Cl) and bromide (Br). Stimulation of CFTR was performed by perfusion of the oocyte with appropriate buffers containing 100 μM IBMX and 10 μM forskolin. ND96 buffer was used throughout the experiment where NaBr replaced NaCl for halide selectivity measurements. The order of buffer perfusion was as follows: (a) ND96 (NaCl); (b) ND96 (NaCl) + forskolin/IBMX; and (c) ND96 (NaBr) + forskolin/IBMX. These results depict average I/V relationships from N experiments for wtCFTR (N = 5), R347E (N = 5), R347P (N = 5), R334W (N = 8), and water (N = 7)-injected oocytes from at least two independent batches of oocytes.
The electrophysiologic properties of wtCFTR and each of the mutants were analyzed in cRNA-injected oocytes by two-electrode voltage clamp. WtCFTR-injected oocytes demonstrated a large increase in slope conductance (ΔGcAMP = 121 ± 35 μs) in response to 10 μM forskolin and 100 μM IBMX (Fig. 6 and Table II). Of note, the conditions used in the ATP efflux experiments (200 μM 8-cpt-cAMP/2.5 μM forskolin) produced approximately tenfold lower conductance changes (data not shown). No cAMP-activated currents were seen in water-injected oocytes under either condition. We therefore used maximum levels of stimulation to facilitate detection of subtle changes in halide selectivities of low-conducting mutants. The stimulated Cl− conductance of R347P was comparable to that of the wtCFTR (ΔGcAMP = 125 ± 28 μs). In contrast, the stimulated Cl− conductances were 4- and 20-fold lower in the R334W and R347E mutants, respectively (Fig. 6 and Table II). The I/V relationships for unstimulated and cAMP-stimulated oocytes in the presence of Cl− and Br− are shown in Fig. 6 and summarized in Table II. The mutants R347P and R334W demonstrated slight alterations in their conductance ratios (GBr/GCl; Br− > Cl−) as compared with wtCFTR (Cl ≅ Br). In contrast, both GBr/GCl and PBr/PCl were significantly altered by the R347E mutation (Table II). These data support previous findings that arginine 347 is important in determining the halide selectivity of the CFTR channel pore (Tabcharani et al., 1993; Anderson et al., 1991).
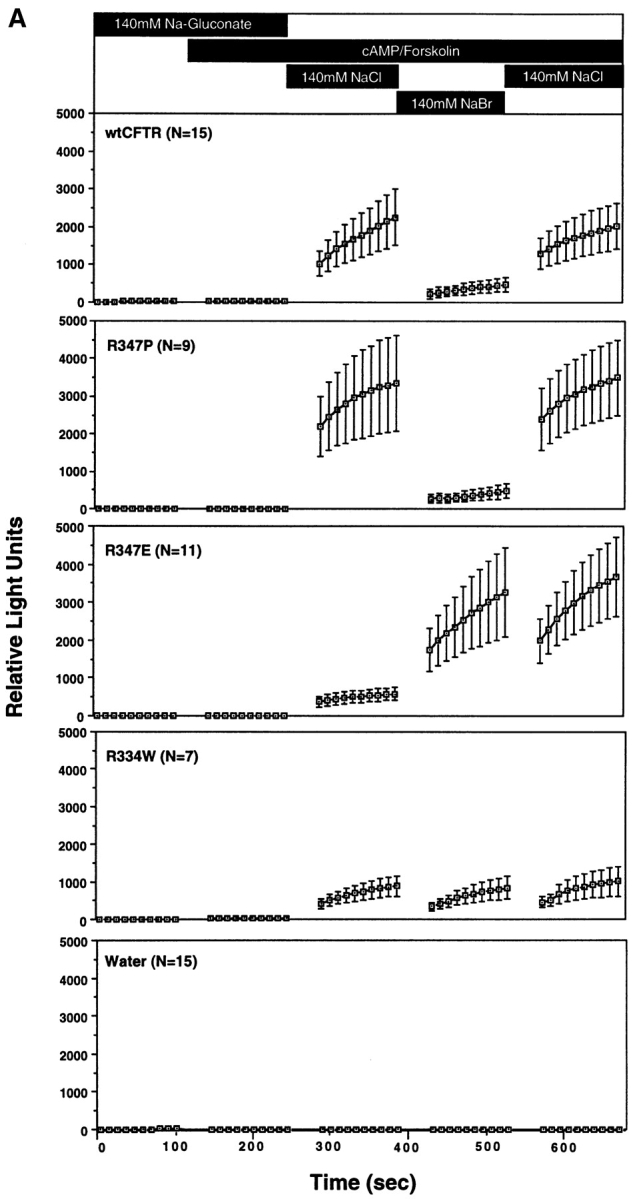
Rates of CFTR-modulated ATP release for the mutant CFTRs are summarized in Fig. 7. These studies were designed to enable comparison of ATP efflux rates in response to extracellular changes in Cl− and Br− within a single oocyte. Frogs that had previously demonstrated CFTR-modulated ATP release in >25% of oocytes were used. As previously demonstrated, exposure of wtCFTR-expressing oocytes to Cl−-free buffers was necessary for activation of ATP efflux following a concentration change to high extracellular Cl− (JATP = 8.7 ± 3.4 pmoles/min). This represents a greater than 400-fold increase in ATP efflux when compared with side-by-side controls in water- injected control oocytes (JATP = 0.021 ± 0.001 pmoles/ min). The typical ATP response curves obtained for wtCFTR demonstrated a halide dependence of Cl− >> Br−. In the presence of extracellular Br−, JATP was approximately fourfold lower (1.9 ± 0.5 pmoles/min) compared with the rate in the presence of Cl−. Again, this inhibition was reversed by replacing extracellular Cl− (Fig. 7 A).
