

Abstract
We used melanophores, cells specialized for regulated organelle transport, to study signaling pathways involved in the regulation of transport. We transfected immortalized Xenopus melanophores with plasmids encoding epitope-tagged inhibitors of protein phosphatases and protein kinases or control plasmids encoding inactive analogues of these inhibitors. Expression of a recombinant inhibitor of protein kinase A (PKA) results in spontaneous pigment aggregation. α-Melanocyte-stimulating hormone (MSH), a stimulus which increases intracellular cAMP, cannot disperse pigment in these cells. However, melanosomes in these cells can be partially dispersed by PMA, an activator of protein kinase C (PKC). When a recombinant inhibitor of PKC is expressed in melanophores, PMA-induced pigment dispersion is inhibited, but not dispersion induced by MSH. We conclude that PKA and PKC activate two different pathways for melanosome dispersion. When melanophores express the small t antigen of SV-40 virus, a specific inhibitor of protein phosphatase 2A (PP2A), aggregation is completely prevented. Conversely, overexpression of PP2A inhibits pigment dispersion by MSH. Inhibitors of protein phosphatase 1 and protein phosphatase 2B (PP2B) do not affect pigment movement. Therefore, melanosome aggregation is mediated by PP2A.
Keywords: microtubules, protein phosphorylation, microtubule motors, kinesin II, cytoplasmic dynein
Melanophores, pigment cells of lower vertebrates, transport organelles containing the black pigment melanin synchronously towards or away from the cell center, providing the mechanism by which fish and amphibia change color. Pigment aggregation and dispersion is dependent on the integrity of cytoplasmic microtubules (Murphy and Tilney, 1974; McNiven and Porter, 1984) and actin filaments (Malawista, 1971; McGuire and Moellmann, 1972; Rodionov et al., 1998; Rogers and Gelfand, 1998), and is accomplished by microtubule motors and by myosin (Clark and Rosenbaum, 1982; Rodionov et al., 1987; Rogers and Gelfand, 1998; Rogers et al., 1997). In pigment cells, as in most other cells, the minus ends of microtubules are connected with the perinuclear centrosome, whereas the plus ends are located at the cell periphery (Euteneuer and McIntosh, 1981). Aggregation is mediated by the minus end–directed microtubule motor cytoplasmic dynein (Nilsson and Wallin, 1997), whereas dispersion is due to the coordinate activities of a plus end microtubule motor and a myosin, most likely kinesin II and myosin V, respectively (Rogers and Gelfand, 1998; Rogers et al., 1997). The signaling pathways and mechanisms for regulating these motors are largely unknown.
Melanophores provide an excellent system for studying molecular motor regulation because the movement of pigment organelles is activated by known physiological signals. In the case of Xenopus laevis melanophores, pigment aggregation is triggered by melatonin, which binds to its membrane receptor and reduces the concentration of cAMP in the cytoplasm through the action of a coupled inhibitory G protein (White et al., 1987; Sugden, 1992). A physiological signal for pigment dispersion is provided by melanocyte-stimulating hormone (MSH)1, which increases the intracellular concentration of cAMP (Daniolos et al., 1990). Thus, the direction of melanosome movement in Xenopus melanophores correlates with the level of cAMP in the cytoplasm. A similar correlation is found for other pigment cells including Tilapia mossambica melanophores (Rozdzial and Haimo, 1986), Pteophyllum scalare melanophores (Sammak et al., 1992), and Carassius auratus xanthophores (Palazzo et al., 1989).
Dispersion of pigment in melanophores can also be induced by activators of PKC, such as phorbol esters, mezerein, and diacylglycerol (Sugden and Rowe, 1992; Graminski et al., 1993), and the hormone endothelin 3 (McClintock et al., 1996). Unlike MSH-induced dispersion, dispersion induced by phorbol esters causes the cell to form fine dendritic processes (Sugden and Rowe, 1992), and does not change the intracellular cAMP concentration (Graminski et al., 1993), indicating that two different signaling pathways are involved in dispersing pigment. We directly addressed this question using specific recombinant inhibitors of protein kinases. These proteins contain peptide sequences derived from regulatory pseudosubstrate regions of protein kinases and act as potent and selective inhibitors of the enzymes in vivo.
Pigment dispersion requires the activity of protein kinases and conversely, pigment aggregation requires the activity of a phosphatase. The phosphatase inhibitor okadaic acid has been shown to inhibit aggregation in Xenopus and angelfish melanophores, implicating PP1 and/or PP2A (Cozzi and Rollag, 1992; Sammak et al., 1992). On the other hand, it has been reported that the Ca2+/calmodulin-dependent protein phosphatase, PP2B (calcineurin), is required for pigment aggregation in melanophores of the African cichlid, Tilapia mossambica (Thaler and Haimo, 1990). To identify the phosphatase involved in aggregation in Xenopus melanophores we used specific inhibitors of PP1, PP2A, and PP2B. In addition, we overexpressed the catalytic subunit of PP2A.
We demonstrate that the MSH-stimulated pathway for melanosome dispersion depends solely on PKA activity and does not require PKC. The PMA-activated PKC pathway, on the other hand, can only partially disperse melanosomes in the absence of PKA activity. In addition, we show that PP2A, but not PP1 or PP2B, is required for melanosome aggregation in Xenopus melanophores. We also demonstrate differences in the pattern of protein phosphorylation on melanosomes purified from cells aggregating and dispersing pigment.
Materials and Methods
Cell Line
An immortalized cell line of melanophores from Xenopus laevis (gift of M. Lerner, University of Texas Southwestern Medical Center, Dallas, TX) was cultured at 27°C in 0.7× L-15 medium (GIBCO BRL, Grand Island, NY) supplemented with 5% fetal bovine serum (HyClone, Logan, UT), 5 μg/ml insulin, and 100 μg/ml each of penicillin and streptomycin as described (Daniolos et al., 1990; Rogers et al., 1997). Cells were transferred to the same medium without serum 24 h before induction of aggregation or dispersion of melanosomes by hormones.
Constructs
We have prepared recombinant constructs that contain inhibitory sequences in the context of larger polypeptides under the control of the Rous sarcoma virus promoter and rabbit β-globin splicing/polyadenylation sites. We matched the structures of the recombinant PKC and Ca2+/ calmodulin-dependent kinase II (CaMK) inhibitors to the sequences of the short conserved peptides located at positions 19–36 in various forms of protein kinase C (PKC) (House and Kemp, 1987) and positions 273–302 of CaMK (Smith et al., 1990, 1992), respectively. The recombinant PKA inhibitors included sequences of the natural heat-stable protein kinase A (PKA) inhibitor PKI (Van Patten et al., 1991). Mutant forms of the inhibitors carrying inactivating amino acid substitutions in the kinase recognition sequence served as controls. We selected the most potent inhibitors based on their ability to antagonize the activation of reporter genes in cultured cells by various stimuli (cAMP, PMA, Ca2+, depolarization). The inhibitory activity of these constructs is high enough to completely prevent the activation of a reporter promoter by the corresponding stimuli; this action is specific, as mutant inactive constructs fail to block reporter induction. Furthermore, the activity of a particular inhibitor does not interfere with the action of the nontarget kinases, reflecting vast differences (100– 10,000-fold) in dissociation constant (Ki) for the cognate and for the unrelated enzymes in vitro. The recombinant inhibitor of protein phosphatase 1 (PP1) includes amino acids 9–54 of the natural rabbit skeletal muscle inhibitor I-1 with a threonine to aspartate mutation at amino acid 35 to mimic phosphorylated threonine (Alberts et al., 1994). The protein phosphatase 2B (PP2B) inhibitor includes amino acids 457–482 of PP2B, the autoinhibitory domain (Hashimoto et al., 1990). Each of the recombinant proteins contains an epitope derived from the influenza virus hemagglutinin protein (HA epitope) (Field et al., 1988) at the COOH-terminal end. This tag allowed visualization of the chimeric products in the transfected cells by fluorescence microscopy using the anti-HA monoclonal antibody 12CA5 (Field et al., 1988). Plasmids pNP210 and pNP211 encode the active and inactive forms of the PKA inhibitor, respectively; pNP212 and pNP214 encode the active and inactive forms of the PKC inhibitor; pNP215 and pNP216 encode the active and inactive forms of the CaMK inhibitor, pNP231 encodes the PP1 inhibitor I-1, and pNP241 encodes the PP2B inhibitor (Table I).
Table I.
List of Plasmids Encoding Inhibitors of Protein Kinases and Protein Phosphatases and Summary of Their Effects on Hormone-induced Melanosome Movement
| Encoded protein | Plasmid | Melatonin (aggregation) | MSH (dispersion) | PMA (dispersion) | Melatonin + PMA | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| PKA inhibitor, active | pNP210 | +* | −‡ | ± § | ± | |||||
| PKA inhibitor, inactive | pNP211 | + | + | + | + | |||||
| PKC inhibitor, active | pNP212 | + | + | + | − | |||||
| PKC inhibitor, inactive | pNP214 | + | + | + | + | |||||
| CAMKII inhibitor, active | pNP215 | + | + | + | + | |||||
| CAMKII inhibitor, inactive | pNP216 | + | + | + | + | |||||
| small t (PP2A inhibitor) | pCEP4-Sm t | − | + | |||||||
| PP1 inhibitor | pNP231 | + | + | |||||||
| PP2B inhibitor | pNP241 | + | + | |||||||
| GFP | pEGFP-C1 | + | + | |||||||
| PP2A | pGRE5-2 (T) 36 | + | − |
Dispersion or aggregation occurs normally.
