

Abstract
While much has been learned in recent years about the movement of soluble transport factors across the nuclear pore complex (NPC), comparatively little is known about intranuclear trafficking. We isolated the previously identified Saccharomyces protein Mlp1p (myosin-like protein) by an assay designed to find nuclear envelope (NE) associated proteins that are not nucleoporins. We localized both Mlp1p and a closely related protein that we termed Mlp2p to filamentous structures stretching from the nucleoplasmic face of the NE into the nucleoplasm, similar to the homologous vertebrate and Drosophila Tpr proteins. Mlp1p can be imported into the nucleus by virtue of a nuclear localization sequence (NLS) within its COOH-terminal domain. Overexpression experiments indicate that Mlp1p can form large structures within the nucleus which exclude chromatin but appear highly permeable to proteins. Remarkably, cells harboring a double deletion of MLP1 and MLP2 were viable, although they showed a slower net rate of active nuclear import and faster passive efflux of a reporter protein. Our data indicate that the Tpr homologues are not merely NPC-associated proteins but that they can be part of NPC-independent, peripheral intranuclear structures. In addition, we suggest that the Tpr filaments could provide chromatin-free conduits or tracks to guide the efficient translocation of macromolecules between the nucleoplasm and the NPC.
Keywords: nucleocytoplasmic transport, nucleoskeleton, nuclear pore complex, nuclear envelope, Saccharomyces cerevisiae
The nuclear envelope (NE)1 defines the boundary between the nucleus and the cytoplasm in eukaryotic cells. The NE is composed of two distinct but continuous membranes enclosing a lumen. The outer nuclear membrane is continuous with the ER membranes and is thought to perform rough ER functions (Fawcett, 1966; Baba and Osumi, 1987; Preuss et al., 1991; Strambio-de-Castillia et al., 1995). Facing the nucleoplasm is the inner nuclear membrane, which in multicellular eukaryotes is often lined by a filamentous network called the nuclear lamina (Gerace et al., 1978; for review see Moir et al., 1995). The inner and the outer nuclear membranes join to form specialized circular apertures of ∼100 nm diameter containing the nuclear pore complexes (NPCs). NPCs provide the only known pathway for the exchange of cellular material across the NE. The core of the NPC consists of a cylindrical assembly of eight identical spoke structures symmetrically arranged around the central transporter (Unwin and Milligan, 1982; Hinshaw et al., 1992; Akey and Radermacher, 1993; Yang et al., 1998). Images of the transporter suggest that it is a gated structure with a roughly cylindrical shape (Akey, 1990; Akey and Radermacher, 1993; Goldberg and Allen, 1996). Peripherally associated nuclear and cytoplasmic filaments project from the core (Feldherr et al., 1984; Dworetzky and Feldherr, 1988; Akey and Goldfarb, 1989). The nuclear filaments conjoin to form the nuclear fishtrap or nuclear basket (Ris, 1991; Goldberg and Allen, 1992). Transport substrates dock to these filaments and translocate through the transporter on their way in and out of the nucleus. The NPCs of Saccharomyces share many features with their vertebrate counterparts, though they are significantly smaller both in mass and in volume (Yang et al., 1998). A variety of immunological, biochemical and genetic techniques have been successfully employed in the past few years to identify NPC component proteins (nucleoporins). Three criteria are commonly used to demonstrate that a novel protein is a bona fide nucleoporin: the protein should (a) immunolocalize to the NPC by immunofluorescence (IF) microscopy or better by immunoelectron microscopy (IEM); (b) cofractionate with the NPC in subcellular fractionation procedures; and (c) interact genetically and/or biochemically with other known nucleoporins. To date 28 yeast proteins and 15 vertebrate proteins have been identified that meet at least two of the above mentioned criteria for a nucleoporin (for review see Rout and Wente, 1994; Bastos et al., 1995; Doye and Hurt, 1997; Fabre and Hurt, 1997).
While it is clear that the NPC is a major regulator of nucleocytoplasmic transport, the knowledge of how this essential superstructure is connected both structurally and functionally to the nuclear interior is still extremely limited. Recently, the investigation of the molecular basis of such connections has received great impetus from the study of vertebrate Tpr (translocated promoter region) and of the Tpr-related Drosophila protein Bx34. The NH2-terminal ∼200 amino acid residues of human Tpr have been detected in various human tumors fused with the kinase domains of the three protooncogenes met, trk, and raf (Park et al., 1986; Soman et al., 1991; Greco et al., 1992; Mitchell and Cooper, 1992). The sequence of Tpr predicts a large protein (∼265 kD) with a bipartite secondary structure (Byrd et al., 1994; Bangs et al., 1996). The NH2-terminal 70% of the polypeptide sequence (∼184 kD) has a high α-helical content and is predicted to give rise to a coiled-coil domain. The remaining 30% of the protein (∼81 kD) is predicted to be acidic. While Tpr had been initially localized exclusively to the cytoplasmic filaments associated with the NPC (Byrd et al., 1994), more recent work has conclusively demonstrated that Tpr is a constitutive component of long nuclear filaments (up to ∼300 nm in Xenopus) that appear to connect the distal ring of the nuclear basket with the nucleolus. These filaments could correspond to the 5–6-nm filaments described on numerous occasions (Franke, 1970; Franke and Scheer, 1970a,b; Kartenbeck et al., 1971; Richardson et al., 1988; Ris and Malecki, 1993; Cordes et al., 1997) and recently found to form extensive networks of branching hollow cables that project from the nuclear baskets towards the nucleoplasm in amphibian oocytes (Ris, 1997). The predicted filamentous structure of Tpr together with its localization led to the proposal that this protein could have a role in providing a structural framework for the transport of material from the NPCs towards the interior and vice versa. This conclusion has received support from results obtained with a Tpr-related protein in Drosophila, called Bx34. This protein was initially described as one of two novel classes of Drosophila NE antigens that had been identified using mAbs obtained against chromosomal protein fractions (Frasch et al., 1988). Strikingly, Bx34 was recently found both at the nuclear periphery in association or near NPCs and in the nuclear interior in extrachromosomal and extranucleolar regions reminiscent of the extrachromosomal channel network described in relation to mRNA processing and transport (Zimowska et al., 1997). Consistent with its localization pattern, Bx34 was found to cofractionate exclusively with biochemical preparations of the nuclear matrix suggesting that it could represent a component of the nuclear matrix filamentous network. Interestingly, Bx34 retains its association with the chromosomes until very late in mitosis leading to the suggestion that it could have a structural role in aiding chromosomal segregation in anaphase. Further support for the involvement of Tpr in nuclear transport has recently come from a report indicating that this protein can be coimmunoprecipitated from Xenopus egg extracts together with karyopherins α and β1 (Shah et al., 1998).
The yeast Saccharomyces presents certain advantages over multicellular eukaryotes as a system to study such proteins. They do not have the complications of nuclear disassembly, having a closed mitosis; in addition, the genetics and molecular biology of yeast are better understood than in any other eukaryote, and the DNA sequence of the entire yeast genome is now known (Clayton et al., 1997). We describe here work aimed at isolating and characterizing non-NPC NE-associated proteins. Isolated NEs prepared as previously described (Strambio-de-Castillia et al., 1995) were used to produce a panel of mAbs against NE-associated components. A novel NPC-clustering assay was devised to specifically isolate anti-NE mAbs that recognized antigens only partially or peripherally associated with the NPCs. Two mAbs were isolated that recognized an ∼220-kD NE antigen that clustered only partially with the NPCs in a NPC-clustering strain. The gene encoding this antigen was molecularly cloned and was found to correspond to the previously isolated myosin-like protein (MLP1; Kolling et al., 1993). This gene encodes a nuclear protein of unknown function that is the closest yeast relative of Tpr. Ultrastructural localization and studies of deletion mutants strongly argues that Mlp1p and its homologue Mlp2p could be involved in providing a structural and functional link between the NPCs and the nuclear interior.
Materials and Methods
Subcellular Fractionation
The yeast strains Saccharomyces uvarum (NCYC 74, ATCC 9080), considered a strain of Saccharomyces cerevisiae (Mortimer and Johnson, 1986), or S. cerevisiae (W303; Thomas and Rothstein, 1989) were used throughout the procedure. Enriched nuclei, NEs and heparin-extracted NEs were prepared as previously described (Rout and Kilmartin, 1990, 1994; Strambio-de-Castillia et al., 1995; Rout and Strambio-de-Castillia, 1998). Enriched NPCs were prepared from nuclei (fraction 7) exactly as described in Rout and Blobel (1993) and Rout and Strambio-de-Castillia (1998).
Proteins contained in 30 ml of the highly enriched NE fraction described above (fraction 10), were precipitated by mixing the sample with 9 vol of methanol and harvested by centrifugation. The methanol pellet was solubilized in 4 ml of 10 mM MES, pH 6.5, 100 mM DTT, 1% SDS at 90°C for 10 min. The resuspended proteins were mixed with 36 ml of 20 mM MES, pH 6.5, 7 M Urea, 1% (vol/vol) Triton X-100, 0.1% SDS, 1 mM DTT (buffer 7). 16 ml of a 1:1 suspension of the cation-exchange S–Sepharose resin (8 ml of resin bed) were loaded on a broad base, 50-ml column and washed three times with 20 ml of buffer 7. The NE sample was loaded onto the column and was allowed to absorb onto the resin by incubating for 1 h at 25°C with gentle rocking. After the binding step, the flow-through from the column was harvested and pooled with a 20-ml wash in buffer 7 (this pooled material was termed unbound fraction). The column was eluted two times with 30 ml each of 1 M NaCl in buffer 7. Proteins from both bound and unbound fractions were harvested by methanol precipitation. Aliquots were separated on SDS-PAGE. After electrophoresis, the fractionation pattern of known NPC components was analyzed by immunoblotting using mAb414 (Davis and Blobel, 1986). The unbound fraction (termed S-NE), was found to be selectively depleted of most nucleoporins recognized by mAb414. Proteins from this fraction were harvested by methanol precipitation, resuspended in PBS, and used to immunize mice.