Figure 7.

Halide dependence of CFTR-modulated ATP release is mediated by a chloride sensor within the channel pore. Results in A demonstrate the mean (± SEM) relative light units produced after activation with 8-cpt-cAMP/forskolin and changes in the extracellular halide concentration. The number of oocytes (N) compiled for each mutant are given with the number of independent experiments for wtCFTR = 3, R347P = 2, R334E = 3, R334W = 2, and water = 3. Although all mutants were not always evaluated at one time, CFTR and water-injected controls were always performed alongside of mutants to confirm the integrity of ATP responses in the particular batch of oocytes. B depicts the mean (± SEM) rate of ATP efflux during the last three buffer conditions as calculated from the slope of relative light units vs. time (for the 100-s reading) for each individual oocyte. The halide selectivity of CFTR-modulated ATP release is compared with the halide permeability (PBr/PCl) and conductance (GBr/GCl) ratios for each of the mutants in C. Results depict the mean (± SEM) for each construct as determined from the individual I/V curves (the number of oocytes analyzed are shown in Table II).
The halide dependence of CFTR-modulated ATP release was similarly analyzed in R334W, R347P, and R347E cRNA-injected oocytes. WtCFTR cRNA-injected oocytes were also examined at the same time to assure the responsiveness of the oocytes. The halide dependence of ATP release for R347P CFTR was similar to that of wtCFTR (Cl− >> Br−); JATP in the presence of Cl− (2.0 ± 0.98 pmoles/min) was eightfold greater than that in the presence of Br− (0.26 ± 0.17 pmoles/min; Fig. 7 and Table II). These results correlate with the similar anion conductance and permeability characteristics of this mutant and wtCFTR. In contrast, the R334W mutant had no halide dependence in the activation of ATP release (JATP[Cl] = 3.4 ± 1.3 pmoles/min, JATP [Br] = 3.7 ± 1.5 pmoles/min; Fig. 7). Most importantly, the halide dependence in the R347E mutant was reversed to Br− >> Cl−; JATP in the presence of Cl− (1.5 ± 0.49 pmoles/min) was 7.3-fold lower than that in the presence of Br− (11 ± 4.8 pmoles/min; Fig. 7 and Table II). Interestingly, exposure of R347E-expressing oocytes to extracellular Br− had a lasting affect on JATP, even after extracellular Br− was replaced with Cl−. In summary, these results suggest that residues R334 and R347 may participate in the extracellular halide dependence of CFTR-modulated ATP release. We hypothesize that these positively charged residues which have been suggested to contribute to the Cl− selectivity of the CFTR pore may also contribute to a chloride sensor at which Cl− binding facilitates structural changes in CFTR necessary to modulate ATP release.
Discussion
The results from this study indicate that CFTR can modulate ATP release in Xenopus oocytes, provided that several requirements are met. First, activation of ATP release requires CFTR expression. Second, CFTR must be activated by cAMP. Finally, CFTR-modulated ATP release was dependent on an incremental increase in the extracellular Cl− concentration. These findings suggest a novel Cl−-dependent mechanism by which CFTR regulates ATP release. The complexities associated with activation of this CFTR-mediated phenomenon may in part explain the present controversies concerning CFTR-modulated ATP release.
Possible Mechanisms of CFTR-modulated ATP Release
We evaluated the possibility that activation of Cl− conductance per se, combined with a change of extracellular Cl− concentration, could account for ATP release as a consequence of hyperpolarizing the membrane potential. However, the absence of ATP release in response to ionomycin, which activated a non-CFTR endogenous Ca2+-activated Cl− conductance, ruled out such a mechanism. We also considered a second mechanism involving perturbations of cell volume. In mammalian cells it has been reported that the release of intracellular ATP can be observed under conditions that cause cell swelling (Greger et al., 1993, Koslowsky et al., 1994). However, direct measurements failed to reveal any alteration of cell volume in oocytes that demonstrated CFTR-modulated ATP release. Furthermore, for changes in cell volume to occur in CFTR cRNA but not water-injected oocytes, one would anticipate that Cl− flux through CFTR would be required for movement of NaCl and water into oocytes. However, the CFTR mutant Δ259-M265V, which failed to conduct Cl−, also conferred near wild-type rates of ATP release. In summary, the lack of correlation between CFTR-modulated ATP release with the magnitude of anion conductance suggests that salt and water movement and changes in cell volume are likely not involved in activating ATP release in this system.