No dispersion or aggregation.
Dispersion is partially inhibited.
Plasmid pCEP4-Sm t (gift of S. Shenolikar, Department of Pharmacology, Duke University Medical Center, Durham, NC) encodes the full-length wild-type SV-40 small t antigen, an inhibitor of protein phosphatase 2A (PP2A) (Alberts et al., 1994). A plasmid encoding green fluorescent protein (GFP), pEGFP-C1 (Clontech, Palo Alto, CA), was used in control transfections for the phosphatase inhibitors (see Results). pGRE5-2 (T) 36 encoding the PP2A C subunit tagged with an HA epitope-derived peptide under the control of a dexamethasone-inducible promoter was a gift of D. Pallas, (Department of Biochemistry and Winship Cancer Center, Emory University School of Medicine, Atlanta, GA) (Ogris et al., 1997).
Transfections
NIH 3T3 fibroblasts were transfected with TransFast transfection reagent (Promega Corp., Madison, WI) according to the manufacturer's protocol and then incubated for 48 h in DME with 10% calf serum to allow protein expression before the experiments were performed. Melanophores were transfected by electroporation at 450 V, 200 ohms, and 250 microfarads using a Bio-Rad Gene Pulser II (Bio-Rad Laboratories, Hercules, CA) according to the protocol of Graminski et al. (1993) and then plated onto 10-cm tissue culture plates. They were replated onto polylysine-coated coverslips 24 h later. The medium was changed to serum-free L-15 on the third day; experiments were performed on the fourth day. Transfection efficiencies varied depending on the plasmid and on the transfection; ∼0.5% of cells transfected with pCEP4-Sm t stained positively for the small t antigen, whereas inhibitor expression was detected in 10–50% of cells transfected with the other plasmids.
Treatment With Drugs
For the experiments with okadaic acid, cells were incubated in serum-free medium containing 0.5 nM-2 μM okadaic acid for 1.5 or 24 h before treatment with 10 nM melatonin. Cyclosporin A (dissolved in 100% ethanol; Calbiochem-Novabiochem, La Jolla, CA) and cypermethrin and fenvalerate (in DMSO; Calbiochem-Novabiochem) were diluted into serum-free medium and added to cells for preincubations of 1 h or overnight before treatment with melatonin or MSH. To aggregate or disperse melanosomes, cells were incubated in 10 nM melatonin (10 μM stock in ethanol), 100 nM MSH (100 μM stock in H2O), 1 mM 3-isobutyl-1-methyl xanthine (IBMX; 500 mM stock in DMSO), or 1 μM PMA (100 μg/ml stock in DMSO) for 60–90 min. Cells were scored as aggregated, partially dispersed, or fully dispersed according to the state of pigment aggregation. Cells scored as fully aggregated had their melanosomes in a dense mass at the center of the cell (for example see Fig. 5, A and G). Cells scored as partially dispersed had melanosomes that were released a short distance from the central pigment mass (see Fig. 5 C). Cells scored as fully dispersed had melanosomes evenly distributed throughout the cytoplasm (see Fig. 5, B–F and H).
Figure 5.
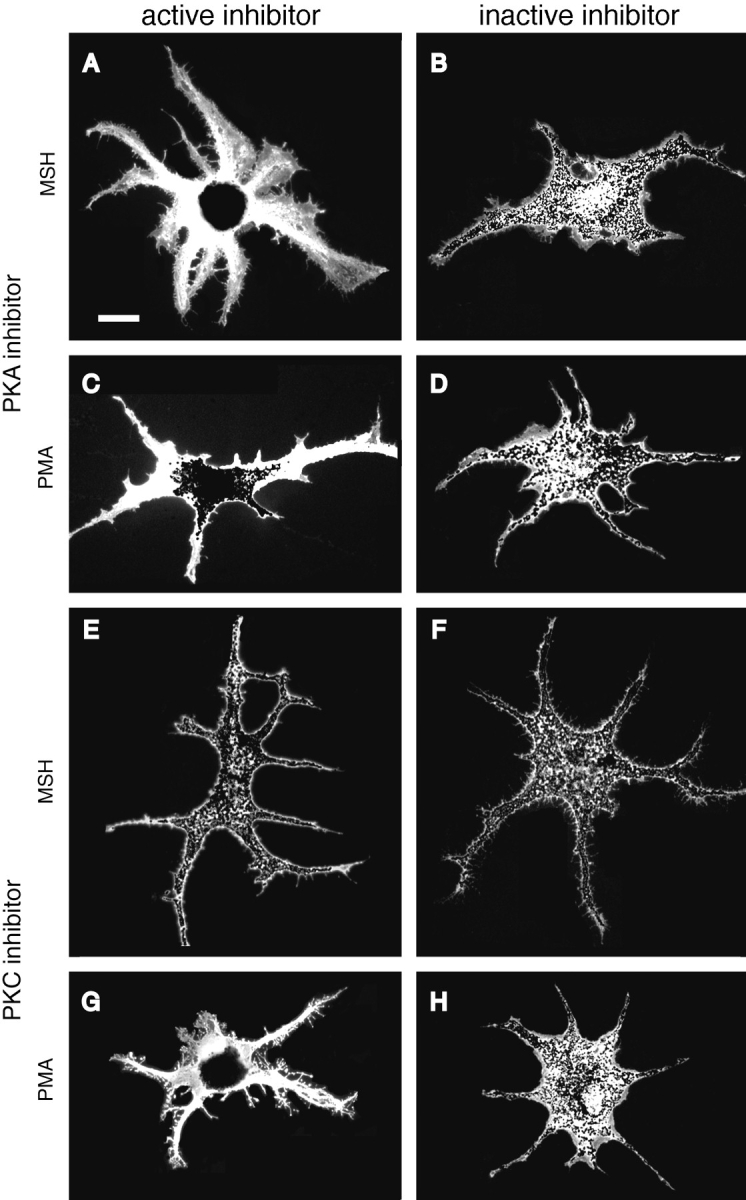
Immunofluorescent staining of cells expressing HA-tagged inhibitor and control peptides and treated with MSH or PMA to induce pigment dispersion. Cells in A–D were transfected with plasmids encoding the active (A and C) or inactive (B and D) PKA inhibitors, and treated with MSH (A and B) or PMA (C and D). Cells in E–H were transfected with plasmids encoding the active (E and G) or inactive (F and H) PKC inhibitors, and treated with MSH (E and F) or PMA (G and H). Cells expressing the PKA inhibitor were blocked from dispersing melanosomes in response to MSH, whereas expression of the control peptide did not prevent dispersion by MSH. Treatment with PMA resulted in only partial pigment dispersion in cells expressing the PKA inhibitor, although cells expressing the control peptide dispersed pigment normally. Expression of either the active or inactive PKC inhibitor did not prevent MSH-induced dispersion of pigment after aggregation by melatonin. PMA-induced dispersion was prevented by the active but not the control inhibitor. Bar, 20 μm.
Immunofluorescence
To stain transfected melanophores, the cells were fixed in freshly prepared 4% paraformaldehyde in 0.7× PBS. A monoclonal antibody that recognizes both large and small t antigen, Ab-1, was obtained from Oncogene Research (Cambridge, MA), and used at 2 μg/ml. A cultured supernatant containing monoclonal antibody 12CA5 that recognizes the HA epitope (Field et al., 1988) was used at a dilution of 1:5. For double-staining with microtubules, cells were fixed with methanol at −20°C. The antibody used for microtubule staining was an affinity-purified rabbit antibody to bovine brain tubulin (gift of A.A. Minin, Institute of Protein Research, Russian Academy of Sciences, Moscow, Russia), and was used at 4.6 μg/ml. Secondary antibodies conjugated with rhodamine or fluorescein were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA), and used at a dilution of 1:100. All antibodies were diluted into 1% BSA, 0.1% Triton X-100 in PBS. Cells were imaged with a Photometrics (Tucson, AZ) CH250 charge-coupled device camera on a Zeiss Axioskop, using a 40×/NA 1.3 Plan-Neofluar oil-immersion lens (both from Carl Zeiss. Inc., Thornwood, NY). Oncor Image (Oncor, Inc., Gaithersburg, MD) was used to acquire bright-field and fluorescence images, and Adobe Photoshop (Adobe Systems, Inc., San Jose, CA) was used to overlay the bright-field image (which clearly shows the distribution of melanosomes) onto the fluorescent image.
For the experiments with NIH 3T3 fibroblasts, cells were treated 48 h after transfection with 1 mM IBMX for 45 min. The IBMX was washed out for 10 min before fixing the cells in 4% paraformaldehyde in PBS. Cells were double-stained with a polyclonal antibody to phosphorylated cAMP response element binding protein (CREB), used at a dilution of 1:200 (New England Biolabs Inc., Beverly, MA), and antibody 12CA5 which recognizes the HA epitope (Field et al., 1988), used at a dilution of 1:50. Transfected cells were scored as having phosphorylated CREB present or absent from the nuclei.