Mice Immunization and Production of mAbs
The production of hybridomas from B-lymphocytes derived from mice spleens was as previously described (Galfre and Milstein, 1981; Rout and Kilmartin, 1990). Supernatants were screened by indirect IF microscopy of whole yeast cells (Rout and Kilmartin, 1990; Wente et al., 1992; Kilmartin et al., 1993). Positive supernatants were also screened by immunoblotting of enriched NEs. Cells from positive lines were cloned up to four times by limiting dilution using a standard protocol (Galfre and Milstein, 1981; Rout and Kilmartin, 1990).
Molecular Cloning and Sequence Analysis
A λgt11 S. cerevisiae genomic expression library (Clontech Laboratories Inc., Palo Alto, CA) was screened by immunoblotting using mAb148G11 following the specifications of the manufacturer. Three positive λ clones containing an identical insert of ∼1.8 kb were obtained and purified to homogeneity by four consecutive rounds of screening. The insert from one of the positive λ clones was subcloned into pBluescript SK(+/−), sequenced from both ends using the T3 and the T7 standard primers and found to contain a sequence identical to the coding region for AA 1,105– 1,700 of MLP1 (Myosin Like Protein 1; Kolling et al., 1993). DNA sequence comparisons were performed using the BLAST algorithm (Altschul et al., 1990). Deducted amino acid sequences were compared with sequences in the SGD (Saccharomyces Genome Database), GenBank and EMBL databases using the FASTA algorithm (Parson and Lipman, 1988). Amino acid sequence alignments were performed using FASTA and CLUSTAL W v. 1.6 (Higgins et al., 1994). Amino acid sequences were analyzed using Protean v. 1.08 (DNAStar Inc.) and MacStripe 1.3.1 (Lupas et al., 1991).
Ultrastructural Studies
IEM analysis of isolated NEs was performed using a modification of a published procedure (Wray and Sealock, 1984; Rout and Kilmartin, 1990; Kraemer et al., 1995). For the IEM of isolated whole nuclei, the nuclear preparations were subjected to mild osmotic shock by diluting them with 9 vol of PVP solution (8% polyvinylpyrrolidone [PVP]; 20 mM potassium phosphate, pH 6.5; 0.75 mM MgCl2). These broken nuclei were then transferred (100 μl of diluted sample per well) to microtiter wells pretreated as described for the IEM of isolated NEs, and centrifuged at 23,500 g for 30 min at 4°C. Nuclei pellets were washed twice with PVP solution at 25°C followed by one wash each for 5 min with the following three solutions: (a) 25% (vol/vol) M buffer (5% dried milk in bt-DMSO [10 mM Bis-Tris-Cl, pH 6.50, 0.1 mM MgCl2, 20% DMSO]), 75% (vol/vol) PVP solution; (b) 50% (vol/vol) M buffer, 50% (vol/vol) PVP solution; and (c) 75% (vol/ vol) M buffer, and 25% (vol/vol) PVP solution. The nuclei pellets were washed once in M buffer for 5 min at 25°C. Subsequent steps were also as described above with the following exceptions: (a) 0.5× PBS-K, 1 mM MgCl2 was substituted with bt-DMSO; and (b) after treatment with osmic acid, samples were postfixed with 1% tannic acid in 50 mM potassium phosphate, pH 7.0, for 30 min at 25°C.
Localization of Green Fluorescent Protein Fusion Proteins
To generate green fluorescent protein (GFP)-tagged proteins we used the plasmid pGFP-N-FUS encoding the GFP gene under the control of the MET25 promoter (Niedenthal et al., 1996). DNA sequences encoding the following Mlp1p fragments: (a) NT1 (AA 1–667; pGFPNT1); (b) NT2 (AA 668–1446; pGFPNT2); (c) CT (AA 1,447–1,875; pGFPCT); and (d) putative nuclear localization sequence (NLS), pNLS (AA 1,486–1,545; pGFPpNLS), were PCR-amplified and cloned in-frame downstream of the GFP gene using the unique cloning sites XbaI and XmaI. pGFPNT1, pGFPNT2, pGFPCT, pGFPpNLS, and the negative control pGFP-N-FUS were transformed into W303 cells. To induce the expression of the GFP-tagged Mlp1p fragments, cells were grown to mid-log phase in selective medium without methionine. GFP fusion proteins were visualized in living cells after mounting on microscope slides with 2% methyl cellulose, using a Zeiss Axiophot microscope. (Carl Zeiss) equipped with a Chroma no. 41014 GFP Filter (Chroma Technology Corp.). Photomicrographic recording was performed using a Sony DKC5000 digital photo camera (Morrel Instruments Inc.) interfaced with Adobe Photoshop v. 4.01 (Adobe Systems, Inc.)
Gene Disruption and Protein A Tagging of MLP1 and MLP2
All yeast strains were derived from W303 (Thomas and Rothstein, 1989; Table I). Gene replacement of MLP1 and MLP2 was accomplished using published methods (Rothstein, 1990; Aitchison et al., 1995a,b). MLP1 was replaced with URA3 by generating a PCR product containing the entire URA3 gene flanked by 75 nucleotides directly upstream and 75 directly downstream of the MLP1 coding region. A similar procedure was followed to disrupt MLP2 with HIS3. A haploid strain carrying disrupted copies of both MLP1 and MLP2 (CSDC09α) was constructed by mating CSDC03α and CSDC05a and subsequent sporulation. Haploid strains of opposite mating types carrying the double deletion of MLP1 and MLP2 were mated to generate a homozygous diploid strain (CSDC13a/α; mlp1&mlp2Δhd). In all cases correct integration and segregation of each individual disruptions were verified by PCR analysis of the genomic DNA. The expression of Mlp1p in wild-type and mutant strains was analyzed by immunoblotting of whole yeast cell lysates and by indirect IF microscopy using mAb148G11.
Table I.
Yeast Strains
| Strain name* | Genotype | Derivation | ||
|---|---|---|---|---|
| W303 | MATa/MATα, leu2-3,112/leu2-3,112, his3-11,15/his3-11,15, trp1-1/trp1-1, can1-100/can1-100, ade2-1/ade2-1, ura3-1/ura3-1 | Thomas and Rothstein, 1989 | ||
| CSDC01a/α | W303 Mata/Matα, mlp1::URA3/MLP1 | This study | ||
| CSDS02a | W303 Mata, mlp1::URA3 | Segregant of CSDC01a/α; this study | ||
| CSDC03α | W303 Matα, mlp1::URA3 | Segregant of CSDC01a/α; this study | ||
| CSDC04a/α | W303 Mata/Matα, mlp2::HIS3/MLP2 | This study | ||
| CSDC05a | W303 Mata, mlp2::HIS3 | Segregant of CSDC04a/α; this study | ||
| CSDC06α | W303 Matα, mlp2::HIS3 | Segregant of CSDC04a/α; this study | ||
| CSDC07a/α | W303 Mata/Matα, mlp1::URA3/MLP1, mlp2::HIS3/MLP2 | This study | ||
| CSDC08α, mlp2Δ | W303 Matα, MLP1, mlp2::HIS3 | Segregant of CSDC07a/α; this study | ||
| CSDC09α, mlp1&mlp2Δ | W303 Matα, mlp1::URA3, mlp2::HIS3 | Segregant of CSDC07a/α; this study | ||
| CDSC10a, mlp1Δ | W303 Mata, mlp1::URA3, MLP2 | Segregant of CSDC07a/α; this study | ||
| CSDC11a, wt | W303 Mata, MLP1, MLP2 | Segregant of CSDC07a/α; this study | ||
| CSDC12a | W303 Mata, mlp1::URA3, mlp2::HIS3 | Segregant of CSDC07a/α; this study | ||
| CSDC13a/α, mlp1&mlp2Δhd | W303 Mata/Matα, mlp1::URA3/mlp1::URA3, mlp2::HIS3/mlp2::HIS3 | This study | ||
| CDSC14a/α | W303 Mata/Matα, mlp1-proteinA::HIS3, URA3/MLP1 | This study | ||
| CSCD15a, mlp1-pA | W303 Mata, mlp1-proteinA::HIS3, URA3 | Segregant of CSDC14a/α; this study | ||
| CSDC17a/α | W303 Mata/Matα, mlp2-proteinA::HIS3, URA3/MLP2 | This study | ||
| CSDC18a, mlp2-pA | W303 Mata, mlp2-proteinA::HIS3, URA3 | Segregant of CSDC17a/α; this study | ||
| W303/pGALMLP1 | W303 Mata/Matα, pGALMLP1::URA3 | This study |
All strains are isogenic to W303. The most important strains for the experiments described in this paper were given a second, more descriptive name that is used in the text and in the figure legends. This designation is indicated in bold characters.
For protein A tagging of Mlp1p and Mlp2p, PCR products were generated that contained the coding region for four and a half IgG-binding repeats of protein A followed by a HIS3, URA3 cassette flanked by 75 nucleotides immediately upstream and 75 nucleotides immediately downstream of the stop codon (i.e., excluding the stop codon itself) of each gene (Aitchison et al., 1995a,b).
In Vivo Import and Diffusion Assays
The in vivo import assay was performed as described (Shulga et al., 1996). In brief, wild-type W303 and mlp1&mlp2Δhd cells expressing GFP (Cody et al., 1993) fused to the SV40 large T antigen NLS were incubated in dextrose-free medium containing 10 mM each of 2-deoxy-d-glucose and sodium azide at 30°C for 45 min. At the end of the incubation, cells were washed once and incubated in dextrose-containing medium at 30°C to allow reimport of the substrate into the nucleus. The number of normal cells showing a clear accumulation of GFP-NLS in the nucleus (nuclear cells) and the number of cells in which the reporter was cytoplasmic were counted at each time point. At least 40 independent cells were scored per time point. At least four independent sets of cells were counted to construct the graph presented in Fig. 8. The results are presented as the percentage of cells presenting nuclear signal as a function of time. Linear regression lines were drawn through the linear portion of each curve using KaleidaGraph and the slope of these straight lines were used to estimate the relative import rates.
Figure 8.