Several lines of evidence suggest that ATP and Cl− permeation occur through distinct pathways in CFTR- expressing oocytes. First, the CFTR mutant Δ259-M265V demonstrated near wild-type rates of CFTR-modulated ATP release despite its inability to conduct Cl−. In contrast, TMD1 mutants gave appreciable Cl− conductances in the absence of activatible ATP release. Furthermore, the R347E had a greater than 20-fold reduced GBr as compared with wtCFTR, yet the magnitude of JATP in the presence of extracellular Br− was sixfold higher for R347E when compared with wtCFTR cRNA-injected oocytes. Taken together, these data demonstrate that the permeation pathway associated with CFTR-modulated ATP release is independent of the Cl− conductance pathway in the channel pore. The observations that only a subset of CFTR-expressing oocytes displayed an ATP permeability despite equivalent levels of cAMP inducible Cl− conductances also supports this conclusion. Of note, however, frogs that produced ATP responder oocytes continued to produce oocytes with CFTR-modulated ATP release in subsequent harvests (see Materials and Methods for more detail). We therefore hypothesize that another factor(s) is required in addition to activated CFTR to confer ATP release in Xenopus oocytes. This conclusion is similar to that reached in MDCK cells, where CFTR-dependent ATP conductances were seen at the single channel level in 30% of patches containing active CFTR Cl− currents (Sugita et al., 1998). Despite the possible need of a cofactor, the rate of CFTR-modulated ATP release correlated (r = 0.74) with the magnitude of the Cl− conductance as measured sequentially in the same oocyte (Fig. 1 D). These data suggest that both the abundance of CFTR and some other factor(s) likely affect the overall magnitude of ATP efflux.
CFTR-mediated ATP Release Depends on Changes in Extracellular Chloride Concentration
Unique to the present study evaluating CFTR-mediated ATP release is the dependence of activation on changes in the extracellular Cl− concentration. Chloride-dependent activation of CFTR-modulated ATP release occurred within a very narrow range of Cl− concentrations (110–120 mM), although the range of chloride concentrations necessary to sustain maximal levels of ATP efflux was slightly broader (85–140 mM) once CFTR-modulated ATP release was activated. We hypothesize that the interaction of Cl− with basic residues within extracellular domains or the channel pore may mediate structural changes in CFTR necessary to activate ATP release. To explore this idea, we examined the halide dependence of ATP release and compared it to the halide conductance properties of CFTR channels. The halide dependence experiments were limited to Cl− and Br−, since other anions, including I−, F−, NO3 −, and SCN−, significantly inhibited luciferin–luciferase activity. Our studies of Wc conductance and permeability of wtCFTR in the presence of external Cl− and Br− were similar to previous findings (GCl ≅ GBr and PBr > PCl; Anderson et al., 1991). Interestingly, these ratios (GBr/GCl = 0.93 ± 0.01, PBr/PCl = 1.13 ± 0.02) were significantly larger than the ratio of halide dependence of CFTR-modulated ATP efflux (JATP[Cl] = 8.7 ± 3.4 pmoles ATP/min, JATP[Br] = 1.9 ± 0.52 pmoles ATP/min). Thus, the activation of CFTR-modulated ATP release appears to have a higher specificity for Cl− when compared with the permeability properties of the CFTR channel pore. We therefore hypothesized that a chloride sensor exposed to the external aqueous environment might exist within a region(s) of the CFTR protein that modulates ATP release by responding to changes in the extracellular concentration of Cl−.
A Chloride Sensor Within the CFTR Channel Pore Regulates ATP Release
Obvious candidate sites in the CFTR molecule that might be capable of sensing the extracellular Cl− concentration are aqueous exposed basic residues within the CFTR channel pore. We therefore characterized the effects of arginine mutations R334W, R347P, and R347E on CFTR-modulated ATP release. R347 has been implicated as a predominant site for Cl− binding that at least in part determines the halide selectivity of the channel pore (Anderson et al., 1991). Mutations in R347 have also been shown to alter the number of anions that can simultaneously occupy the pore (Tabcharai et al., 1993). Comparison of halide conductance and permeability of CFTR to the rate of CFTR-mediated ATP release for these mutants suggests that the properties of anion binding in the channel pore may directly affect the activation of ATP efflux. For example, the R347P mutant demonstrated minimal changes in ΔGCl(cAMP), GBr/GCl, and PBr/PCl when compared with wtCFTR (Table II). Although the rate of ATP release in the presence of external Cl− was approximately fourfold lower for this mutant when compared with the wild type, the halide dependence ratio of ATP release rates (JATP[Br]/JATP[Cl] = 0.13) was quite similar to that of wtCFTR (JATP[Br]/JATP[Cl] = 0.22). Thus, a mutation that resulted in minimal alterations in the anion conductance properties of the CFTR channel pore was associated with minimal changes in the halide dependence of ATP release. In contrast, the R347E mutation caused pronounced alterations of both electrophysiologic as well as ATP release properties of CFTR. The significant increase in GBr/GCl when compared with wtCFTR is in agreement with previous studies (Anderson et al., 1991). Concomitantly, the R347E mutation dramatically altered the halide dependence of ATP efflux (JATP[Br]/JATP[Cl] = 7.3) when compared with wtCFTR (JATP[Br]/JATP[Cl] = 0.22). These analyses of the R347P and R347E mutations suggest that the molecular interactions of Cl− and Br− within the channel pore likely influence activation of ATP efflux. The mutant R334W demonstrated no halide dependence in the ATP efflux rates (JATP[Br]/JATP[Cl] = 1.1) when compared with wtCFTR, suggesting that multiple residues within the CFTR channel pore may be involved in halide binding and subsequent conformational changes necessary for activation of CFTR-modulated ATP release.