Radioactive Labeling
Cells were grown to confluence on 10-cm plates and transferred into 10 ml of phosphate-free DME (Sigma Chemical Co., St. Louis, MO) without serum containing 0.025 mCi/ml 32Pi for 22–25 h. Pigment was induced to aggregate with 10 nM melatonin or to disperse with 100 nM MSH, 1 mM IBMX, or 0.1 μM PMA for 30 min. Melanosomes were purified in IMB50 buffer (50 mM imidazole, pH 7.4, 1 mM EGTA, 0.5 mM EDTA, 5 mM magnesium acetate, 175 mM sucrose) supplemented with protease inhibitors (1 mM PMSF, and 10 μg/ml each of chymostatin, leupeptin, and pepstatin A [CLP]), according to Rogers et al. (1998), with the following modifications. To inhibit phosphatases and kinases, the IMB50 was supplemented with 1 μM microcystin-LR (GIBCO BRL), 0.1 mM sodium orthovanadate, 0.5 μM staurosporine (Sigma Chemical Co.), 1 μM PKI (GIBCO BRL), and 1 mM EDTA. After lysing cells in 2 ml of buffer by passing them through a 25-gauge hypodermic needle, unlysed cells were removed by centrifugation at 2,000 rpm (650 g) for 2 min in a rotor (model HB-6; Sorvall, Inc., Newtown, CT). The supernatant was layered onto 80% Percoll in IMB50 containing microcystin, staurosporine, PKI, PMSF, and CLP, and then spun for 20 min at 10,000 rpm (16,300 g) in the HB-6 rotor. The pelleted melanosomes were resuspended directly into SDS sample buffer.
Western Blots
Proteins were run on 7% SDS-PAGE gels and transferred to nitrocellulose (Towbin et al., 1979). Blots were blocked overnight in 5% normal goat serum, 0.1% Tween 20 in TBS. A mouse monoclonal antibody to phoshothreonine, used at 0.5 μg/ml, and a polyclonal antibody to phosphoserine, used at 0.25 μg/ml, were from Zymed Laboratories (South San Francisco, CA). Monoclonal antibody PY20 to phosphotyrosine (Transduction Laboratories, Lexington, KY) was used at 2 μg/ml. Monoclonal antibody K2.4 to the sea urchin kinesin II 85-kD subunit (gift of J. Scholey, University of California, Davis, CA) (Cole et al., 1993) was used at a 1:200 dilution, and monoclonal antibody 70.1 to dynein intermediate chain (Sigma Chemical Co.) was used at a dilution of 1:500. The secondary antibodies were HRP goat anti–mouse or HRP goat anti–rabbit (Jackson ImmunoResearch Laboratories, Inc.), diluted 1:10,000. All antibodies were diluted into blocking buffer. SuperSignal (Pierce Chemical Co., Rockford, IL) was used for secondary antibody detection.
Immunoprecipitations
Cells were labeled with 32Pi and induced to aggregate or disperse pigment as described above. The cells were rinsed briefly in 10 mM Hepes, 150 mM NaCl, pH 7.3, to remove the medium, then lysed with 150 μL lysis buffer per 10-cm plate. The lysis buffer contained 1% Triton X-100, 50 mM Tris-HCL, pH 8.0, 50 mM NaCl, 2 mM EGTA, 2 mM EDTA, 1 mM β-mercaptoethanol, and was supplemented with the protease, kinase, and phosphatase inhibitors described above for purification of melanosomes. Extracts were spun at 14,000 rpm for 10 min in an Eppendorf tabletop centrifuge (model 5415C; Hamburg, Germany), then at 70,000 rpm for 20 min in a TLA100.3 rotor in a TL-100 ultracentrifuge (Beckman Instrs., Palo Alto, CA). Extracts were precleared for 1.5 h with 10 μL of normal mouse serum or normal rabbit serum prebound to protein A beads. The precleared extracts were incubated for 12 h at 4°C with 2.5 μL of monoclonal antibody 74.1 to the dynein intermediate chain (gift of K. Pfister, University of Virginia Health Science Center, Charlottesville, VA) (Dillman and Pfister, 1994) or 4 μL of rabbit polyclonal antibody to KIF3B (the mouse homologue of the kinesin II 95-kD subunit; gift of V. Muresan and B. Schnapp, Harvard University Medical School, Boston, MA) (Muresan et al., 1998). Pelleted beads were washed twice with lysis buffer, once with lysis buffer containing 0.5 M NaCl, and once more with lysis buffer. Beads were resuspended in 25 μL 2× Laemmli sample buffer (Laemmli, 1970).
Results
Effects of Protein Phosphatase Inhibitors on Melanosome Movement
We used inhibitors of protein phosphatases to determine which phosphatases are involved in pigment aggregation by melatonin. Melanosomes in the Xenopus cell line are normally dispersed throughout the cytoplasm in the absence of treatment with hormones, and aggregate in response to melatonin. Treatment of the cells with okadaic acid blocked melanosome aggregation by melatonin at a concentration of 500 nM when the cells were incubated for 1.5 h with the drug before treatment, and at 125 nM when they were preincubated for 25 h with the drug. We considered it necessary to incubate the cells with okadaic acid overnight because melanophores are cultured at 27°C, and it has been reported that the half-time for okadaic acid influx through the cell membrane is over 1 h at 37°C and over 4 h at 25°C (Namboodiripad and Jennings, 1996). However, our results, like those of previous studies (Cozzi and Rollag, 1992; Sammak et al., 1992) could not distinguish between PP1 and PP2A for playing a role in melanosome aggregation (the Ki for okadaic acid inhibition for PP1 is 20–315 nM, and for PP2A it is 0.1–2 nM). We therefore sought to distinguish between these phosphatases by the use of more specific inhibitors.
To test the involvement of PP1 in pigment aggregation, we constructed plasmid pNP231 encoding constitutively active inhibitor I (Alberts et al., 1994), an endogenous pseudosubstrate inhibitor of PP1, tagged with an epitope from the influenza virus HA so that transfected cells could be visualized by immunofluorescent staining with antibody 12CA5 (Field et al., 1988). We tested the activity of the HA-tagged inhibitor of PP1 by taking advantage of the fact that PP1 mediates dephosphorylation of CREB (Alberts et al., 1994; Hagiwara et al., 1992). When constitutively active inhibitor 1 is overexpressed in NIH 3T3 fibroblasts, it increases CREB phosphorylation and prevents its dephosphorylation after stimulation of cells with 8-bromo-cAMP and IBMX (a phosphodiesterase inhibitor) (Alberts et al., 1994). We therefore transfected 3T3 cells with pNP231 encoding the HA-tagged inhibitor 1, or with pNP211 encoding an HA-tagged inactive peptide (described in detail below) to control for the effects of transfection and expression of an HA-tagged peptide on CREB phosphorylation. Transfected cells were incubated with 1 mM IBMX to induce CREB phosphorylation, then rinsed with PBS and incubated an additional 10 min in medium without IBMX to allow CREB to become dephosphorylated. Immunofluorescent staining with an antibody against phosphorylated CREB showed that phosphoCREB levels in the nuclei declined in both control cells and cells expressing the PP1 inhibitor after washing out the IMBX, but the disappearance of phosphoCREB from the nucleus was inhibited in cells expressing the inhibitor (data not shown). Having verified the activity of the HA-tagged PP1 inhibitor, we transfected melanophores with the plasmid encoding the inhibitor and found that its expression had no effect on pigment aggregation or dispersion induced by melatonin or MSH, respectively. We therefore conclude that PP1 is not involved in the regulation of pigment movement in melanophores.
To test the involvement of PP2A in pigment aggregation, we transfected melanophores with a plasmid encoding the SV-40 small t antigen, which binds to PP2A and inhibits its activity (Yang et al., 1991; Sontag et al., 1993). Expression of the small t antigen resulted in nearly a complete block of pigment aggregation by melatonin (Fig. 1). As a control we transfected cells with a plasmid encoding GFP to score the amount of aggregation and dispersion in transfected cells (Fig. 1). We observed that the transfection procedure itself induced some cells to aggregate melanosomes, but virtually all such control cells were able to disperse pigment in the presence of MSH. To independently confirm that PP2A is necessary for pigment aggregation, we performed a reverse experiment overexpressing the epitope-tagged catalytic subunit of PP2A in melanophores to determine if an excess of PP2A could bias pigment towards an aggregated state. Cells overexpressing PP2A, identified by immunofluorescent staining with the HA antibody, had relatively high levels of expressed protein in the nucleus and a mixture of diffuse and punctate staining in the cytoplasm, a localization previously described for PP2A (Turowski et al., 1995). If PP2A is required for pigment aggregation, overexpression of PP2A might be expected to cause cells to aggregate pigment. However, cells overexpressing PP2A did not spontaneously aggregate pigment, perhaps because PP2A activity requires activation by melatonin. After inducing pigment aggregation with melatonin in cells overexpressing PP2A, we observed an inhibition of pigment dispersion by 1 nM MSH (Fig. 2). However, this inhibition can be overcome by increasing MSH to 10 nM. The level of PP2A overexpression previously achieved with this plasmid was only 10–50% (Ogris et al., 1997), so the relatively modest effect on inhibition of dispersion is not surprising. Since the intracellular concentration of cAMP in melanophores increases with increasing concentrations of MSH applied to the cell (Potenza and Lerner, 1992) it is likely that this low amount of overexpressed PP2A was sufficient only to counteract a low level of intracellular cAMP.