Mlp1p and Mlp2p are involved in facilitating nuclear import of a NLS-GFP reporter. (A) Graphical representation of the results of an in vivo nuclear import assay performed as described in Materials and Methods. During recovery from an incubation in the presence of the metabolic inhibitors deoxyglucose and sodium azide, wild-type cells reimported a NLS-GFP reporter very rapidly and the reaction was complete in 15 min. The mutant (mlp1&mlp2Δhd) on the other hand, displayed a much slower rate of reimport and took up to twice as much as the wild-type to reach steady state levels of nuclear signal. (B) Graphical representation of the results of an in vivo nuclear diffusion assay analogous to the import assay described above (see Materials and Methods for details). During incubation in the presence of the metabolic inhibitors deoxyglucose and sodium azide, aliquots were collected from both wild-type and mlp1&mlp2Δhd and the percentage of normal cells displaying clear nuclear localization of the reporter protein NLS-GFP was scored as a function of time. The kinetic of nuclear egress is significantly faster in mutant cells relative to wild-type, consistent with the reduced import rate of the mutant observed in the top panel. (C) Graphical representation of a model explaining the results presented in A and B. Gray arrows depict the rate of passive diffusion of the NLS-GFP substrate from the nucleus which is constant both in the presence (+ Inhibitor) and in the absence (− Inhibitor) of metabolic inhibitors. Black arrows indicate the rate of active import of the substrate into the nucleus.
Passive diffusion assays were performed as follows. pGFP-LEU transformed wild-type and mutant cells were grown as described (Shulga et al., 1996). Cells were harvested by centrifugation and resuspended in one-fifth of the initial volume of selective medium. Resuspended cells were held at 4°C until use. The diffusion assay was started by centrifuging the cells, washing them once in sterile water, resuspending them in 1 vol of 10 mM sodium azide and 10 mM deoxyglucose in dextrose-free selective medium and finally placing them at 30°C. Aliquots were taken at each time point and scored as described above. At least 3 independent sets of 40 cells were scored at each time point. The relative passive equilibration rates were estimated as described for the import rates.
Overexpression of MLP1
To achieve the overexpression of Mlp1p in yeast cells, the MLP1 gene was inserted into the pYES2 yeast expression plasmid (Invitrogen Corp.) downstream of the GAL1/10 inducible promoter. The unique BspHI site located at nucleotide position 4958 of pYES2 was disrupted using the Klenow fragment of DNA polymerase I to generate pYES2-no BspHI. DNA from the λ clone λPM-5620 (Olson et al., 1986; ATCC catalog number 70598; America Type Culture Collection) containing a yeast genomic fragment of ∼16,800 bp from chromosome XI (chromosome bp coordinates: 611843–628638), was used as the template to generate a ∼650 bp PCR product containing the 5′ region of MLP1 from nucleotide position −12 (relative to the first bp of the coding region) to nucleotide position +634 flanked by a BamHI site at the 5′ end. This PCR product was cut with BamHI and EcoRI (site located at nucleotide position +572 of the MLP1 coding region) and inserted into the BamHI and EcoRI sites of pYES2-no BspHI, to generate pYES2-570MLP1. Finally, an ∼5,900-bp BspHI fragment of λPM-5620 that contains the entire MLP1 coding region except the first 8 bp, was inserted into pYES2-570MLP1 that had been linearized with BspHI (site located at nucleotide position +8 of the MLP1 coding region) to generate pGALMLP1.
pGALMLP1 was transformed into W303 cells by electroporation and transformants were selected taking advantage of the URA3 selectable marker present on the plasmid. Only freshly transformed cells were used for each experiment. To induce the expression of MLP1, W303/ pGALMLP1 cells were grown to mid-logarithmic phase in selective medium containing 2% D(+)-raffinose (referred to as raffinose throughout the text), transferred to selective medium containing 1% raffinose and 2% galactose and incubated at 30°C for up to 4 h. For repression of MLP1 expression, W303/pGALMLP1 cells were grown in raffinose as above, transferred to selective medium containing 1% raffinose and 2% dextrose and incubated for 4 h at 30°C.
Miscellaneous
SDS-PAGE and immunoblotting were performed essentially as described (Rout and Blobel, 1993). The intensity of bands on immunoblots was quantified using the ImageQuant v.1.1 software in the PhosphorImager system (Molecular Dynamics), when a Cy5-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories) was used. Occasionally, computer images of enhanced chemiluminescence signals on photographic films were quantified using the gel plotting macro of NIHImage v.1.60 (Research Services Branch, National Institutes of Health, Bethesda, MD).
Cells were prepared for indirect IF microscopy using the procedure of Kilmartin and Adams (1984) with the modifications of Wente et al. (1992) and Kilmartin et al. (1993). Double labeling with mAb148G11 and a polyclonal rabbit anti-Nup159p antibody (Del Priore et al., 1997) was visualized using Cy3-labeled polyclonal donkey anti–rabbit IgG (cross absorbed against rabbit IgG) and DTAF-labeled polyclonal donkey anti–mouse IgG (cross absorbed against mouse IgG; Jackson ImmunoResearch Laboratories). In all single labeling experiments, the bound antibody was visualized using Cy3-labeled polyclonal donkey anti–mouse or anti–rabbit IgG (Jackson ImmunoResearch Laboratories). The staining conditions were as described (Wente et al., 1992). Photomicrographic recording was performed using a Zeiss Axioplan2 microscope (Carl Zeiss) equipped with a Photometrics SenSys A2S digital photo camera (Photometrics) interfaced with IPLab Spectrum p v. 3.1.1c (Signal Analytics).
The growth competition experiment was performed following published procedures (Smith et al., 1996; Rout et al., 1997; Thatcher et al., 1998).
Results
A Screen for Non-NPC Proteins Associated with the NE
Mutations or deletions of the genes that encode for certain nucleoporins (for example, Nup133p, Nup120p, Nup145p, and Nup159p) can cause the NPCs to accumulate at one side of the NE, giving rise to tight clusters that are easily identified as spots or patches when cells from such strains are stained with a nucleoporin specific antibody (Doye et al., 1994; Wente and Blobel, 1994; Aitchison et al., 1995a; Heath et al., 1995; Kraemer et al., 1995; Li et al., 1995; Pemberton et al., 1995). It was reasoned that proteins only partially associated with the NPC, or localized to areas of the NE that are not in close contact with the NPC, would either fail to cluster or would only partially cluster with the NPCs in these strains. Isolated yeast NEs prepared as previously described (Strambio-de-Castillia et al., 1995) were used to produce a panel of 114 anti-NE mAbs that were screened by an assay based on this NPC clustering phenomenon. The indirect IF pattern generated by each of the individual mAbs on fixed whole wild-type yeast cells was compared with the staining pattern obtained on a yeast strain carrying a gene disruption in the NUP133 gene (Pemberton et al., 1995). The anti-NE antibody, mAb148G11, was identified by this screen as recognizing an antigen that appeared to only partially cluster in the NUP133 disrupted strain. This mAb recognized a protein that runs as a single band of ∼220 kD (apparent molecular mass; p220) on immunoblots (see Fig. 3).
Figure 3.

Mlp1p is peripherally associated with NEs but only partially cofractionates with NPCs. Yeast cells were fractionated to produce nuclei (Nuclear Prep, fractions 1–8), NEs (NE prep, fractions 9–12) and NPCs (NPC Prep, fractions 9–17; Rout and Kilmartin, 1990; Rout and Blobel, 1993; Strambio-de-Castillia et al., 1995). Peripherally associated NE components were stripped from the NE fraction by heparin-extraction (NE Prep, fractions 13–16; Strambio-de-Castillia et al., 1995). Proteins from each subcellular fraction were resolved on SDS-PAGE, transferred to nitrocellulose and immunoblotted with the anti-Mlp1p mAb148G11. Fractions that belong to each of the enrichment steps are grouped as indicated by brackets at the top of the gels. Figures on top of the gels refer to the fraction numbers as previously described (Rout and Blobel, 1993; Strambio-de-Castillia et al., 1995). The total spheroplasts lysate (Cells) and subsequent NE- and NPC-containing fractions are indicated on top of each gel. In all cases C- stands for Crude and E- stands for Enriched. H-NEs denotes heparin-extracted NEs. A small arrow points to a band that disappears together with Mlp1p in strains harboring a deletion of the gene and is therefore an Mlp1p breakdown product. Numbers at the bottom of the gels (% Total Cellular Amount) represent an estimate of the amount of Mlp1p specific signal present in each fraction based on quantitative immunoblotting, expressed as a percentage of the total cellular amount. The figures below the bottom brackets (Loading Equivalents) indicate the number of cell equivalents (n) that were used as the starting material to prepare each fraction (Rout and Blobel, 1993; Strambio-de-Castillia et al., 1995).
Both wild-type yeast and NUP133 deleted cells were double stained with mAb148G11 and a rabbit polyclonal antibody against the nucleoporin Nup159p (Fig. 1). In wild-type cells Nup159p and p220 colocalize to a great extent although this colocalization is not absolute. In particular, some areas of the NE are devoid of one or the other signals and some cells show some nucleoplasmic staining with mAb148G11 (Fig. 1, wt, p220 and merge, e.g., the three top cells). This raises the possibility that p220 may also be found in the nuclear interior. In the clustering strain the difference in the localization of Nup159p and p220 is striking (Fig. 1, ΔNUP133). In these cells Nup159p clearly forms tight clusters localized at the nuclear periphery consistent with its localization at the NPC, while p220 appears to be localized in large patches or even in continuous rims at the nuclear periphery that only partially overlap with the NPC clusters. Furthermore, Nup159p signal is closer to the cytoplasm than the p220 signal (Fig. 1, ΔNUP133, merge). This is consistent with the localization of p220 by IEM (see below) and is in accord with results recently obtained in Xenopus oocytes (Shah et al., 1998). In addition, similarly to data obtained with different spindle associated markers (Rout and Kilmartin, 1990), these findings demonstrate that it is possible to obtain IF localization to subregions of the NE even in yeast. Similar results were obtained when the localization of Mlp1p and Nup159p were compared in a NUP120 knock-out strain (data not shown; Aitchison et al., 1995a).
Figure 1.

Partial colocalization of p220 and NPCs in yeast cells by indirect IF microscopy. Logarithmically growing wild-type and ΔNUP133 cells were double labeled with mAb148G11 (anti-p220) and a rabbit anti-Nup159p polyclonal antibody. Bar, 2 μm.