In summary, mutational analyses implicate basic residues R347 and R334 within the CFTR channel pore as important sites that contribute to the effects of extracellular halides on CFTR-modulated ATP release. Because CFTR is insufficient to promote ATP release, we hypothesize that the properties of the Cl− and Br− binding sites within the channel pore reflect those of a chloride sensor that determines the extent of functional interaction between CFTR and a second cofactor necessary to mediate ATP release (Fig. 8). In wtCFTR, this sensor has a dependence of Cl > Br. We hypothesize that mutations within the channel pore that alter the affinity of Cl− and/or Br− binding are also responsible for changes in the halide dependence of ATP efflux. An unexplained feature is the need for both cAMP agonist stimulation and a change in extracellular Cl− concentration (from low to high external Cl−). We believe that these conditions may be necessary to trap CFTR in a particular conformation that is favorable for activating the ATP release machinery (Fig. 8), and that Cl− binding within the channel pore may facilitate attainment of this conformation. Why CFTR-modulated ATP release is not seen in cAMP-stimulated oocytes bathed in high external Cl− without prior exposure to Cl−-free solutions is currently unknown. However, this feature suggests that CFTR can sense changes of external Cl− concentration and respond by invoking structural changes necessary for interaction with an ATP release pathway (Fig. 8). The magnitude of such concentration changes may be extremely small (10–15 mM Cl−) as indicated by the sharp relationship between ATP efflux and extracellular Cl− concentration (Fig. 5).
Figure 8.
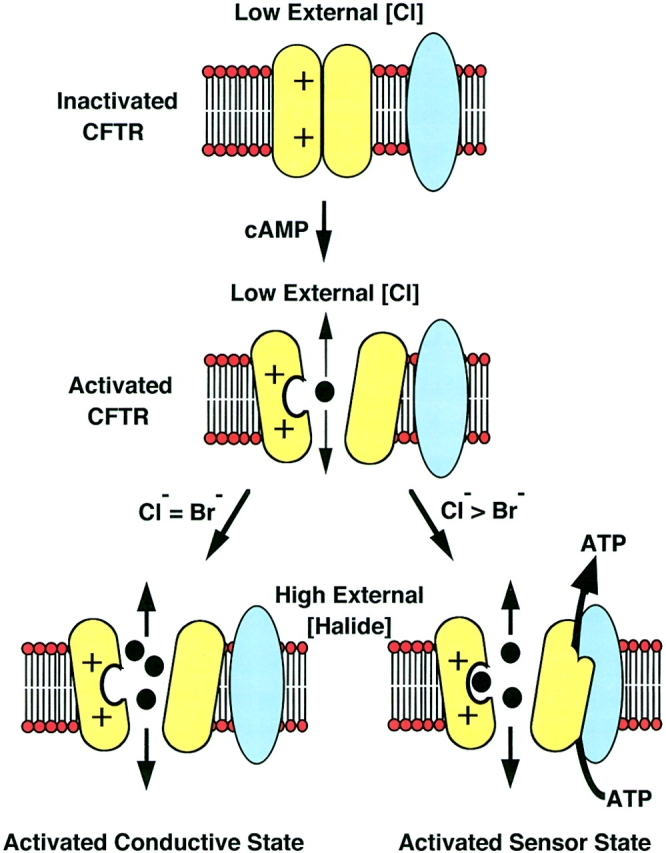
Chloride sensor model for CFTR activation of ATP release. The proposed mechanism for CFTR-modulated ATP release in Xenopus oocytes must fit several criteria. First, extracellular Cl− and cAMP agonist stimulation are necessary but not sufficient to activate CFTR-modulated ATP release. Second, the fact that not all batches of oocytes that demonstrated CFTR-modulated Cl− conductance were capable of ATP efflux, suggests that a cofactor(s) (blue) is needed for CFTR-modulated ATP release (CFTR is yellow). The dependency of CFTR-modulated ATP release on the external Cl− concentration and the effects of mutagenesis of putative pore residues in CFTR suggest that Cl− may bind within the channel pore to activate structural changes in CFTR necessary for the appropriate interactions with a cofactor(s)- dependent ATP release pathway. This model also incorporates the altered halide selectivity of CFTR mediated conductance (Cl = Br) and ATP efflux (Cl > Br) as dependent on the affinity for halide binding at the chloride sensor within the channel pore.