Figure 1.

Inhibition of PP2A by the small t antigen blocks melanosome aggregation. (A) Melanophores transfected with plasmids encoding the small t antigen or GFP were treated with 10 nM melatonin for 90 min and fixed with formaldehyde. Cells expressing the small t antigen were identified by immunofluorescent staining with an antibody against the SV-40 small t antigen. The vertical axis shows the percentage of cells that were scored as aggregated (white), partially dispersed (grey), or dispersed (black). Scoring was done as described in Materials and Methods. Note the almost complete inhibition of pigment aggregation in the small t-expressing cells. The data are from over 400 small t-expressing and over 500 GFP-expressing cells from three separate experiments. (B) A cell expressing small t maintained its pigment in a dispersed state after melatonin treatment (left), whereas a GFP control cell aggregated pigment normally (right). The cells are shown by overlaying bright-field images onto fluorescent images. Bar, 20 μm.
Figure 2.

Overexpression of PP2A inhibits melanosome dispersion at low levels of cAMP. Cells transfected with plasmids encoding the HA-tagged PP2A catalytic subunit or GFP were treated with 25 μM dexamethasone for 24 h to induce PP2A expression. Cells were then treated with 10 nM melatonin for 1 h to aggregate pigment, followed by 1 nM or 10 nM MSH for 30 min. PP2A-expressing cells were identified by immunofluorescent staining with an antibody to HA, and 100 cells were scored per treatment. The data is from a typical experiment; similar results were obtained in three independent experiments.
Since PP2A colocalizes with microtubules (Sontag et al., 1995) and is involved in the control of microtubule dynamics during mitosis (Tournebize et al., 1997), we wanted to eliminate the possibility that inhibition or overexpression of PP2A might be altering microtubule number or stability. Cells expressing the small t antigen or overexpressing PP2A were treated with melatonin or MSH, fixed, and then double-stained for microtubules with a polyclonal tubulin antibody and either the small t antigen or the HA-derived epitope tagging the PP2A catalytic subunit. Cells expressing the small t antigen and cells overexpressing PP2A had a normal density and distribution of microtubules (data not shown). We therefore conclude that PP2A is required in the signaling pathway for melanosome aggregation.
We also tested the involvement of PP2B in pigment aggregation in Xenopus melanophores since it has been reported that PP2B is required for aggregation in fish melanophores (Thaler and Haimo, 1990). We treated cells with two types of pharmacological inhibitors to PP2B. Cyclosporin A binds to the intracellular protein cyclophilin to form a complex which binds to and inactivates PP2B with nanomolar affinity (Liu et al., 1991). Treatment of melanophores with 1 μM of cyclosporin A neither affected the ability of cells to aggregate pigment in the presence of 10 nM of melatonin nor to disperse pigment in the presence of 100 nM MSH. Therefore, even a high cyclosporin A concentration had no effect on the movement of pigment in melanophores. We also used two inhibitors of PP2B in the type II pyrethroid class of drugs, cypermethrin and fenvalerate (Enan and Matsumura, 1992). Doses ranging from 1–50 nM cypermethrin (50% inhibitory concentration [IC50] is 40 pM) and 20 nM–2 μM fenvalerate (IC50 is 2–4 nM) failed to have an effect on aggregation or dispersion of pigment induced by melatonin or MSH, respectively. In addition, we transfected melanophores with a plasmid encoding a recombinant PP2B inhibitor based on the pseudosubstrate domain of PP2B (Hashimoto et al., 1990). Expression of this inhibitor peptide had no effect on pigment aggregation or dispersion induced by melatonin or MSH. We therefore conclude that PP2B is not involved in the regulation of pigment movement in Xenopus melanophores.
Effects of Protein Kinase Inhibitors on Pigment Dispersion
To study the role of protein kinases in melanosome movement, we used highly selective recombinant inhibitors of protein kinases. These inhibitors were derived from the pseudosubstrate inhibitory regions of PKC and CaMK, and the endogenous heat stable PKA inhibitor, PKI (Soderling, 1990; Kemp et al., 1991; Walsh and Glass, 1991). The recombinant inhibitors are highly effective and specific, and were used to dissect the signaling cascades activated by Ca2+ influx in neuronal cells (Peunova, N.I., and G.N. Enikolopov, manuscript in preparation). Mutant forms of the inhibitors carrying inactivating amino acid substitutions in the kinase recognition sequence (Grove et al., 1987; House and Kemp, 1987; Waldmann et al., 1990) served as controls for the effects of the transfection procedure and the accumulation of large amounts of peptide in the cells.
Expression of the PKA Inhibitor
Melanosomes disperse when the cAMP level is high, as when cells are treated with MSH, and aggregate when cAMP is low, which occurs when cells are treated with melatonin. In the absence of melatonin or MSH, melanophores grow in culture with melanosomes in a dispersed state (Fig. 3 A). To determine whether this steady-state dispersion depends solely on the level of active PKA, we transfected melanophores with plasmid pNP210 encoding the epitope-tagged specific PKA inhibitor. After 48–72 h, a significant number of cells in the transfected culture had fully aggregated melanosomes (Fig. 3 B). Nontransfected cells and cells transfected with plasmid pNP211 encoding an epitope-tagged inactive analogue of PKI maintained their melanosomes in the dispersed state (Fig. 3, A and C). We confirmed that the aggregated cells in the pNP210 culture were expressing the inhibitor by immunofluorescent staining for the HA tag. Therefore, the steady-state dispersion of melanosomes depends on PKA activity.
Figure 3.
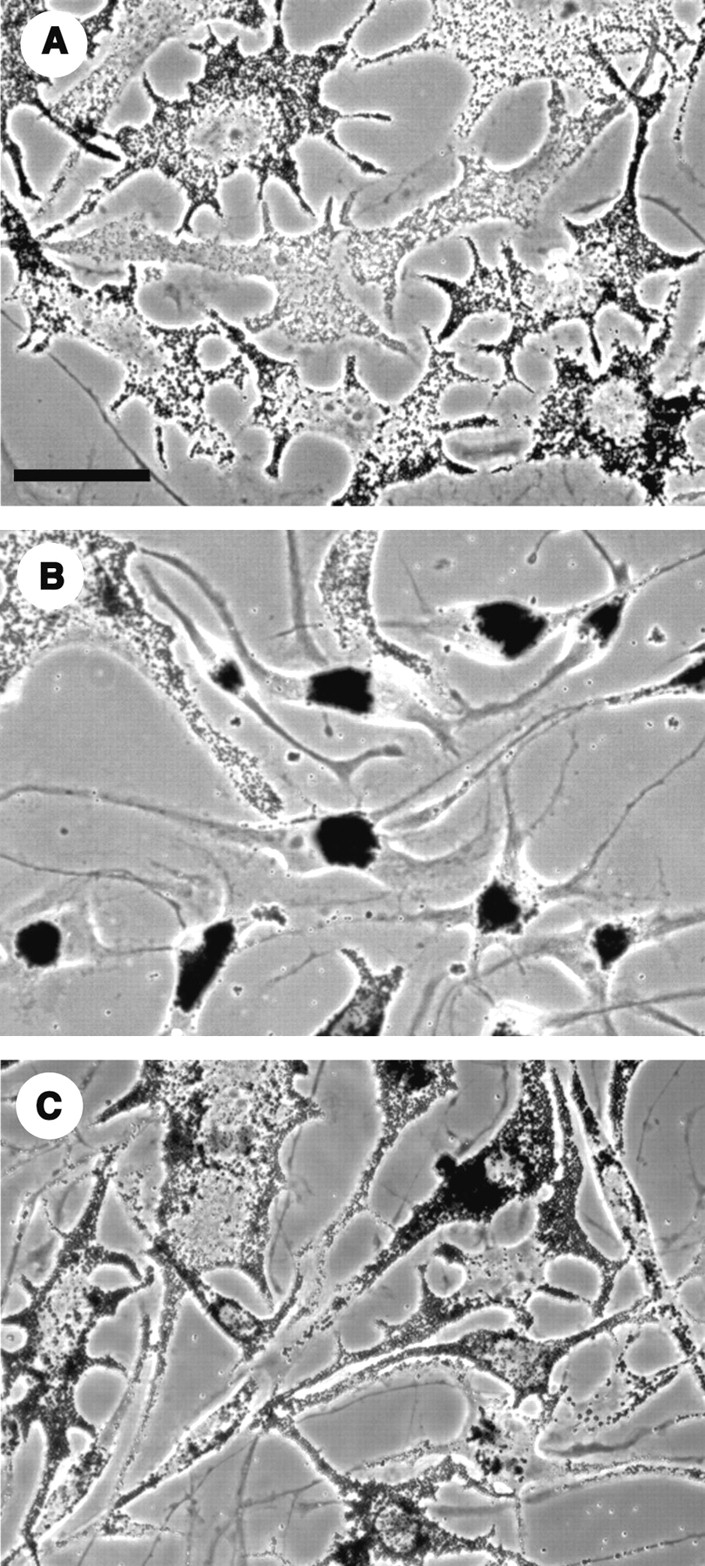
Phase-contrast images of nontransfected cells (A), and cells transfected with the PKA inhibitor plasmid (B), or with the plasmid encoding the inactive analogue of the inhibitor (C). After 72 h, a large percentage of cells in the culture expressing the active inhibitor had fully aggregated pigment. Control cells maintained their melanosomes in the dispersed state. Bar, 50 μm.