Isolation of the Gene Encoding p220
The gene encoding p220, mAb148G11, was identified as MLP1 (Kolling et al., 1993). MLP1 encodes for a protein of 1875 AA with a predicted molecular mass of 218 kD cloned on the basis of its cross-reactivity with a mAb recognizing human platelet myosin (Kolling et al., 1993). This nonessential protein was hypothesized to have a nuclear function on the basis of its subcellular localization (see Discussion). The major structural features of Mlp1p, the position of the cloned fragment of the gene and of the epitope of mAb148G11, are shown in Fig. 2 A. The NH2-terminal ∼80% of the protein is predicted to have a high α-helical content and contains the heptad-repeats pattern characteristic of coiled-coil proteins. The COOH-terminal ∼400 amino acid residues of the protein are predicted to form a globular tail rich in phenylalanine and proline residues (Fig. 2 A, P/F rich region). The sequence with the highest degree of similarity to MLP1 based on FASTA analysis (28% identical and 66% similar) was the uncharacterized yeast open reading frame (ORF), YIL149c (referred to as MLP2 in Fig. 2 B). YIL149c is expected to encode a protein of 1,680 AA with a predicted molecular mass of 195 kD. MLP1 and YIL149c belong to a duplicated chromosomal region of the yeast genome (Block 38; Wolfe and Shields, 1997) present both on Chromosome XI and on Chromosome IX. The similarity between Mlp1p and Yil149p extends over the whole amino acid sequence and is underscored by the similarity of the overall predicted secondary structure of the proteins. Yil149p also contains a phenylalanine and proline rich COOH terminus. Based on its similarity to Mlp1p and the fact that MLP1 and YIL149c appear to have arisen from a genome duplication event, we propose the name MLP2 for YIL149c (see Discussion). The complete yeast genomic database (Clayton et al., 1997) contains no other putative homologues of MLP1.
Figure 2.


Schematic diagram representing some of the primary and secondary structural features of Mlp1p and a map of the Mlp1p fragments that were NH2-terminally tagged with GFP; and alignment between Mlp1p, Mlp2p and Tpr. (A) Shown as a function of the position in the sequence are: the probability of forming coiled-coil displayed as the probability value; the position of the region of the protein that is encoded by the clone isolated from the expression library using mAb148G11 and therefore contains the epitope of this mAb; the position of the proline/phenylalanine rich blocks together with the frequency of these residues within each block (the overall frequency of these two residues along the entire amino acid sequence is 3%). Coiled-coil predictions were performed as described in Materials and Methods. (B) Comparison of amino acid sequences of Mlp1p, Mlp2p (the yeast ORF YIL149c) and the human protein Tpr. Identical amino acid residues are shaded in white on black; similar amino acid residues are shaded in gray.
Mlp1p and Mlp2p are also similar to the vertebrate and Drosophila Tpr proteins (Frasch et al., 1988; Mitchell and Cooper, 1992; Byrd et al., 1994; Zimowska et al., 1997; Fig. 2 B). These proteins are associated with structures found on the nuclear side of the NPC and may be involved in facilitating nucleocytoplasmic transport (Cordes et al., 1997; Zimowska et al., 1997; Shah et al., 1998). The main difference between Tpr and Mlp1p is that the COOH-terminal tail (excluding the predicted coiled-coil region) of Tpr is relatively much longer, spanning the final 30% of the protein, and has a much more marked acidic character (Byrd et al., 1994; Zimowska et al., 1997). A recently identified S. pombe uncharacterized ORF (SPC162.08c) also shows significant similarities to MLP1, MLP2, and Tpr.
Mlp1p Is Associated with Intranuclear Filaments that Are Localized at the Interface between the NPCs and the Nuclear Interior
Results from the indirect IF analysis shown in Fig. 1 demonstrated that Mlp1p is localized at the NE in areas that are only partly occupied by NPCs, and that it cannot be only a NPC constituent, even a peripheral one (e.g., a nuclear basket component). To better understand its localization, the fractionation pattern of Mlp1p was followed during the preparation of NEs and NPCs (Fig. 3; Rout and Blobel, 1993; Strambio-de-Castillia et al., 1995). As expected, the majority of the Mlp1p fractionated with the nuclei and highly enriched NE fraction, in agreement with the indirect IF data (Fig. 1). After heparin-extraction of NEs, nearly all of the NE-associated Mlp1p pool (68% of the total) was found in the heparin supernatant (Fig. 3, NE Prep, fractions 13 and 14), demonstrating it is not strongly associated with the membrane. Only some of Mlp1p cofractionated with the enriched NPC preparation (Fig. 3, E-NPCs), suggesting a partial or weak association of this protein with NPCs, again consistent with the indirect IF results (Fig. 1). It is important to note at this point that Mlp1p represents the first NE-associated protein to display such a subcellular fractionation pattern (see Discussion).
The ultrastructural localization of Mlp1p was investigated by pre-embedding labeling IEM using mAb148G11 on both isolated NEs (Fig. 4 A, B, and B′) and isolated whole nuclei that had been subjected to mild osmotic shock to expose the nuclear interior (Fig. 4 C). It should be remembered that using this procedure only the position of the epitope of the antigen is being localized rather then the position of the protein as a whole. On the scale of the immunostained structures here this may not actually reflect the extent of the localization of the entire protein. A second important caveat of many immunolocalization techniques is that the dimensions of the antibody-gold conjugate have to be taken into account in determining the precise localization of the epitope. Nonetheless, on isolated NEs labeled with mAb148G11, gold particles were found almost exclusively on the nuclear side of the NE often though not always in the vicinities of NPCs. The average of the distance between gold particles and the nearest NPC was 66 ± 19 nm on the y axis and 41 ± 34 nm on the x axis (n = 50; Fig. 5, B and C). On numerous occasions the gold appeared to be associated with fibrillar structures stretching from the nuclear side of the NE towards the nucleoplasm (Fig. 4 B′, arrow). This was in marked contrast to the labeling of Nup159p, a nucleoporin known to be associated with the cytoplasmic fibrils attached to the outer ring of the NPC (Fig. 5 A). In this case, and consistent with published results (Kraemer et al., 1995), the gold particles were found significantly closer to the NPC (Y = 33 ± 13 nm; X = 8 ± 8 nm; n = 50; Fig. 5 C). The results obtained with the Nup159p control demonstrate that the absence of Mlp1p signal on the cytoplasmic side of the NE can not be due either to a lack of accessibility or to gross alterations or damage of the NE.
Figure 4.
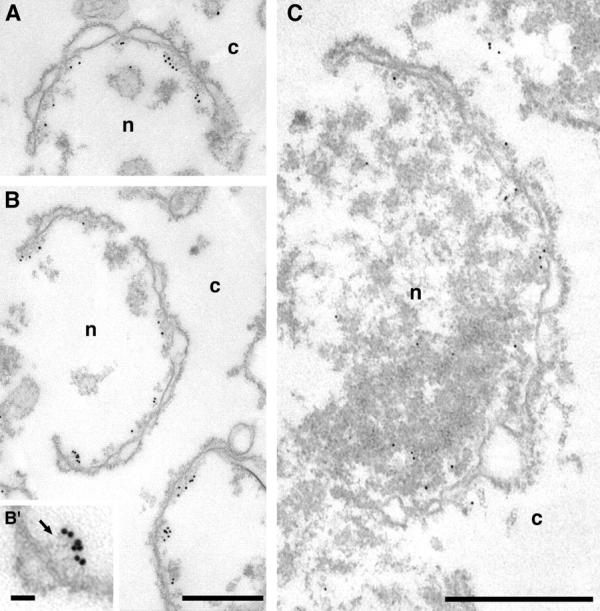
Mlp1p is found on the nuclear side of the NE and is often associated with fibrils stretching from the vicinities of NPCs towards the nuclear interior. Isolated NEs (A, B, and B′) and osmotically shocked isolated nuclei (C) were incubated with mAb148G11 and the labeling was visualized with either 10 nm (A, B, and B′) or 5 nm (C) gold-conjugated secondary antibodies. Labeled NEs were subsequently prepared for transmission EM analysis. The inset presented in B′ represents a threefold magnification of an area presented in B. The arrow points to nucleoplasmic fibrils immunostained with mAb148G11. c, cytoplasm; n, nucleoplasm. Bars, 500 nm (A and B, C); 50 nm (B′).
Figure 5.

Mlp1p is peripherally localized relative to the NPC. (A and B) Composites of selected NPC images from isolated NEs immunolabeled either with mAb165C10 that binds specifically to Nup159p (A) or with the anti-Mlp1p antibody, mAb148G11 (B) as in Fig. 4. 14 images of individual NPCs that had been clearly sectioned orthogonally to the mid-plane of the NE were selected from each sample. A radius of ∼150 nm around the center of each NPC was selected as the cut-off point for each image. The 14 images derived from each sample were superimposed using Adobe Photoshop v. 3.05 (Adobe Systems, Inc.) to produce the composites presented here. (C) The distances from the center of each gold particle to the mid-plane of the NE (y) and to the cylindrical axis of the NPC (x) were measured for isolated NEs (NEs) and broken nuclei (Nuclei) labeled either with mAb165C10 (Nup159p) or mAb148G11 (Mlp1p). Cyt, cytoplasm; Nuc, nucleoplasm.
The distribution of the Nup159p control in the isolated whole nuclei was unchanged with respect to the NEs (Y = 37 ± 11 nm; X = 12 ± 14 nm; n = 14; Fig. 5 C, Nuclei). While the majority of the Mlp1p signal in whole nuclei was found in the immediate vicinity of the NE, a significant fraction was found at a considerable distance into the nuclear interior, sometimes as much as ∼300 nm from the mid-plane of the NE (Fig. 5 C, Nuclei). This resulted in the average distribution of Mlp1p being further from the mid-plane of the NE than in isolated NEs (Y = 84 ± 56 nm; X = 33 ± 16 nm; n = 50; using the nearest NPC as a referent). The shorter distance in the case of isolated NEs could be due to the collapse of filaments during the NE isolation procedure (see Discussion). Gold was found even further into the nucleoplasm but it was difficult to establish whether this signal was significantly above background (Fig. 4 C). No obvious structure was observed in association with gold localized in the interior of the nucleus, although fibrils could again be seen associated with gold particles found near the NE. Interestingly, in both isolated NEs and isolated whole nuclei the Mlp1p signal appeared to extend a maximum of ∼120 nm from the cylindrical axis of the NPCs, which corresponds to the proposed minimum in vivo inter-NPC distance (Winey et al., 1997). The results of the IEM localization studies are consistent with the indirect IF and immunoblot results presented above (Figs. 1 and 3), and suggest that Mlp1p is associated with filaments localized at an interface between the nuclear interior and the NPC.