Implications in CF Airways Disease
The mechanism(s) by which CFTR regulates ionic composition of surface airway fluid are only beginning to be understood. Several studies have demonstrated regulatory affects of CFTR on other epithelial ion channels such as ENaC and the ORCC (Egan et al., 1992; Gabriel et al., 1993; Schweibert et al., 1995; Jovov et al., 1995; Grubb et al., 1994; Chinet et al., 1994; Johnson et al., 1995; Hyde et al., 1993; Ismailov et al., 1996). Defects in the functional properties of both the ORCC and ENaC have been observed in CF airways disease (Guggino, 1993; Gabriel et al., 1993; Stutts et al., 1995; Kunzelmann et al., 1997; Iwase et al., 1994). Previous studies have implicated CFTR-modulated ATP release as likely mechanisms by which CFTR regulates the ORCC through P2U purinergic receptors pathways (Schwiebert et al., 1995). The relevance of this dysregulation in CF airways disease is currently unknown, but may reflect one potential mechanism by which CFTR regulates Cl− transport in the airways. Findings from the present study suggest that CFTR can sense the extracellular Cl− concentration in the airways and respond by activating ATP release pathways, which may in turn be responsible for regulating other channels in the apical membrane of epithelial cells. Because the concentration of Cl− necessary for activation of CFTR-modulated ATP release is within the physiologic range of the known Cl− concentration in the airway (85– 130 mM; Smith et al., 1996; Gilljam et al., 1989; Joris et al., 1993), we speculate that this regulatory function of CFTR may act as a salt concentration sensor important in maintaining airway electrolyte balance. Such mechanisms that maintain the appropriate salt concentration in the airway have important consequences on the antibacterial activity of surface airway fluid (Smith et al., 1996).
Table III.
Summary of Electrophysiologic Measurements on CFTR Mutants
| Wild type | R347P | R347E | R334W | Mock | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ΔGCl (cAMP) (μs) | 121 ± 35 | 125 ± 28 | 5.3 ± 2.1 | 32 ± 20 | −0.61 ± 0.62 | |||||
| (N = 5) | (N = 5) | (N = 5) | (N = 8) | (N = 7) | ||||||
| GBr /GCl | 0.93 ± 0.01 | 1.12 ± 0.01 | 2.36 ± 0.23 | 1.12 ± 0.03 | ||||||
| (N = 5) | (N = 5) | (N = 5) | (N = 8) | − | ||||||
| PBr /PCl | 1.13 ± 0.02 | 1.02 ± 0.01 | 1.00 ± 0.04 | 1.22 ± 0.02 | ||||||
| (N = 5) | (N = 5) | (N = 5) | (N = 8) | − | ||||||
| JATP (Cl) | 8.7 ± 3.4 | 2.0 ± 0.98 | 1.5 ± 0.49 | 3.4 ± 1.3 | 0.021 ± 0.001 | |||||
| (N = 15) | (N = 9) | (N = 11) | (N = 7) | (N = 15) | ||||||
| JATP (Br) | 1.9 ± 0.51 | 0.26 ± 0.17 | 11.0 ± 4.8 | 3.7 ± 1.5 | 0.013 ± 0.006 | |||||
| (N = 15) | (N = 9) | (N = 11) | (N = 7) | (N = 15) | ||||||
| JATP (Br)/JATP (Cl) | 0.22 | 0.13 | 7.3 | 1.1 | − |
CFTR cRNAs were injected into Xenopus oocytes and evaluated for both electrophysiologic and ATP release characteristics. ΔGCl (cAMP), change in Cl− conductance in response to IBMX/forskolin; GBr/GCl, halide conductance ratio; PBr/PCl, halide permeability ratio, JATP (Cl), rate of ATP efflux in the presence of extracellular Cl− (pmoles ATP/min); JATP (Br), rate of ATP efflux in the presence of extracellular Br− (pmoles ATP/min); JATP (Br)/JATP (Cl), halide dependence ratio of ATP efflux. Values represent the Mean ± SEM with N = number of oocytes analyzed from two to three independent frogs.
Acknowledgments
We thank Dr. Welsh for his mutant CFTR cDNAs R334W, R347E, and R347P, and his thoughtful review of this manuscript.
We gratefully acknowledge the support of the Cystic Fibrosis Foundation for its fellowship support of Dr. Jiang, grant support to J.K. Foskett, W.B. Guggino, and J.F. Engelhardt (9720), and National Institutes of Health DK49136 (J.F. Engelhardt), DK48977 (W.B. Guggino), and HL47122 (W.B. Guggino).