When cells expressing the PKA inhibitor were treated with MSH, virtually all the transfected cells stayed completely aggregated even after prolonged incubation in MSH, demonstrating that PKA was effectively inhibited (Fig. 4 and Fig. 5 A). In control experiments in which melanophores were transfected with plasmid pNP211 encoding the inactive inhibitor, typically 50% of the HA-positive cells had partially-dispersed pigment after transfection, and virtually all of them dispersed pigment after treatment with MSH (Fig. 4 and Fig. 5 B). The same result was observed in GFP-transfected cells; therefore, pigment aggregation is the result of the transfection procedure itself rather than expression of the inactive inhibitor. To rule out the possibility that microtubule disassembly was responsible for the inability of cells expressing the active PKA inhibitor to disperse pigment, we fixed cells and performed double staining for microtubules and the HA epitope. Both transfected and nontransfected cells contained an elaborate array of microtubules radiating from the cell center (data not shown). Furthermore, the density of the microtubule network in transfected and nontransfected cells did not differ.
Figure 4.
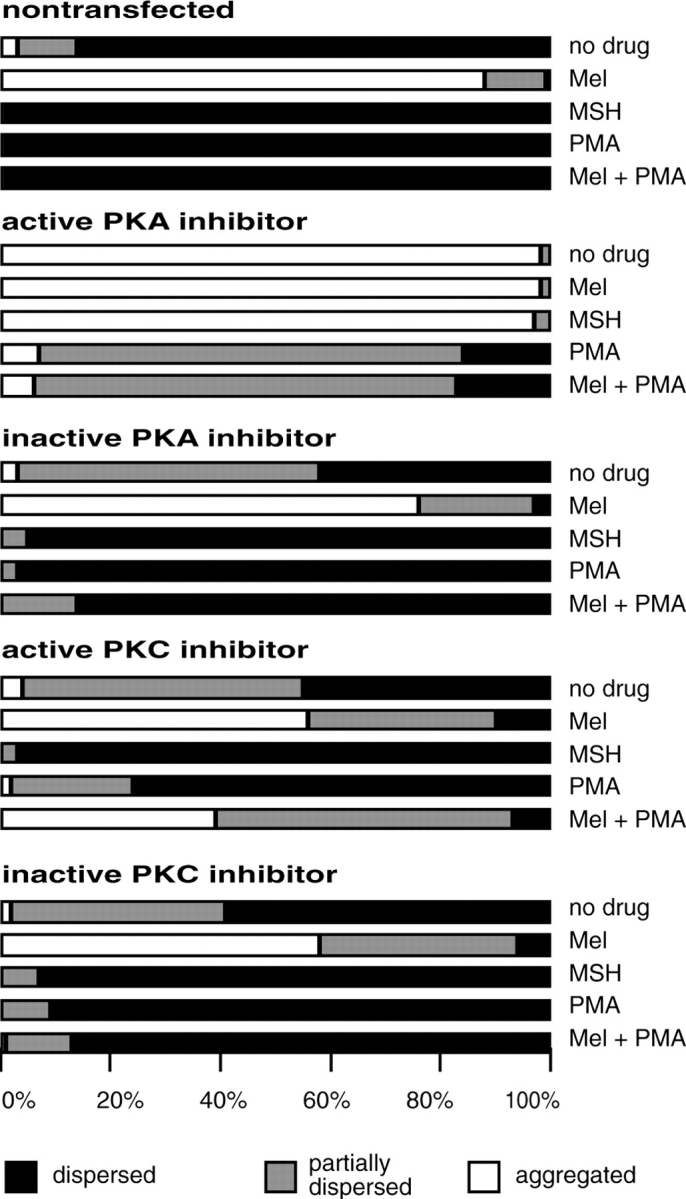
Quantitative analysis of the effects of the active and inactive PKA and PKC inhibitor peptides on melanosome aggregation and dispersion. Cells expressing the PKA inhibitor aggregated their pigment and could not disperse it when treated with MSH. These cells partially dispersed melanosomes in response to PMA. Cells expressing the inactive PKA inhibitor dispersed melanosomes normally in response to MSH and PMA. Cells expressing the PKC inhibitor dispersed pigment when treated with MSH, but were inhibited from dispersing pigment after aggregation with melatonin and treatment with PMA. Percentages are from scoring 200 cells per treatment. Similar results were obtained in at least three other independent experiments with each inhibitor.
Having shown that expression of the PKA inhibitor completely inhibited dispersion by MSH, we tested whether activation of PKC could disperse melanosomes in the absence of PKA activity. The PKC activator PMA can override melatonin-induced aggregation and disperse pigment even in the presence of melatonin (Sugden and Rowe, 1992). Since melatonin acts by reducing cAMP (Daniolos et al., 1990), PKC may be signaling pigment dispersion through an alternate pathway. When cells expressing the PKA inhibitor were aggregated with melatonin and then treated with 1 μM of PMA to activate PKC, in most cells the melanosomes were released from the central aggregate a short distance to become partially dispersed (Fig. 5 C), and in 17% of the cells pigment dispersed to the tips of the processes. Identical treatment of cells transfected with the control plasmid pNP211 resulted in full dispersion of 86% of cells (Fig. 4 and Fig. 5 D). We conclude that PKC can partially disperse pigment in the absence of active PKA, but requires a basal level of PKA activity to induce full dispersion.
Expression of the PKC and CaMK Inhibitors
It is possible that a component in the pathway for melanosome movement might require phosphorylation by both PKA and PKC for full activation. Having found that PKC requires some PKA activity for full dispersion, we also considered the reverse question, whether the PKA pathway requires any PKC activity. Previous studies have shown that a pharmacological inhibitor of PKC, Ro 31-8220, did not block MSH-induced dispersion (Sugden and Rowe, 1992; McClintock et al., 1996). The very high specificity of the peptide inhibitor of PKC used together with its inactive control provided a tool to address this question more definitively.
Unlike expression of the PKA inhibitor, expression of the active inhibitor of PKC from plasmid pNP212 did not induce aggregation of pigment (Fig. 4), and the appearance of cultures transfected with active and inactive inhibitor constructs was identical. pNP212-transfected cells could still aggregate melanosomes in the presence of melatonin, but PMA-induced dispersion of cells treated with melatonin was inhibited compared with dispersion in cells transfected with the inactive control plasmid pNP214 (Fig. 4 and Fig. 5, G and H). All cells expressing the PKC inhibitor dispersed pigment in the presence of MSH (Fig. 5, E and F), confirming the studies showing that PKA-mediated dispersion is PKC independent.
We also wanted to test the involvement of CaMK in pigment movement because calcium levels in fish melanophores have been shown to rise with aggregation (Sammak et al., 1992). We transfected cells with plasmid pNP215 encoding a peptide inhibitor to CaMK, and with a control plasmid, pNP216. The CaMK inhibitor has been shown to specifically block CaMK activity in cultured cells (Peunova, N.I., and G.N. Enikolopov, manuscript in preparation); however, it had no effect on pigment motility in melanophores.
Phosphorylation Targets in Cells Aggregating and Dispersing Pigment
Ultimately, the signaling pathways regulating pigment transport should activate or inhibit motor proteins responsible for organelle movement. To identify phosphorylation targets of PKA and PKC, we purified melanosomes from cells labeled with 32Pi. Cells were induced to disperse pigment with MSH, IBMX (a phosphodiesterase inhibitor) or PMA (an activator of PKC), or aggregate pigment with melatonin.
Labeled melanophore cell extract has a complex pattern of phosphorylated bands (Fig. 7). When melanosomes are purified from labeled cells, the pattern of phosphorylation is much simpler than in the total cell extract. Most bands are equally phosphorylated on aggregating and dispersing melanosomes except for a region of mol wt 87–95 kD (Fig. 6 A). Melatonin-treated melanosomes show a heavily-phosphorylated band at 95 kD, whereas IBMX- and MSH-treated melanosomes show phosphorylation at 87 kD, and untreated and PMA-treated melanosomes have a band of broad phosphorylation from 88–92 kD.
Figure 7.