The COOH Terminus of Mlp1p Is Responsible for Its Nuclear Localization
To determine which region of Mlp1p is responsible for its nuclear localization, the coding sequence of the protein was roughly divided in three thirds (NH2-terminal 1, NT1; NH2-terminal 2, NT2; and COOH-terminal, CT; Fig. 2 A) and each third was GFP-tagged. In addition, a short sequence localized at the non-coiled-coil COOH terminus was selected because of its high lysine and arginine content and hence its similarities to known NLSs, and was also GFP-tagged (putative NLS, pNLS; Fig. 2 A). The intracellular localization of GFP was determined in living cells expressing either GFP alone or the GFP-tagged fragments of Mlp1p (Fig. 6). As expected, untagged GFP had a predominantly cytoplasmic distribution even though it was not excluded from the nucleus. A similar distribution was observed with cells expressing GFP fused to the both of the NH2-terminal thirds of Mlp1p and to the putative NLS. Interestingly however, the COOH terminus of Mlp1p was able to direct the targeting of GFP to the nucleus but not to the NPCs. This suggests that it contains a bona fide NLS that has no obvious homologies to other NLSs, and is not sufficient for NPC association.
Figure 6.

The COOH-terminal, non-coiled-coil region of Mlp1p is responsible for its nuclear localization. W303 cells expressing pGFP-N-FUS (GFP), pGFPNT1, pGFPNT2, pGFPCT, and pGFPpNLS were observed by fluorescence microscopy to determine the subcellular localization of the GFP-tagged fragments of Mlp1p (see Materials and Methods and text for details). The top panels show the GFP signal and the bottom panels show the corresponding cells by Nomarsky optics. Bar, 4 μm.
Mlp2p Resembles Mlp1p in Its Fractionation Behavior and Cellular Localization
To determine the subcellular localization of Mlp2p, the gene was genomically tagged with an in-frame COOH-terminal fusion of the IgG binding domains of protein A (pA; Aitchison et al., 1995a). As a control, MLP1 was similarly tagged. Highly enriched NEs fractions were prepared from the Mlp1p- and Mlp2p-tagged strains (Strambio-de-Castillia et al., 1995) and the fractionation pattern of the proteins was assessed on immunoblots. As expected, the fractionation pattern of Mlp1p-pA was indistinguishable from the one observed in Fig. 3 (data not shown). Similarly, Mlp2p-pA cofractionated with the highly enriched NE fraction (Fig. 7 A, fraction 10) but was almost entirely stripped off by the heparin treatment (Fig. 7 A, fractions 13 and 14). The tagged strains were used to determine the subcellular localization of the proteins by indirect IF microscopy (Fig. 7 B). Both proteins were found localized predominantly at patches found at the nuclear periphery similar to that previously observed for Mlp1p (Fig. 1). The localization of Mlp1p and Mlp2p in tagged strains was also determined by preembedding IEM on isolated NEs. Again these two proteins displayed a very similar localization pattern to that observed for Mlp1p (data not shown). These observations are consistent with the hypothesis that Mlp1p and Mlp2p are functional homologues in accordance with their structural similarities and their genetic interaction (see below).
Figure 7.


Mlp2p is a Mlp1p homologue. (A) Highly enriched NE fractions from a yeast haploid strain expressing a pA tagged version of Mlp2p were prepared and probed as described in Fig. 3. (B) The localization of Mlp1p-pA or Mlp2p-pA (as indicated on the top of the images) was examined by indirect IF detection of the pA tag. The staining of DNA with DAPI is also shown. Bar, 2 μm.
Double Deletions of MLP1 and MLP2 Cause a Marked Decrease in the Yeast Comparative Fitness
The entire coding regions of both MLP1 and MLP2 were individually disrupted in the diploid yeast strain W303 by integrative transformation of the URA3 and HIS3 genes respectively. Each heterozygous diploid strain (mlp1:: URA3/+ and mlp2::HIS3/+) was sporulated and tetrads were dissected. In both cases four viable spores from most tetrads were observed, demonstrating that neither of these genes is essential and confirming and extending published results (Kolling et al., 1993). Immunostaining with mAb148G11 revealed that the punctate IF staining pattern and the ∼220-kD immunoblot band recognized by both antibodies was absent in mlp1::URA3 cells but was present in mlp2::HIS3 cells (data not shown). This confirmed that mAb148G11 binds specifically to Mlp1p and demonstrated that mAb148G11 does not cross-react with Mlp2p. Segregants of opposite mating types carrying the individual disruptions as confirmed by both phenotypic and genotypic analyses were mated and sporulated. Spores were isolated that carried both selectable markers demonstrating that the double knock-out of MLP1 and MLP2 is viable.
To assess the degree of selective disadvantage conferred by individual and double disruptions in the MLP1 and MLP2 genes, the mlp1Δ, mlp2Δ, and mlp1&mlp2Δ strains were each grown competitively with their wild-type counterpart in rich medium (Smith et al., 1996; Rout et al., 1997; Thatcher et al., 1998). While mlp1Δ and mlp2Δ competed successfully with wild-type, the strain harboring the double deletion lost ground rapidly even though it was initially added in twofold excess and appeared to be eliminated from the population after 30 generations (data not shown). These results demonstrated that mlp1&mlp2Δ had a fitness defect relative to the parental stock equal to 24% (selection coefficient 0.235 ± 0.021; Thatcher et al., 1998) and was effectively non-viable outside the protected laboratory environment. These results also indicated that MLP1 and MLP2 are homologous.
Deletion of MLP1 and MLP2 Affects the Efficiency of Nuclear Import
To investigate the possibility that Mlp1p and Mlp2p may be involved in transport of molecules in and out of the nucleus, an in vivo import assay was performed as described by Shulga et al. (1996). This assay allows the detection of kinetic defects in the import rates of a NLS-GFP reporter (Fig. 8 A). In this assay logarithmically growing yeast cells constitutively expressing NLS-GFP are harvested and poisoned in order to block the production of energy. Under these conditions the active import of the NLS-GFP reporter into the nucleus is dramatically reduced resulting in the equilibration of the GFP signal between the nucleus and the cytoplasm by passive diffusion across the NPC. When the metabolic inhibitors are removed and the cells are allowed to recover, NLS-GFP is once again rapidly imported in the nucleus. The relative rates of accumulation of the mutant strain are compared with the ones found with wild-type, revealing any defect in the mutant's efficiency of nuclear import. The steady state distribution of NLS-GFP was indistinguishable in homozygous diploid cells carrying a double deletion of MLP1 and MLP2 as compared with wild-type (data not shown). Nevertheless, mlp1&mlp2Δhd (homozygous diploid) cells displayed a markedly slower relative accumulation rate of NLS-GFP into the nucleus with respect to their wild-type counterpart, 13.5 ± 0.2%/min (wild-type) versus 8.9 ± 0.1%/min (mlp1&mlp2Δhd). Significantly, when double mutant cells were allowed to recover for extended periods of time (up to double the time required for wild-type), the initial equilibrium distribution of reporter protein was regained, again indicating that the efficiency of import and not its steady state balance was affected by the absence of Mlp1p and Mlp2p. The rates of passive equilibration of the NLS-GFP reporter during the incubation with the metabolic inhibitors were also measured in both wild-type and mutant cells (Fig. 8 B). In this case, mlp1&mlp2Δhd displayed a significant increase in the relative passive nuclear egress rates of the NLS-GFP reporter as compared with wild-type, −8.75 ± 1.0%/min (wild-type) versus −13.1 ± 0.6%/ min (mlp1&mlp2Δhd). A model that could explain both the import and the diffusion assay results is presented in Fig. 8 C. The basic assumption of this model is that the rates of passive diffusion of the reporter across the NPC are constant, both in the presence and in the absence of metabolic inhibitors (i.e., they do not require NTP hydrolysis). Consequently, during recovery, after the removal of the metabolic inhibitors (− Inhibitor), the rate of active import becomes greater than the diffusion rate and the net effect is accumulation of the transport substrate into the nucleus. If the rate of import is lower in mlp1&mlp2Δhd cells with respect to wild-type the prediction is that mutant cells will take a longer period of time to reach the steady state levels of nuclear accumulation of the substrate, and this is exactly what is observed. On the other hand, during the incubation with the inhibitors (+ Inhibitor), the rate of active import is drastically reduced in both wild-type and mutant cells. However, in the mutant cells this residual import is even further compromised as compared with the wild-type cells. As the diffusion rate remains constant this will appear as an overall faster egress rate from the nucleus in the mutant cells.
The involvement of Mlp1p and Mlp2p in active nuclear export was investigated using two steady state assays (data not shown). In the first assay, the subcellular distribution of poly(A)+ RNA was analyzed by in situ hybridization using digoxigenin-labeled oligo(dT)30 as a probe (Wente and Blobel, 1993). In the second assay, the steady state distribution of a nuclear export sequence (NES)-GFP was studied by direct fluorescent microscopy (Stade et al., 1997). In both cases no effect on export was detected at steady state and the double mutant cells appeared indistinguishable from wild-type.