Abbreviations used in this paper
- ABC
ATP-binding cassette
- CF
cystic fibrosis
- ORCC
outwardly rectifying Cl− channel
- TEV
two-electrode voltage clamp
- Vm
transmembrane potential
- Wc
whole cell
Footnotes
Address all correspondence to Dr. John F. Engelhardt, Ph.D., Associate Professor, University of Iowa, Department of Anatomy and Cell Biology, 1-111 BSB, 51 Newton Road, Iowa City, IA 52242-1109. Tel.: (319) 335-7753. Fax: (319) 335-6581. E-mail: john-engelhardt@uiowa.edu
References
- Abraham EH, Prat AG, Gerweck L, Seneveratne T, Arceci RJ, Kramer R, Guidotti G, Cantiello HF. The multidrug resistance (mdr1) gene product functions as an ATP channel. Proc Natl Acad Sci USA. 1993;90:312–316. doi: 10.1073/pnas.90.1.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Awqati Q. Regulation of ion channels by ABC transporters that secrete ATP. Science. 1995;269:805–806. doi: 10.1126/science.7543697. [DOI] [PubMed] [Google Scholar]
- Ames GF. Bacterial periplasmic transport systems: structure, mechanism, and evolution. Annu Rev Biochem. 1986;55:397–425. doi: 10.1146/annurev.bi.55.070186.002145. [DOI] [PubMed] [Google Scholar]
- Anderson MP, Gregory RJ, Thompson S, Souza DW, Sucharita P, Mulligan RC, Smith AE, Welsh MJ. Demonstration that CFTR is a Cl−channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Bear CE, Li CH, Kartner N, Bridges RJ, Jensen TJ, Ramjeesingh M, Riordan JR. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR) Cell. 1992;68:809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- Boton R, Dascal N, Gillo B, Lass Y. Two calcium-activated Cl− conductances in Xenopus laevisoocytes permeabilized with the ionophore A23187. J Physiol. 1989;408:511–534. doi: 10.1113/jphysiol.1989.sp017473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantiello HF, Jackson GR, Jr, Grosman CF, Prat AG, Borkan SC, Wang Y, Reisin IL, O'Riordan CR, Ausiello DA. Electrodiffusional ATP movement through the cystic fibrosis transmembrane conductance regulator. Am J Physiol. 1998;274:C799–C809. doi: 10.1152/ajpcell.1998.274.3.C799. [DOI] [PubMed] [Google Scholar]
- Chinet TC, Fullton JM, Yankaskas JR, Boucher RC, Stutts MJ. Mechanism of sodium hyperabsorption in cultured cystic fibrosis nasal epithelium: a patch-clamp study. Am J Physiol. 1994;266:C1061–C1068. doi: 10.1152/ajpcell.1994.266.4.C1061. [DOI] [PubMed] [Google Scholar]
- Drumm ML, Pope HA, Cliff WH, Rommens JM, Marvin SA, Tsui LC, Collins FS, Frizzell RA, Wilson JM. Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell. 1990;62:1227–1233. doi: 10.1016/0092-8674(90)90398-x. [DOI] [PubMed] [Google Scholar]
- Egan M, Flotte T, Afione S, Solow R, Zeitlin PL, Carter BJ, Guggino WB. Defective regulation of outwardly rectifying Cl−channels by protein kinase A corrected by insertion of CFTR. Nature. 1992;358:581–584. doi: 10.1038/358581a0. [DOI] [PubMed] [Google Scholar]
- Gabriel SE, Clarke LL, Boucher RC, Stutts MJ. CFTR and outward rectifying Cl−channels are distinct proteins with a regulatory relationship. Nature. 1993;363:263–268. doi: 10.1038/363263a0. [DOI] [PubMed] [Google Scholar]
- Gilljam H, Eilin A, Strandvik B. Increased bronchial chloride concentration in cystic fibrosis. Scand J Clin Lab Invest. 1989;49:121–124. doi: 10.3109/00365518909105409. [DOI] [PubMed] [Google Scholar]
- Greger R, Allert N, Frobe U, Normann C. Increase in cytosolic Ca2+ regulates exocytosis and Cl−conductance in HT29 cells. Pflugers Arch. 1993;424:329–334. doi: 10.1007/BF00384360. [DOI] [PubMed] [Google Scholar]
- Gregory RJ, Cheng SH, Rich DP, Marshall J, Paul S, Hehir K, Ostedgaard L, Klinger KW, Welsh MJ, Smith AE. Expression and characterization of the cystic fibrosis transmembrane conductance regulator. Nature. 1990;347:382–386. doi: 10.1038/347382a0. [DOI] [PubMed] [Google Scholar]
- Grubb BR, Vick RN, Boucher RC. Hyperabsorption of Na+ and raised Ca(2+)-mediated Cl−secretion in nasal epithelia of CF mice. Am J Physiol. 1994;266:C1478–C1483. doi: 10.1152/ajpcell.1994.266.5.C1478. [DOI] [PubMed] [Google Scholar]
- Grygorczyk R, Tabcharani JA, Hanrahan JW. CFTR channels expressed in CHO cells do not have detectable ATP conductance. J Membr Biol. 1996;151:139–148. doi: 10.1007/s002329900065. [DOI] [PubMed] [Google Scholar]
- Guggino WB. Outwardly rectifying chloride channels and CF: a divorce and remarriage. J Bioenerg Biomembr. 1993;25:27–35. doi: 10.1007/BF00768065. [DOI] [PubMed] [Google Scholar]