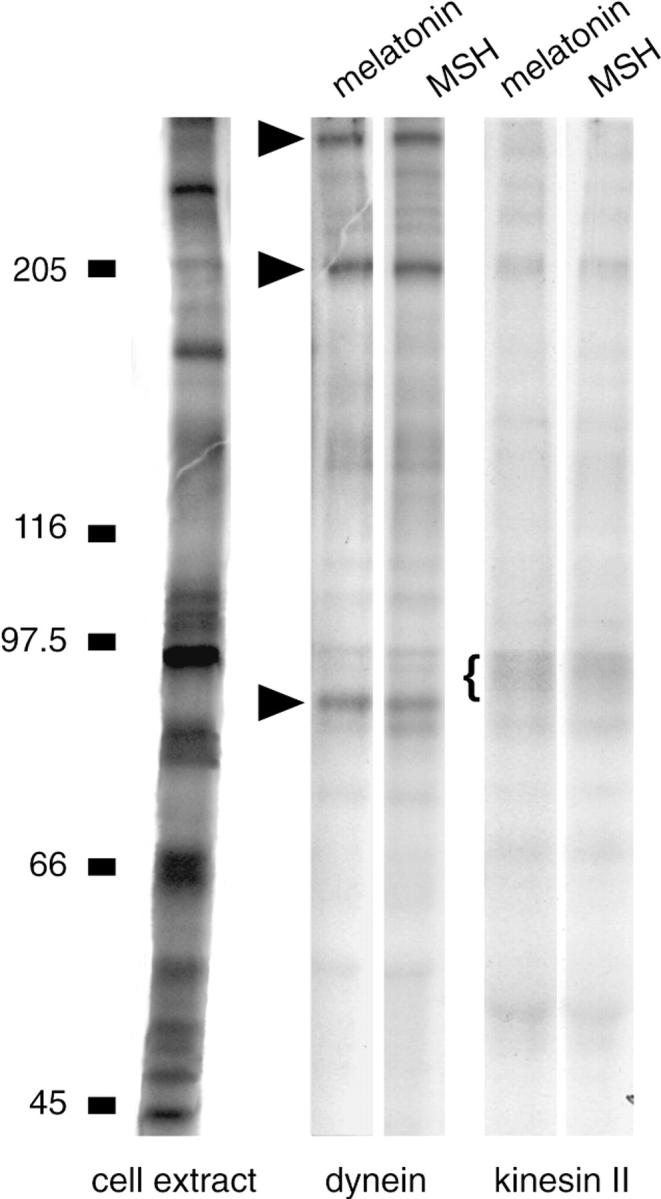
Immunoprecipitations of kinesin II and cytoplasmic dynein from 32Pi- labeled melanophore extract. Left lane, cell extract. Immunoprecipitates of dynein from melatonin-treated and MSH-treated cells show strong phosphorylation at the molecular weights of the dynein intermediate chain (83 kD) and the dynein heavy chain, and at an unidentified band at 200 kD (arrowheads). Immunoprecipitates of kinesin II from melatonin-treated and MSH-treated cells have phosphorylation in a broad band at 95 kD (bracket). No consistent phosphorylation differences have been observed in immunoprecipitations of dynein or kinesin II.
Figure 6.

Phosphorylation of melanosome proteins in cells treated with 10 nM melatonin, 100 nM MSH, 0.1 μM PMA, or 1 mM IBMX. Phosphorylation differences were reproduced in three separate experiments. (A) Cells were labeled with 32Pi for 21 h before melanosome purification. The differences in phosphorylation are found in the 85–95 kD area. Melanosomal proteins from melatonin-treated cells show phosphorylation at ∼86, 92, and 95 kD (arrows), whereas proteins from MSH- and IBMX-treated cells are phosphorylated at 87 and 94 kD. Proteins from untreated and PMA-treated cells show phosphorylation in a broad band from 88–95 kD. (B) Western blot of melanosomal proteins with the anti-phosphothreonine antibody. The pattern of threonine phosphorylation looks very similar in the mol wt range 85–95 kD to the pattern of total phosphorylation in A. There is less phosphorylation on melanosomes purified from cells treated with IMBX and MSH than on melanosomes purified from cells treated with melatonin, whereas it appears that untreated and PMA-treated cells are in a phosphorylation state intermediate between that of aggregating and dispersing cells. In addition, phosphorylation of the band at 50 kD is reduced with melatonin treatment.
Blots of melanosomes probed with an anti-phosphothreonine antibody show a similar pattern of phosphorylation in the mol wt region of 87–95 kD, and the heavier phosphorylation in melatonin-treated cells is even more obvious (Fig. 6 B). Blots with an antibody to phosphoserine show no obvious phosphorylation differences, and little phosphorylation in the region of 87–95 kD (data not shown). Blots probed for phosphotyrosine show no appreciable phosphorylation at 87–95 kD, but show a band of heavier phosphorylation at 50 kD in MSH-treated cells compared with melatonin-treated cells (data not shown). This difference at 50 kD can also sometimes be seen in blots probed for phosphothreonine (Fig. 6 B).
As cytoplasmic dynein and kinesin II are present on melanosomes (Rogers et al., 1997), and since the dynein intermediate chain has a mol wt of 83 kD in Xenopus (Allan, 1995), and the motor subunits of kinesin II have mol wts of 85 and 95 kD, respectively, we considered the possibility that the phosphorylated proteins on melanosomes are the motor proteins themselves. Kinesin II and dynein were immunoprecipitated from labeled extracts of cells induced to aggregate or disperse melanosomes. The presence of kinesin II in the immunoprecipitate was verified by Western blotting with monoclonal antibody K2.4 to the 85-kD subunit of kinesin II, and the presence of dynein was verified by Western blotting with monoclonal antibody 70.1 to the dynein intermediate chain (data not shown). No consistent phosphorylation differences were seen in the immunoprecipitations, nor was differential phosphorylation of the pattern seen on the purified melanosomes observed (Fig. 7). We also probed the immunoprecipitates with antibodies to phosphothreonine and phosphoserine and again found neither consistent phosphorylation differences nor any phosphorylation like that seen on melanosomes. Therefore, it is likely that the phosphorylation on melanosomes is neither kinesin II nor the dynein intermediate chain. It is possible, however, that differential phosphorylation of the motor proteins occurs only locally on melanosomes, and it may therefore be necessary to immunoprecipitate the motors from a melanosomal protein fraction rather than from the cell extract. These experiments, however, have proven difficult to perform due to the small amount of protein that we obtain from a melanosomal fraction, and because extraction of melanosomes with detergents other than SDS results in binding of proteins to melanin.
Discussion
The state of aggregation of melanosomes is determined by the balance between phosphorylation and dephosphorylation of the proteins that regulate the activity of the motor proteins on the organelles. Using specific inhibitors to kinases and phosphatases, we investigated the signaling pathways for melanosome aggregation and dispersion. We identified PP2A as the phosphatase necessary for pigment aggregation by expressing the small t antigen and obtaining a nearly complete block of aggregation, and by overexpressing PP2A, which inhibited pigment dispersion. These results are consistent with our results of those of others (Cozzi and Rollag, 1992; Sammak et al., 1992) showing that okadaic acid blocks pigment aggregation at concentrations that inhibit PP1 or PP2A. The PP2B inhibitors cyclosporin A, cypermethrin, and fenvalerate had no effect on melanosome movement, a finding supported by previous results showing that pigment aggregation in melanophores of angelfish (Pterophyllum scalare) and squirrel fish (Holocentrotus ascensionus) does not require elevated Ca2+ (Sammak et al., 1992; Kotz and McNiven, 1994). In addition, we expressed specific peptide inhibitors to PP2B and PP1 and found no effect on aggregation or dispersion.
Previous studies have used pharmacological inhibitors to analyze the role of PKA and PKC in melanosome motility. However, most of these inhibitors are capable of inhibiting several types of protein kinases, thus limiting the interpretation of results. We therefore examined pathways for melanosome aggregation and dispersion using highly specific recombinant inhibitors of protein kinases; the inactive variants were used as controls. We found that inhibition of PKA causes melanosomes to spontaneously aggregate and remain at the cell center, indicating that basal PKA activity is responsible for the normally dispersed state of the pigment. Moreover, the PKA pathway does not require PKC, since inhibition of PKC with the recombinant inhibitor does not prevent full dispersion of melanosomes upon activation of PKA by MSH.
The PKC pathway can also initiate dispersion, but unlike the PKA pathway, is insufficient on its own to fully disperse melanosomes. PKA may be needed for full activation of a component of the PKC pathway upstream of the motor proteins. Another possibility is that the PKC pathway may activate a plus end–directed motor to a submaximal level, allowing partial but not complete dispersion. Unlike our observations of partial dispersion induced by PMA when PKA is inhibited, McClintock et al. (1996) reported that the PKA inhibitor H89 completely blocked dispersion through the PKC pathway. Inhibition by H89 may be due to nonspecific inhibition of PKC, as the authors suggest. However, it may be necessary to activate PKC to a high level to observe partial pigment dispersion in the absence of PKA activity. McClintock et al. (1996) activated PKC through hormonal activation of phospholipase C, which may not stimulate PKC to the same extent as the high concentration of PMA that we used.
Our results agree with previous results suggesting that the PKA and PKC pathways for pigment dispersion are distinct. PMA-treated cells assume a morphology different from cells treated with MSH (Sugden and Rowe, 1992). Furthermore, in primary cultures of Xenopus melanophores, the PKC inhibitor Ro 31-8220 blocked PMA- induced, but not MSH-induced dispersion of melanosomes (Sugden and Rowe, 1992). Similarly, in an immortalized cell line of Xenopus melanophores, Ro 31-8220 blocked dispersion by an exogenous transfected bombesin receptor which uses the phospholipase C pathway, but did not block dispersion by agonists shown to raise intracellular cAMP, including MSH (McClintock et al., 1996).