Overexpression of Mlp1p
When Mlp1p was expressed from a high copy number 2 μm plasmid, the anti-Mlp1p antibody used by Botstein and coworkers recognized intensely staining dots and sometimes rings localized adjacent to the nucleus (Kolling et al., 1993). This localization does not correspond to the native localization of Mlp1p discussed in this manuscript (see above). Mlp1p was overexpressed in W303 cells in order to investigate whether this discrepancy could be accounted for by different levels of expression of this protein. The entire coding region of MLP1 was subcloned in a 2 μm–based yeast expression vector under the control of the GAL inducible promoter (see Materials and Methods). Using this plasmid (pGALMLP1), it was possible to overexpress Mlp1p at least ∼100-fold over wild-type levels as demonstrated by semi-quantitative immunoblotting performed with mAb148G11 (Fig. 9 A). Strikingly, overexpression of Mlp1p at these levels was not lethal as demonstrated by the ability of cells carrying pGALMLP1 to grow for days on galactose (data not shown). Cells containing this construct were either induced with galactose for various periods of time or repressed with dextrose for 4 h and analyzed by indirect IF microscopy using mAb148G11 to reveal the localization of Mlp1p (Fig. 9 B). As expected, cells in which the expression of the exogenous copy of MLP1 was repressed with dextrose (Fig. 9 B, 4 h [Glu]) showed a staining pattern very similar to the one observed in wild-type cells. In contrast, induced cells showed an increase of the Mlp1p-specific signal as a function of the induction time. Initially, small dots (1–4 per cell) could be seen at the nuclear periphery and a clear punctate rim pattern could still be distinguished in most cells (Fig. 9 B, 0 h [Gal]). Subsequently, these dots appeared to coalesce and generally gave rise to one prominent circular patch per cell and the nuclear rim pattern became increasingly more diffuse (Fig. 9 B, 0.5–2 h [Gal]). Finally, the large patch grew to occupy most of the nuclear interior (Fig. 9 B, 4 h [Gal]). This staining pattern closely resembled the one described by Kolling et al. (1993), thus reconciling the apparent difference. Careful examination of the overexpressing cells showed that the intense nuclear DAPI signal was either much reduced of excluded from the overexpression spots. It often appeared that the growing Mlp1p spheroids had pushed the chromatin aside, leaving clear indentations in the chromatin or in some cases even compressing it between two spheroids (Fig. 9 C). Cells overexpressing Mlp1p were observed by thin-section transmission EM to reveal whether any novel structure could be detected (data not shown). As a comparison, cells that had been grown in dextrose to repress the expression of the non-chromosomal copy Mlp1p were also analyzed using the same technique. Extensive electron-dense fibrillogranular networks that extended from different areas of the NE towards the nuclear interior were observed in cells grown in galactose but were absent in cells grown in dextrose. To determine if the electron-dense networks present in induced cells were indeed formed of large accumulations of Mlp1p, isolated nuclei from cells grown in both galactose (Fig. 10) and dextrose (data not shown) were immunostained with mAb148G11 and prepared for IEM using the same pre-embedding labeling technique used above. As expected, the dense fibrillogranular networks present in induced cells specifically stained with mAb148G11 demonstrating that they contain large quantities of Mlp1p and that the Mlp1p spheroids are highly permeable to large macromolecules such as the antibody-gold conjugate. In cells grown in dextrose, Mlp1p appeared to have a localization that was indistinguishable from wild-type.
Figure 9.

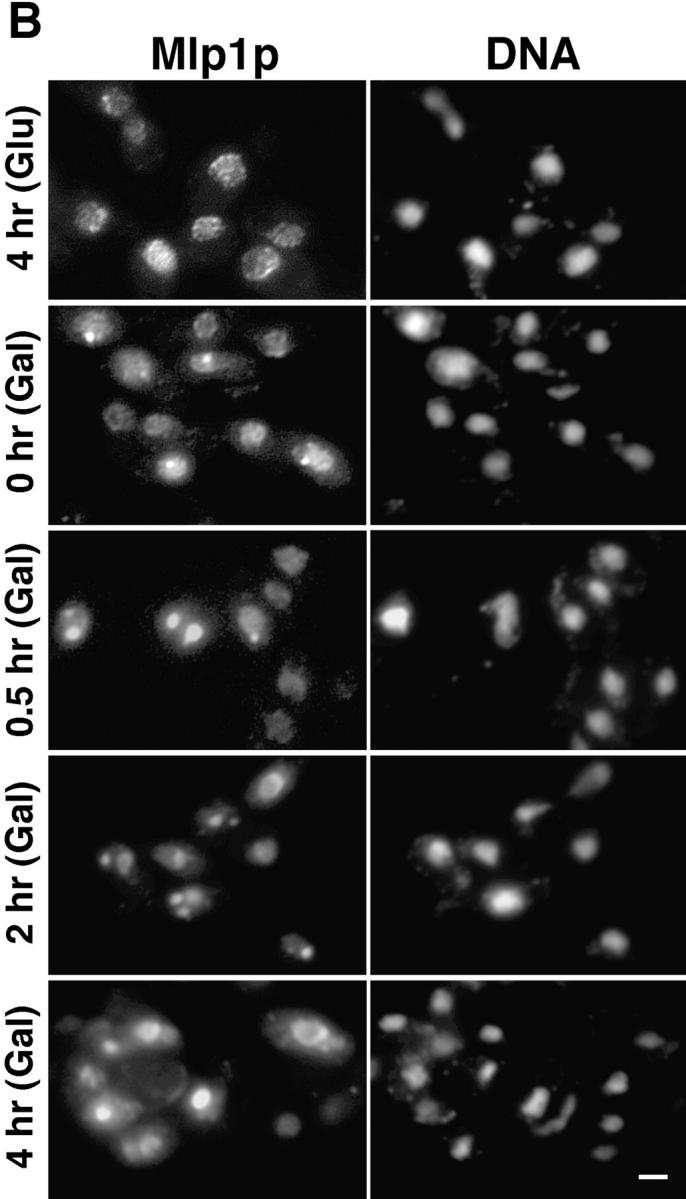

Upon overexpression, Mlp1p forms multiple peripheral nuclear dots that subsequently coalesce and take over the majority of the nuclear volume while pushing the chromatin to one side. (A) Wild-type W303 cells were transformed with pGALMLP1, expressing the entire MLP1 gene under the control of the inducible GAL1-10 promoter (see Materials and Methods for details of the cloning procedure). Transformants were exponentially grown in selective medium containing raffinose before induction with galactose. Samples of the culture were taken at the indicated time points (0, 0.5, 1, 2, and 4 h) and proteins form total cell lysates were separated by SDS-PAGE and immunoblotted with mAb148G11. Numbers below the gel represent an estimate of the relative expression level of Mlp1p at each time point. (B) W303 cells containing pGALMLP1 were grown to mid-logarithmic phase in selective medium containing raffinose before induction with galactose or repression with dextrose. Cells were sampled at the indicated time points either after repression (4 h [Glu]) or induction (0, 0.5, 2, and 4 h [Gal]), fixed, and immunostained with mAb148G11 to reveal the position of Mlp1p. As a comparison, the position of the DNA was revealed using DAPI. (C) W303 cells expressing pGALMLP1 were induced with galactose for 1 h before immunostaining with mAb148G11. Shown here are clear examples of Mlp1p spheroids creating indentations in the chromatin as revealed by DAPI staining. Bar, 2 μm.
Figure 10.
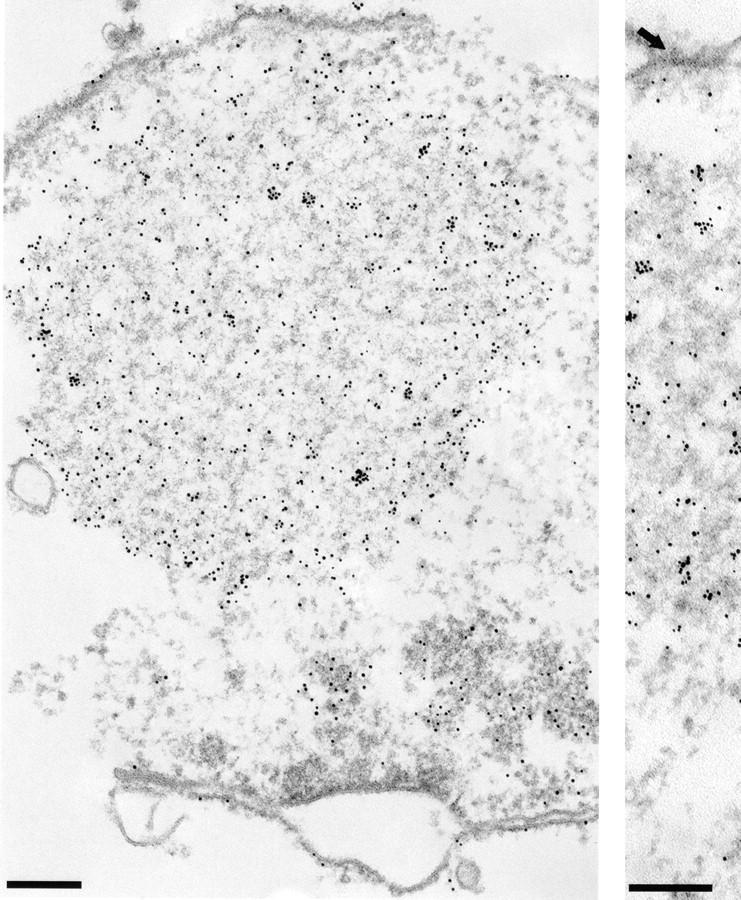
When overexpressed in yeast, Mlp1p forms extensive fibrillogranular networks that project form the NE towards the nucleoplasm. Exponentially growing cells containing pGALMLP1 were transferred to selective medium containing galactose and the incubation was continued for 4 h. Nuclei were isolated from cells grown in galactose and subjected to IEM analysis using mAb148G11. An arrow in the right panel points to a clearly distinguishable NPC. Bar, 200 nm.