- Higgins CF, Hyde SC, Mimmack MM, Gileadi U, Gill DR, Gallagher MP. Binding protein-dependent transport systems. J Bioenerg Biomembr. 1990;22:571–592. doi: 10.1007/BF00762962. [DOI] [PubMed] [Google Scholar]
- Hyde SC, Emsley P, Hartshorn MJ, Mimmack MM, Gileadi U, Pearce SR, Gallagher MP, Gill DR, Hubbard RE, Higgins CF. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature. 1990;346:362–365. doi: 10.1038/346362a0. [DOI] [PubMed] [Google Scholar]
- Hyde SC, Gill DR, Higgins CF, Trezise AE, MacVinish LJ, Cuthbert AW, Ratcliff R, Evans MJ, Colledge WH. Correction of the ion transport defect in cystic fibrosis transgenic mice by gene therapy. Nature. 1993;362:250–255. doi: 10.1038/362250a0. [DOI] [PubMed] [Google Scholar]
- Ho K. The ROMK-cystic fibrosis transmembrane conductance regulator connection: new insights into the relationship between ROMK and cystic fibrosis transmembrane conductance regulator channels. Curr Opin Nephrol Hypertens. 1998;7:49–58. doi: 10.1097/00041552-199801000-00009. [DOI] [PubMed] [Google Scholar]
- Ismailov II, Awayda MS, Jovov B, Berdiev BK, Fuller CM, Dedman JR, Kaetzel MA, Benos DJ. Regulation of epithelial sodium channels by the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271:4725–4732. doi: 10.1074/jbc.271.9.4725. [DOI] [PubMed] [Google Scholar]
- Iwase N, Sasaki T, Shimura S, Yamamoto M, Suzuki S, Shirato K. ATP-induced Cl−secretion with suppressed Na+ absorption in rabbit tracheal epithelium. Respir Physiol. 1997;107:173–180. doi: 10.1016/s0034-5687(96)02516-9. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Chloride channels: a molecular perspective. Curr Opin Neurobiol. 1996;6:303–310. doi: 10.1016/s0959-4388(96)80112-7. [DOI] [PubMed] [Google Scholar]
- Johnson LG, Boyles SE, Wilson J, Boucher RC. Normalization of raised sodium absorption and raised calcium-mediated Cl−secretion by adenovirus-mediated expression of cystic fibrosis transmembrane conductance regulator in primary human cystic fibrosis airway epithelial cells. J Clin Invest. 1995;95:1377–1382. doi: 10.1172/JCI117789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joris L, Dab I, Quinton PM. Elemental composition of human airway surface fluid in healthy and diseased airways. Am Rev Respir Dis. 1993;148:1633–1637. doi: 10.1164/ajrccm/148.6_Pt_1.1633. [DOI] [PubMed] [Google Scholar]
- Jovov B, Ismailov II, Berdiev BK, Fuller CM, Sorscher EJ, Dedman JR, Kaetzel MA, Benos DJ. Interaction between cystic fibrosis transmembrane conductance regulator and outwardly rectified Cl−channels. J Biol Chem. 1995;270:29194–29200. doi: 10.1074/jbc.270.49.29194. [DOI] [PubMed] [Google Scholar]
- Kartner N, Hanrahan JW, Jensen TJ, Naismith AL, Sun SZ, Ackerley CA, Reyes EF, Tsui LC, Rommens JM, Bear CE, et al. Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell. 1991;64:681–691. doi: 10.1016/0092-8674(91)90498-n. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Widdicombe JH. Halide transport in Xenopusoocytes. J Physiol. 1991;443:587–599. doi: 10.1113/jphysiol.1991.sp018853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslowsky T, Hug T, Ecke D, Klein P, Greger R, Gruenert DC, Kunzelmann K. Ca(2+)- and swelling-induced activation of ion conductances in bronchial epithelial cells. Pflugers Arch. 1994;428:597–603. doi: 10.1007/BF00374583. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Kiser GL, Schreiber R, Riordan JR. Inhibition of epithelial Na+ currents by intracellular domains of the cystic fibrosis transmembrane conductance regulator. FEBS Lett. 1997;400:341–344. doi: 10.1016/s0014-5793(96)01414-7. [DOI] [PubMed] [Google Scholar]
- Li C, Ramjeesingh M, Bear CE. Purified cystic fibrosis transmembrane conductance regulator (CFTR) does not function as an ATP channel. J Biol Chem. 1996;271:11623–11626. doi: 10.1074/jbc.271.20.11623. [DOI] [PubMed] [Google Scholar]
- Liu CM, Harmann TE. Characterization of ionomycin as a calcium innophore. J Biol Chem. 1978;253:5892–5896. [PubMed] [Google Scholar]