PKA appears to mediate the default pathway for pigment dispersion. The dispersed state of melanosomes in untreated cells does not depend on PKC activity since cells became completely and permanently aggregated when transfected with the peptide inhibitor of PKA, but not when transfected with the peptide inhibitor of PKC. Similarly, angelfish melanophores were induced to aggregate by microinjection of PKI5–24 (Sammak et al., 1992), and Xenopus melanophores were induced to partially aggregate with the competitive PKA inhibitor Rp-cAMPS, but not with the PKC inhibitor Ro 31-8220 (Sugden and Rowe, 1992).
We transfected melanophores with an inhibitor to CaMK because a Ca2+ elevation accompanying pigment aggregation has been demonstrated in fish melanophores (Sammak et al., 1992). However, we found no effect on melanosome aggregation or dispersion with inhibition of CaMK. The function of the Ca2+ increase in fish melanophores is unclear, since aggregation was not blocked when this increase was suppressed by withdrawal of extracellular Ca2+ or loading of an intracellular Ca2+ chelator, nor was aggregation induced with the calcium ionophore ionomycin (Sammak et al., 1992). Similarly, Kotz and McNiven (1994) showed that a calcium increase is not required for pigment aggregation in melanophores of squirrel fish. Considering that a calcium elevation may not be essential for pigment aggregation, it is not surprising that we detected no effect of CaMK inhibition on melanosome movement.
The ultimate target for the regulation of melanosome motility should be on the melanosomes themselves. The target protein or set of proteins should be involved in switching the direction the melanosomes travel on microtubules. We looked for differential phosphorylation of proteins on melanosomes purified from cells treated with melatonin, MSH, IBMX, and PMA, and we found phosphorylation differences in a region of mol wt 87–95 kD. Considering that dispersion requires a kinase whereas aggregation requires a phosphatase, we were surprised by the heavier phosphorylation on melanosomes from melatonin-treated cells. This result indicates that PKA, PKC, and PP2A cannot be the only players in the signaling cascades resulting in phosphorylation of melanosomal proteins, and that other kinases and phosphatases in the pathways have yet to be identified. It is also interesting to note that the pattern of phosphorylation on melanosomes from cells treated with PMA and MSH is different, and supports the idea that there is a different mechanism of dispersion by these two pathways. The phosphorylation differences occur on threonine, since blots with a phosphothreonine antibody, but not phosphoserine or phosphotyrosine antibodies, showed the same pattern of differential phosphorylation seen in cells labeled with 32Pi.
Potential targets of regulation include the motor proteins themselves, motor-associated proteins such as dynactin, receptors for the motor proteins on the melanosomes, and microtubule-associated proteins. We did not find differential phosphorylation of two motor proteins known to be present on melanosomes, dynein and kinesin II, when we immunoprecipitated these proteins from the cell extract. A myosin motor, myosin V, is present on melanosomes (Rogers and Gelfand, 1998) and could also be a potential target for regulation, but this motor does not have any subunits in the 80–90 kD range. The plus and minus end–directed motors may exist together with the kinases and phosphatases in a regulatory complex on the melanosomes, and may therefore be regulated only when present on the organelles, an arrangement that would maximize the efficiency and specificity of their regulation. Evidence for the existence of complexes containing motor proteins and associated kinases or phosphatases is provided by many recent studies (Alphey et al., 1997; Blangy et al., 1995; Karki et al., 1997; Robbins et al., 1997; Nagata et al., 1998). We hope that by identifying differentially phosphorylated proteins on melanosomes, we will gain some insight into the mechanism of regulation of melanosome movement.
Acknowledgments
The authors would like to thank M. Lerner for the Xenopus melanophore cell line, S. Shenolikar for plasmid pCEP4-Sm t and advice on the phosphoCREB experiments, D. Pallas for plasmid pGRE5-2 (T) 36, and S. Madhu (University of Illinois, Urbana-Champaign, IL) for help with protein phosphorylation experiments.
This work was supported by grants to V.I. Gelfand from the National Science Foundation (MCB 9513388) and the National Institutes of Health (GM 52111).
Abbreviations used in this paper
- CaMK
Ca2+/calmodulin-dependent kinase
- CREB
cAMP-response element binding protein
- GFP
green fluorescent protein
- HA
hemagglutinin
- IBMX
3-isobutyl-1-methyl xanthine
- MSH
melanocyte-stimulating hormone
- PKA
protein kinase A
- PKC
protein kinase C
- PP1
protein phosphatase 1
- PP2A
protein phosphatase 2A
- PP2B
protein phosphatase 2B
Footnotes
Address all correspondence to Vladimir Gelfand, University of Illinois, Department of Cell and Structural Biology, B107 Chemical and Life Sciences Laboratory, 601 South Goodwin Ave., Urbana, IL 61801. Tel.: (217) 333-5972. Fax: (217) 333-5982. E-mail: vgelfand@uiuc.edu
I.S. Tint's present address is Department of Anatomy and Cell Biology, Temple University School of Medicine, Philadelphia, PA 19140.
References
- Alberts AS, Montminy M, Shenolikar S, Feramisco JR. Expression of a peptide inhibitor of protein phosphatase 1 increases phosphorylation and activity of CREB in NIH 3T3 fibroblasts. Mol Cell Biol. 1994;14:4398–4407. doi: 10.1128/mcb.14.7.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan V. Protein phosphatase 1 regulates the cytoplasmic dynein-driven formation of endoplasmic reticulum networks in vitro. J Cell Biol. 1995;128:879–891. doi: 10.1083/jcb.128.5.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alphey L, Parker L, Hawcroft G, Guo Y, Kaiser K, Morgan G. KLP38B: A mitotic kinesin-related protein that binds PP1. J Cell Biol. 1997;138:395–409. doi: 10.1083/jcb.138.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangy A, Lane HA, Dherin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34(cdc2) regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- Clark TG, Rosenbaum JL. Pigment particle translocation in detergent-permeabilized melanophores of Fundulus heteroclitus. . Proc Natl Acad Sci USA. 1982;79:4655–4659. doi: 10.1073/pnas.79.15.4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DG, Chinn SW, Wedaman KP, Hall K, Vuong T, Scholey JM. Novel heterotrimeric kinesin-related protein purified from sea urchin eggs. Nature. 1993;366:268–270. doi: 10.1038/366268a0. [DOI] [PubMed] [Google Scholar]
- Cozzi B, Rollag MD. The protein-phosphatase inhibitor okadaic acid mimics MSH-induced and melatonin-reversible melanosome dispersion in Xenopus laevismelanophores. Pigment Cell Res. 1992;5:148–154. doi: 10.1111/j.1600-0749.1992.tb00011.x. [DOI] [PubMed] [Google Scholar]
- Daniolos A, Lerner AB, Lerner MR. Action of light on frog pigment cells in culture. Pigment Cell Res. 1990;3:38–43. doi: 10.1111/j.1600-0749.1990.tb00260.x. [DOI] [PubMed] [Google Scholar]
- Dillman JF, Pfister KK. Differential phosphorylation in vivo of cytoplasmic dynein associated with anterogradely moving organelles. J Cell Biol. 1994;127:1671–1681. doi: 10.1083/jcb.127.6.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enan E, Matsumura F. Specific inhibition of calcineurin by type II synthetic pyrethroid insecticides. Biochem Pharmacol. 1992;43:1777–1784. doi: 10.1016/0006-2952(92)90710-z. [DOI] [PubMed] [Google Scholar]
- Euteneuer U, McIntosh JR. Polarity of some motility-related microtubules. Proc Natl Acad Sci USA. 1981;78:372–376. doi: 10.1073/pnas.78.1.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J, Nikawa J, Broek D, MacDonald B, Rodgers L, Wilson IA, Lerner RA, Wigler M. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiaeby use of an epitope addition method. Mol Cell Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graminski GF, Jayawickreme CK, Potenza MN, Lerner MR. Pigment dispersion in frog melanophores can be induced by a phorbol ester or stimulation of a recombinant receptor that activates phospholipase C. J Biol Chem. 1993;268:5957–5964. [PubMed] [Google Scholar]
- Grove JR, Price DJ, Goodman HM, Avruch J. Recombinant fragment of protein kinase inhibitor blocks cyclic AMP-dependent gene transcription. Science. 1987;238:530–533. doi: 10.1126/science.2821622. [DOI] [PubMed] [Google Scholar]
- Hagiwara M, Alberts A, Brindle P, Meinkoth J, Feramisco J, Deng T, Karin M, Shenolikar S, Montminy M. Transcriptional attenuation following cAMP induction requires PP-1-mediated dephosphorylation of CREB. Cell. 1992;70:105–113. doi: 10.1016/0092-8674(92)90537-m. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Perrino BA. Identification of an autoinhibitory domain in calcineurin. J Biol Chem. 1990;265:1924–1927. [PubMed] [Google Scholar]