Discussion
The clustering of yeast NPCs within characteristic patches in the NE induced by the deletion of certain nucleoporins has previously been used to confirm the association of various proteins with NPCs (Grandi et al., 1995; Aitchison et al., 1996; Simos et al., 1996; Siniossoglou et al., 1996; Rout et al., 1997). Here we used this clustering assay to screen a bank of monoclonal antibodies raised against NE proteins, and isolate those which recognize NE components not associated with the NPC from those recognizing NPC components and nuclear transport factors. We expected to find mainly integral nuclear membrane proteins, analogous to the LAPs or LBR proteins in vertebrate NEs (Senior and Gerace, 1988; Worman et al., 1988, 1990; Foisner and Gerace, 1993). Instead, a monoclonal antibody was found which identified a protein, Mlp1p, associated with the nuclear periphery of the NPC and NE and extending into the nuclear interior. Sequence comparisons of the predicted amino acid sequence of Mlp1p with the entire GenBank database revealed numerous proteins with strong similarities. The most significant was a second yeast ORF predicted to have arisen as a chromosomal duplication event (Wolfe and Shields, 1997). We showed this to encode a redundant homologue of Mlp1p which we therefore named Mlp2p. Next, the vertebrate protein Tpr, its probable Drosophila homologue, and an uncharacterized S. pombe ORF showed strong similarity to Mlp1p and Mlp2p along their entire lengths. Though sequence similarity alone is not necessarily definitive, given the similarities in their localization and possible roles in nuclear transport, we conclude that Mlp1p and Mlp2p are the yeast homologues of Tpr (see below; Cordes et al., 1997; Zimowska et al., 1997; Bangs et al., 1998; Cordes et al., 1998; Shah et al., 1998). This represents the first identification and characterization of Tpr homologues in yeast, setting the stage for studies that will hopefully elucidate the functions of this protein in a genetically and molecularly tractable system.
The Tpr homologues are predicted to consist of a coiled-coil NH2 terminus occupying most of the primary sequence. It seems likely that the putative coiled-coil domain forms an extended structure that may in turn be involved in organizing higher order homopolymers (for example, filaments). In contrast, the COOH terminus may interact with heterologous factors and anchor these polymeric structures to the NPCs or to the nuclear interior (see also below). Despite its name, based on its cross reactivity with a mAb against myosin (most likely accounted for by the large coiled-coil NH2-terminal domain; Kolling et al., 1993), Mlp1p does not belong to the myosin family due to the different organization of the structural domains along the primary sequence of the protein.
IF microscopy of the NPC clustering strains shows a partial coclustering of Mlp1p with nucleoporin markers, indicating a significant fraction of Mlp1p is associated with the NPCs. Our IEM experiments localizing Mlp1p and Mlp2p to the nuclear face of the nuclear envelope in yeast agree with the recent results obtained with vertebrate Tpr (Cordes et al., 1997). Our IEM localization also suggests that, like Tpr, Mlp1p and Mlp2p form extensive filamentous structures radiating into the interior of the nucleus from foci at the NPC periphery, perhaps attached at the distal ends of the NPC baskets or fishtraps (Ris and Malecki, 1993; Cordes et al., 1997; Zimowska et al., 1997). Also like Tpr, Mlp1p penetrates deep into the yeast nucleus, as far as half of the nuclear radius, and is thus potentially in contact with at least 80% of the chromatin. Interestingly, a significant fraction of Mlp1p failed to cocluster with NPCs, remaining instead as numerous punctate foci distributed around the nuclear periphery. However, virtually all of Mlp1p cofractionated with the NE fraction, and the majority of Mlp1p signal was in close proximity to the NE inner membrane by IEM. Taken together, this suggests that there is also a portion of Mlp1p not associated with NPCs but distributed around the interporous regions of the NE inner face. This may differ from Tpr, which was reported to be absent from these interporous regions, although other studies have found structures believed to be composed of Tpr interconnecting between NPCs over the inner NE surface (Ris and Malecki, 1993; Cordes et al., 1997). The punctate IF staining pattern of Mlp1p in regions of NE devoid of nucleoporin signal (and hence NPCs) may indicate that it can also organize from non- NPC-associated foci distributed around the nuclear face of the inner nuclear membrane. In an alternative scenario, structures formed of Mlp1p would absolutely depend on NPCs for their nucleation at the NE but would be subsequently able to spread all around the nuclear rim in a non-NPC-dependent manner.
It has long been observed that the NPCs are structurally continuous with the nuclear interior (Monneron and Bernhard, 1969; Franke and Falk, 1971). Interconnecting open channels have been observed radiating from the nuclear interior towards NPCs (Fawcett, 1981; Berezney et al., 1995), and the movement of proteins and RNAs along distinct intranuclear pathways can be studied both in vivo and in vitro (Lawrence et al., 1989; Xing et al., 1993; Zachar et al., 1993; Misteli et al., 1997). These and other similar observations have led to speculations that efficient exchange of material between the nuclear periphery and the nuclear interior could occur along a filamentous network of tracks. (Blobel, 1985; Lawrence et al., 1989, 1993; Meier and Blobel, 1992, 1994; Xing et al., 1993; Berezney et al., 1995; Ris, 1997; Pederson, 1998). Indeed, one model proposes the existence of a nucleoskeleton (defined as a nonchromatin intranuclear structural framework) composed of a filamentous network organized from the nuclear periphery and playing a major role in the direction of nuclear trafficking to and from the NPCs (Berezney et al., 1995). Until recently, the existence of tracks had remained unproven, as no clear candidates for track components had emerged.
A case can be made for the Mlp1p/Tpr family of proteins meeting some of the characteristics expected of nucleoskeletal track components. As one would expect for track proteins they are localized further away from the NPCs than the most peripherally described structures of the NPCs (the nuclear baskets and cytoplasmic filaments) and penetrate most of the volume occupied by chromatin (as discussed above; Ris and Malecki, 1993; Cordes et al., 1997; Ris, 1997). Their peripheral association with the NPC is emphasized by the partial fractionation of Mlp1p with isolated NPCs, though an independent tight association with the nuclear periphery is suggested by its cofractionation with enriched NEs and continuous distribution around the NE even in strains where the NPCs have clustered. Furthermore, such components would not necessarily be stoichiometric with respect to NPC proteins, and not coassemble with them during NPC assembly. Instead, they could carry an NLS to be imported separately into the nucleus. Thus, the domain necessary for NPC association could be separated from that for nuclear localization, as indeed we showed for Mlp1p and was also recently shown for vertebrate Tpr (Bangs et al., 1998; Cordes et al., 1998). The Tpr family seems to form filamentous structures of considerable length (unsurprising considering their predicted coiled-coil structure) that are likely polymeric (Cordes et al., 1997). Interestingly, the most distal of the intranuclear signal appeared to collapse towards the inner nuclear membrane during the preparation of isolated NEs. Preliminary data indicate an extreme version of this collapse occurs when NEs are isolated from cells overexpressing Mlp1p (data not shown; see below). Thus it would seem, as expected of a track component, that interaction with intact chromatin is required to maintain the normal distribution of Mlp1p within the nucleus and even opens the possibility of a role for this protein in the maintenance of the nuclear architecture. As lamins are absent from yeast, it may even substitute in part for their function. The ability of Mlp1p to self-assemble and form regular polymers that could account for such structural functions remains to be demonstrated and will be the subject of future studies.
As with Tpr, the copious labeling of the COOH-terminal epitope recognized by our antibody indicates that this part of the protein is free, perhaps to interact with chromatin or transport factors (Cordes et al., 1997). A possible interaction has been found between Tpr and the vertebrate karyopherin β1 nuclear import factor (Shah et al., 1998). Here we show that Mlp1p and Mlp2p are required for the efficient nuclear import of a substrate of the homologous yeast karyopherin β1 plus karyopherin α. Although this represents the first in vivo evidence for a role of this protein family in nuclear transport, it must still be regarded with caution given the potential for pleiotropic effects in this experiment (see Results). Nonetheless, together such data support the idea of a direct role for the Tpr family in nuclear transport by active transport of substrates between binding sites positioned opportunely along the filaments. However, Tpr also coincides with clear channels extending from the NPC into the nuclear interior (Zimowska et al., 1997). Overexpression of Mlp1p forms spheroidal fibrillogranular structures which can occupy large portions of the nucleus. Overexpression aggregates such as these usually only display some of the characteristics of the protein under normal conditions, and other characteristics may be anomalous. However, contrary to many other proteinaceous aggregates (e.g., Capsey et al., 1990) the overexpression spheroids of Mlp1p are surprisingly open to large molecules, as indicated by the apparently unhindered access and accumulation of antibody conjugated ∼10 nm gold (added before fixation, embedding and sectioning for IEM) throughout them. In addition, it is interesting that the Mlp1p spheroids displaced chromatin as they grew to form large chromatin free regions within the nucleus. One could imagine a polymeric cylinder of Mlp1p with the properties of these spheroids projecting from the NPC into the nucleus. Such a structure would maintain a chromatin free channel that remains highly permeable to even large proteinaceous particles. These data are consistent with binding tracks for transport factors, but also raise another possible and less direct role for this protein family; they may maintain efficient nucleocytoplasmic transport by holding open diffusion channels, for the unhindered intranuclear movement of transport substrates.
It came as a considerable surprise that the deletion of both yeast Tpr homologues was not lethal. The function of both proteins must therefore not be essential in yeast, or other functionally redundant proteins must exist. Either way, this result holds important consequences for the increasing number of studies underway on the vertebrate Tpr proteins. Thus despite a reported association with transport factors (Shah et al., 1998) and a deleterious effect of vertebrate Tpr overexpression on mRNA export (Bangs et al., 1998), Tpr homologues are not necessary to support basic nuclear transport. In vivo experiments should be conducted to determine if vertebrate cells have a greater requirement than yeast for Tpr, and what factors (such as cell size) might contribute to any differences. Further work is also required on the Mlp1p/Mlp2p deficient strain to find conditions where at least one of these proteins is required for viability. Such experiments may lead to interacting proteins and processes; yeast studies have been key in showing that many nucleocytoplasmic transport operatives are redundant with numerous subtly overlapping functions, and the Tpr family are proving no exception (Rout et al., 1997; Wozniak et al., 1998). The behavior of these proteins should also be studied in living yeast cells (such as by using GFP-tagged Mlp1p), taking advantage of available conditional mutant strains, to avoid some of the problems previously associated with studying potential nucleoskeletal proteins biochemically (Singer and Green, 1997; Pederson, 1998).