- McNicholas CM, Guggino WB, Schwiebert EM, Hebert SC, Giebisch G, Egan ME. Sensitivity of a renal K+ channel (ROMK2) to the inhibitory sulfonylurea compound glibenclamide is enhanced by coexpression with the ATP-binding cassette transporter cystic fibrosis transmembrane regulator. Proc Natl Acad Sci USA. 1996;23:8083–8088. doi: 10.1073/pnas.93.15.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas CM, Nason MW, Jr, Guggino WB, Schwiebert EM, Hebert SC, Giebisch G, Egan ME. A functional CFTR-NBF1 is required for ROMK2-CFTR interaction. Am J Physiol. 1997;273:F843–F848. doi: 10.1152/ajprenal.1997.273.5.F843. [DOI] [PubMed] [Google Scholar]
- Morral N, Llevadot R, Casals T, Gasparini P, Macek M, Jr, Dork T, Estivill X. Independent origins of cystic fibrosis mutations R334W, R347P, R1162X, and 3849 + 10kbC → T provide evidence of mutation recurrence in the CFTR gene. Am J Hum Genet. 1994;55:890–898. [PMC free article] [PubMed] [Google Scholar]
- Pasyk EA, Foskett JK. Cystic fibrosis transmembrane conductance regulator-associated ATP and adenosine 3′-phosphate 5′-phosphosulfate channels in endoplasmic reticulum and plasma membranes. J Biol Chem. 1997;272:7746–7751. doi: 10.1074/jbc.272.12.7746. [DOI] [PubMed] [Google Scholar]
- Piazza Carroll, T., M.M. Morales, S.B. Fulmer, S.S. Allen, T.R. Flotte, G.R. Cutting, and W.B. Guggino. Alternate translation initiation codons can create functional forms of cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1995;270:11914–11946. doi: 10.1074/jbc.270.20.11941. [DOI] [PubMed] [Google Scholar]
- Prat AG, Reisin IL, Ausiello DA, Cantiello HF. Cellular ATP efflux by the cystic fibrosis transmembrane conductance regulator. Am J Physiol. 1996;270:C538–C545. doi: 10.1152/ajpcell.1996.270.2.C538. [DOI] [PubMed] [Google Scholar]
- Reddy MM, Quinton PM, Haws C, Wine JJ, Grygorczyk R, Tabcharani JA, Hanrahan JW, Gunderson KL, Kopito RR. Failure of the cystic fibrosis transmembrane conductance regulator to conduct ATP. Science. 1996;271:1876–1879. doi: 10.1126/science.271.5257.1876. [DOI] [PubMed] [Google Scholar]
- Reisin IL, Prat AG, Abraham EH, Amara JF, Gregory RJ, Ausiello DA, Cantiello HF. The cystic fibrosis transmembrane conductance regulator is a dual ATP and Cl−channel. J Biol Chem. 1994;269:20584–20591. [PubMed] [Google Scholar]
- Schwiebert EM, Egan ME, Hwang TH, Fulmer SB, Allen SS, Cutting GR, Guggino WB. CFTR regulates outwardly rectifying Cl− channels through an autocrine mechanism involving ATP . Cell. 1995;81:1063–1073. doi: 10.1016/s0092-8674(05)80011-x. [DOI] [PubMed] [Google Scholar]
- Schwiebert EM, Morales MM, Devidas S, Egan ME, Guggino WB. Chloride channel and chloride conductance regulator domains of CFTR, the cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA. 1998;95:2674–2679. doi: 10.1073/pnas.95.5.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Rich DP, Ostedgaard LS, Gregory RJ, Smith AE, Welsh MJ. Mutations in CFTR associated with mild-disease-form Cl−channels with altered pore properties. Nature. 1993;362:160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85:229–236. doi: 10.1016/s0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- Stuhmer W. Electrophysiological recording from Xenopusoocytes. Methods Enzymol. 1992;207:319–339. doi: 10.1016/0076-6879(92)07021-f. [DOI] [PubMed] [Google Scholar]
- Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- Stutts MJ, Rossier BC, Boucher RC. Cystic fibrosis transmembrane conductance regulator inverts protein kinase A-mediated regulation of epithelial sodium channel single channel kinetics. J Biol Chem. 1997;272:14037–14040. doi: 10.1074/jbc.272.22.14037. [DOI] [PubMed] [Google Scholar]
- Sugita M, Yue Y, Foskett JK. CFTR Cl-channel and CFTR-associated ATP channel: distinct pores regulated by common gates. EMBO (Eur Mol Biol Organ) J. 1998;17:898–908. doi: 10.1093/emboj/17.4.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabcharani JA, Rommens JM, Hou Y-X, Chang X-B, Tsui L-C, Riordan JR, Hanrahan JW. Multi-ion pore behavior in the CFTR Cl−channel. Nature. 1993;366:79–82. doi: 10.1038/366079a0. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Watkins SC, Howard M, Frizzell RA. CFTR- dependent membrane insertion is linked to stimulation of the CFTR chloride conductance. Am J Physiol. 1996;271:C1887–C1894. doi: 10.1152/ajpcell.1996.271.6.C1887. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Plant S. Mechanism of release of Ca2+ from intracellular stores in response to ionomycin in oocytes of the frog Xenopus laevis. . J Physiol. 1992;458:307–318. doi: 10.1113/jphysiol.1992.sp019419. [DOI] [PMC free article] [PubMed] [Google Scholar]