- House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototype in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- Karki S, Tokito MK, Holzbaur EL. Casein kinase II binds to and phosphorylates cytoplasmic dynein. J Biol Chem. 1997;272:5887–5891. doi: 10.1074/jbc.272.9.5887. [DOI] [PubMed] [Google Scholar]
- Kemp BE, Pearson RB, House CM. Pseudosubstrate-based peptide inhibitors. Methods Enzymol. 1991;201:287–304. doi: 10.1016/0076-6879(91)01026-x. [DOI] [PubMed] [Google Scholar]
- Kotz KJ, McNiven MA. Intracellular calcium and cAMP regulate directional pigment movements in teleost erythrophores. J Cell Biol. 1994;124:463–474. doi: 10.1083/jcb.124.4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Malawista S. Cytochalasin B reversibly inhibits melanin granule movement in melanocytes. Nature. 1971;234:354–355. doi: 10.1038/234354a0. [DOI] [PubMed] [Google Scholar]
- McClintock TS, Rising JP, Lerner MR. Melanophore pigment dispersion responses to agonists show two patterns of sensitivity to inhibitors of cAMP-dependent protein kinase and protein kinase C. J Cell Physiol. 1996;167:1–7. doi: 10.1002/(SICI)1097-4652(199604)167:1<1::AID-JCP1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- McGuire J, Moellmann G. Cytochalasin B: effects on microfilaments and movement of melanin granules within melanocytes. Science. 1972;175:642–644. doi: 10.1126/science.175.4022.642. [DOI] [PubMed] [Google Scholar]
- McNiven MA, Porter KR. Chromatophores—models for studying cytomatrix translocations. J Cell Biol. 1984;99(Suppl.):152–158. doi: 10.1083/jcb.99.1.152s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan V, Abramson T, Lyass A, Winter D, Porro E, Hong F, Chamberlin NL, Schnapp BJ. KIF3C and KIF3A form a novel neuronal heteromeric kinesin that associates with membrane vesicles. Mol Biol Cell. 1998;9:637–652. doi: 10.1091/mbc.9.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DB, Tilney LG. The role of microtubules in movement of pigment granules in teleost melanophores. J Cell Biol. 1974;61:757–779. doi: 10.1083/jcb.61.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata KI, Puls A, Futter C, Aspenstrom P, Schaefer E, Nakata T, Hirokawa N, Hall A. The MAP kinase MLK2 co-localizes with activated JNK along microtubules and associates with kinesin superfamily motor KIF3. EMBO (Eur Mol Biol Organ) J. 1998;17:149–158. doi: 10.1093/emboj/17.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namboodiripad AN, Jennings ML. Permeability characteristics of erythrocyte membrane to okadaic acid and calyculin A. Amer J Physiol. 1996;270:C449–C456. doi: 10.1152/ajpcell.1996.270.2.C449. [DOI] [PubMed] [Google Scholar]
- Nilsson H, Wallin M. Evidence for several roles of dynein in pigment transport in melanophores. Cell Motil Cytoskeleton. 1997;38:397–409. doi: 10.1002/(SICI)1097-0169(1997)38:4<397::AID-CM9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Ogris E, Gibson DM, Pallas DC. Protein phosphatase 2A subunit assembly: the catalytic subunit carboxy terminus is important for binding cellular B subunit but not polyomavirus middle tumor antigen. Oncogene. 1997;15:911–917. doi: 10.1038/sj.onc.1201259. [DOI] [PubMed] [Google Scholar]
- Palazzo RE, Lynch TJ, Taylor JD, Tchen TT. cAMP-independent and cAMP-dependent protein phosphorylations by isolated goldfish xanthophore cytoskeletons: evidence for the association of cytoskeleton with a carotenoid droplet protein. Cell Motil Cytoskeleton. 1989;13:21–29. doi: 10.1002/cm.970130104. [DOI] [PubMed] [Google Scholar]
- Potenza MN, Lerner MR. A rapid quantitative bioassay for evaluating the effects of ligands upon receptors that modulate cAMP levels in a melanophore cell line. Pigment Cell Res. 1992;5:372–378. doi: 10.1111/j.1600-0749.1992.tb00565.x. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Nybakken KE, Kobayashi R, Sisson JC, Bishop JM, Therond PP. Hedgehog elicits signal transduction by means of a large complex containing the kinesin-related protein costal 2. Cell. 1997;90:225–234. doi: 10.1016/s0092-8674(00)80331-1. [DOI] [PubMed] [Google Scholar]
- Rodionov VI, Vardanyan AG, Kamalov VF, Gelfand VI. The movement of melanosomes in melanophore fragments obtained by laser microbeam irradiation. Cell Biol Int Rep. 1987;11:565–572. doi: 10.1016/0309-1651(87)90136-6. [DOI] [PubMed] [Google Scholar]
- Rodionov VI, Hope AJ, Svitkina TM, Borisy GG. Functional coordination of microtubule-based and actin-based motility in melanophores. Curr Biol. 1998;8:165–168. doi: 10.1016/s0960-9822(98)70064-8. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Gelfand VI. Myosin cooperates with microtubule motors during organelle transport in melanophores. Curr Biol. 1998;8:161–164. doi: 10.1016/s0960-9822(98)70063-6. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Tint IS, Fanapour PC, Gelfand VI. Regulated bidirectional motility of melanophore pigment granules along microtubules in vitro. Proc Natl Acad Sci USA. 1997;94:3720–3725. doi: 10.1073/pnas.94.8.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SL, Tint IS, Gelfand VI. In vitromotility assay for melanophore pigment organelles. Methods Enzymol. 1998;298:361–372. doi: 10.1016/s0076-6879(98)98032-6. [DOI] [PubMed] [Google Scholar]
- Rozdzial MM, Haimo LT. Bidirectional pigment granule movements of melanophores are regulated by protein phosphorylation and dephosphorylation. Cell. 1986;47:1061–1070. doi: 10.1016/0092-8674(86)90821-4. [DOI] [PubMed] [Google Scholar]
- Sammak PJ, Adams SR, Harootunian AT, Schliwa M, Tsien RY. Intracellular cyclic AMP, not calcium, determines the direction of vesicle movement in melanophores: direct measurement by fluorescence ratio imaging. J Cell Biol. 1992;117:57–72. doi: 10.1083/jcb.117.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MK, Colbran RJ, Brickey DA, Soderling TR. Functional determinants in the autoinhibitory domain of calcium/calmodulin-dependent protein kinase II. J Biol Chem. 1992;267:1761–1768. [PubMed] [Google Scholar]
- Smith MK, Colbran RJ, Soderling TR. Specificities of autoinhibitory peptides for four protein kinases. J Biol Chem. 1990;265:1837–1840. [PubMed] [Google Scholar]
- Soderling TR. Protein kinases: regulation by autoinhibitory domains. J Biol Chem. 1990;265:1823–1826. [PubMed] [Google Scholar]
- Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M. The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell. 1993;75:887–897. doi: 10.1016/0092-8674(93)90533-v. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Bloom GS, Mumby MC. A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J Cell Biol. 1995;128:1131–1144. doi: 10.1083/jcb.128.6.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden D, Rowe SJ. Protein kinase C activation antagonizes melatonin-induced pigment aggregation in Xenopus laevismelanophores. J Cell Biol. 1992;119:1515–1521. doi: 10.1083/jcb.119.6.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler CD, Haimo LT. Regulation of organelle transport in melanophores by calcineurin. J Cell Biol. 1990;111:1939–1948. doi: 10.1083/jcb.111.5.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournebize R, Andersen SSL, Verde F, Doree M, Karsenti E, Hyman AA. Distinct roles of PP1 and PP2A-like phosphatases in control of microtubule dynamics during mitosis. EMBO (Eur Mol Biol Organ) J. 1997;16:5537–5549. doi: 10.1093/emboj/16.18.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turowski P, Fernandez A, Favre B, Lamb NJC, Hemmings BA. Differential methylation and altered conformation of cytoplasmic and nuclear forms of protein phosphatase 2A during cell cycle progression. J Cell Biol. 1995;129:397–410. doi: 10.1083/jcb.129.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Patten SM, Ng DC, Th'ng JP, Angelos KL, Smith AJ, Walsh DA. Molecular cloning of a rat testis form of the inhibitor protein of cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1991;88:5383–5387. doi: 10.1073/pnas.88.12.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann R, Hanson PI, Schulman H. Multifunctional Ca2+/ calmodulin-dependent protein kinase made Ca2+independent for functional studies. Biochemistry. 1990;29:1679–1684. doi: 10.1021/bi00459a002. [DOI] [PubMed] [Google Scholar]
- Walsh DA, Glass DB. Utilization of the inhibitor protein of adenosine cyclic monophosphate-dependent protein kinase, and peptides derived from it, as tools to study adenosine cyclic monophosphate-mediated cellular processes. Methods Enzymol. 1991;201:304–316. doi: 10.1016/0076-6879(91)01027-y. [DOI] [PubMed] [Google Scholar]
- White BH, Sekura RD, Rollag MD. Pertussis toxin blocks melatonin-induced pigment aggregation in Xenopusdermal melanophores. J Comp Physiol B. 1987;157:153–159. doi: 10.1007/BF00692359. [DOI] [PubMed] [Google Scholar]
- Yang S, Lickteig RL, Estes R, Rundell K, Walter G, Mumby MC. Control of protein phosphatase 2A by simian virus 40 small-t antigen. Mol Cell Biol. 1991;11:1988–1995. doi: 10.1128/mcb.11.4.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]