In conclusion, we believe that Mlp1p and Mlp2p, together with their multicellular eukaryotic counterparts, satisfy all the criteria expected of non-nucleoporin nucleoskeletal components. We further suggest they represent the strongest candidates to date for the molecular components of the long hypothesized nuclear tracks connecting the NPCs with the nuclear interior. The characterization of the homologues of Tpr in a genetically tractable organism complements the studies of these proteins in vertebrate systems and opens new avenues in the study of nuclear cell biology.
Abbreviations used in this paper
- GFP
green fluorescent protein
- IEM
immunoelectron microscopy
- IF
immunofluorescence
- MLP
myosin-like protein
- NE
nuclear envelope
- NES
nuclear export sequence
- NPC
nuclear pore complex
- NUP
nucleoporin
- PVP
polyvinylpyrrolidone
Footnotes
Address correspondence to Dr. G. Blobel, Laboratory of Cell Biology, Howard Hughes Medical Institute, The Rockefeller University, New York, New York 10021. Tel.: (212) 327-8096. Fax: (212) 327-7880. E-mail: blobel@rockvax.rockefeller.edu
We are very grateful to J. Aitchison, C. Akey, R. Beckmann, N. Bonifaci, Y. Chook, E. Coutavas, U. O'Doherty, R. Erdmann, B. Fontoura, J. Helmers, M. Hurwitz, E. Johnson, J. Kilmartin, M. Matunis, L. Pemberton, and S. Smith for many helpful suggestions and discussions throughout the course of this study. We are deeply indebted to J. Aris, C. Cole, D. Goldfarb, E. Johnson, J. Kilmartin, M. Lewis, L. Pemberton, M. Sogaard, K. Weis, and R. Wozniak, for providing us with antibodies and other reagents without which this work would not have been possible and to T. De Lange and J. Karlsreder for assistance in collecting the immunofluorescence data presented here. Many thanks also to R. Beckmann, Y. Chook, and J. Luban for critical reading of the manuscript in whole or in part. Special thanks go to H. Shio and E. Sphicas for excellent technical assistance in the electron microscopic studies. Our sincerest gratitude goes to E. Ellison, H. Ijikata, and Y. Oh for invaluable and skillful technical support that was essential for the completion of various parts of the work presented here.
References
- Aitchison JD, Blobel G, Rout MP. Nup120p: a yeast nucleoporin required for NPC distribution and mRNA transport. J Cell Biol. 1995a;131:1659–1675. doi: 10.1083/jcb.131.6.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison JD, Rout MP, Marelli M, Blobel G, Wozniak RW. Two novel related yeast nucleoporins Nup170p and Nup157p: complementation with the vertebrate homologue Nup155p and functional interactions with the yeast nuclear pore-membrane protein Pom152p. J Cell Biol. 1995b;131:1133–1148. doi: 10.1083/jcb.131.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison JD, Blobel G, Rout MP. Kap104p: a karyopherin involved in the nuclear transport of messenger RNA binding proteins. Science. 1996;274:624–627. doi: 10.1126/science.274.5287.624. [DOI] [PubMed] [Google Scholar]
- Akey CW. Visualization of transport-related configurations of the nuclear pore transporter. Biophys J. 1990;58:341–355. doi: 10.1016/S0006-3495(90)82381-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey CW, Goldfarb DS. Protein import through the nuclear pore complex is a multistep process. J Cell Biol. 1989;109:971–982. doi: 10.1083/jcb.109.3.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey CW, Radermacher M. Architecture of the Xenopusnuclear pore complex revealed by three-dimensional cryo-electron microscopy. J Cell Biol. 1993;122:1–19. doi: 10.1083/jcb.122.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Baba M, Osumi M. Transmission and scanning electron microscope examination of intracellular organelles in freeze-substituted Kloeckera and Saccharomyces cerevisiaeyeast cells. J Elec Microsc Tech. 1987;5:249–261. [Google Scholar]
- Bangs PL, Sparks CA, Odgren PR, Fey EG. Product of the oncogene-activating gene Tpr is a phosphorylated protein of the nuclear pore complex. J Cell Biochem. 1996;61:48–60. doi: 10.1002/(sici)1097-4644(19960401)61:1<48::aid-jcb7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Bangs P, Burke B, Powers C, Craig R, Purohit A, Doxsey S. Functional analysis of Tpr: identification of nuclear pore complex association and nuclear localization domains and a role in mRNA export. J Cell Biol. 1998;143:1801–1812. doi: 10.1083/jcb.143.7.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos R, Pante N, Burke B. Nuclear pore complex proteins. Int Rev Cytol. 1995;162:B257–B302. doi: 10.1016/s0074-7696(08)62619-4. [DOI] [PubMed] [Google Scholar]
- Berezney R, Mortillaro MJ, Ma H, Wei X, Samarabandu J. The nuclear matrix: a structural milieu for genomic function. Int Rev Cytol. 1995;162:A1–A65. doi: 10.1016/s0074-7696(08)61228-0. [DOI] [PubMed] [Google Scholar]
- Blobel G. Gene gating: a hypothesis. Proc Natl Acad Sci USA. 1985;82:8527–8529. doi: 10.1073/pnas.82.24.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd DA, Sweet DJ, Pante N, Konstantinov KN, Guan T, Saphire AC, Mitchell PJ, Cooper CS, Aebi U, Gerace L. Tpr, a large coiled coil protein whose amino terminus is involved in activation of oncogenic kinases, is localized to the cytoplasmic surface of the nuclear pore complex. J Cell Biol. 1994;127:1515–1526. doi: 10.1083/jcb.127.6.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capsey LJ, Williamson DH, Banks GR. Ty virus-like particles in the Saccharomyces cerevisiaestrain NCYC74. Curr Genet. 1990;18:485–491. doi: 10.1007/BF00327018. [DOI] [PubMed] [Google Scholar]
- Clayton RA, White O, Ketchum KA, Venter JC. The first genome from the third domain of life. Nature. 1997;387:459–462. doi: 10.1038/387459a0. [DOI] [PubMed] [Google Scholar]
- Cody CW, Prasher DC, Wester WM, Prendergast FG, Ward WW. Chemical structure of the hexapeptide chromophore of the Aequoreagreen fluorescent protein. Biochemistry. 1993;32:1212–1218. doi: 10.1021/bi00056a003. [DOI] [PubMed] [Google Scholar]
- Cordes VC, Reidenbach S, Rackwitz HR, Franke WW. Identification of protein p270/Tpr as a constitutive component of the nuclear pore complex-attached intranuclear filaments. J Cell Biol. 1997;136:515–529. doi: 10.1083/jcb.136.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes VC, Hase ME, Müller L. Molecular segments of protein Tpr that confer nuclear targeting and association with the nuclear pore complex. Exp Cell Res. 1998;245:43–56. doi: 10.1006/excr.1998.4246. [DOI] [PubMed] [Google Scholar]
- Davis LI, Blobel G. Identification and characterization of a nuclear pore complex protein. Cell. 1986;45:699–709. doi: 10.1016/0092-8674(86)90784-1. [DOI] [PubMed] [Google Scholar]
- Del Priore V, Heath C, Snay C, MacMillan A, Gorsch L, Dagher S, Cole C. A structure/function analysis of Rat7p/Nup159p, an essential nucleoporin of Saccharomyces cerevisiae. . J Cell Sci. 1997;110:2987–2999. doi: 10.1242/jcs.110.23.2987. [DOI] [PubMed] [Google Scholar]
- Doye V, Hurt E. From nucleoporins to nuclear pore complexes. Curr Opin Cell Biol. 1997;9:401–411. doi: 10.1016/s0955-0674(97)80014-2. [DOI] [PubMed] [Google Scholar]
- Doye V, Wepf R, Hurt EC. A novel nuclear pore protein Nup133p with distinct roles in poly(A)+ RNA transport and nuclear pore distribution. EMBO (Eur Mol Biol Organ) J. 1994;13:6062–6075. doi: 10.1002/j.1460-2075.1994.tb06953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworetzky SI, Feldherr CM. Translocation of RNA-coated gold particles through the nuclear pores of oocytes. J Cell Biol. 1988;106:575–584. doi: 10.1083/jcb.106.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre E, Hurt E. Yeast genetics to dissect the nuclear pore complex and nucleocytoplasmic trafficking. Annu Rev Genet. 1997;31:277–313. doi: 10.1146/annurev.genet.31.1.277. [DOI] [PubMed] [Google Scholar]
- Fawcett, D.W. 1981. The Cell. W.B. Saunders Company, Philadelphia, PA. 862 pp.
- Feldherr CM, Kallenbach E, Schultz N. Movement of a karyophilic protein through the nuclear pores of oocytes. J Cell Biol. 1984;99:2216–2222. doi: 10.1083/jcb.99.6.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisner R, Gerace L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell. 1993;73:1267–1279. doi: 10.1016/0092-8674(93)90355-t. [DOI] [PubMed] [Google Scholar]
- Franke WW. On the universality of nuclear pore complex structure. Z Zellforsch Mikrosk Anat. 1970;105:405–429. doi: 10.1007/BF00335464. [DOI] [PubMed] [Google Scholar]
- Franke WW, Scheer U. The ultrastructure of the nuclear envelope of amphibian oocytes: a reinvestigation. I. The mature oocyte. J Ultrastruct Res. 1970a;30:288–316. doi: 10.1016/s0022-5320(70)80064-8. [DOI] [PubMed] [Google Scholar]
- Franke WW, Scheer U. The ultrastructure of the nuclear envelope of amphibian oocytes: a reinvestigation. II. The immature oocyte and dynamic aspects. J Ultrastruct Res. 1970b;30:317–327. doi: 10.1016/s0022-5320(70)80065-x. [DOI] [PubMed] [Google Scholar]
- Franke WW, Falk H. Appearance of nuclear pore complexes after Bernhard's staining procedure. Histochemie. 1971;24:266–278. doi: 10.1007/BF00304196. [DOI] [PubMed] [Google Scholar]
- Frasch M, Paddy MR, Saumweber H. Developmental and mitotic behavior of two novel groups of nuclear envelope antigens. J Cell Sci. 1988;90:247–264. doi: 10.1242/jcs.90.2.247. [DOI] [PubMed] [Google Scholar]
- Galfre G, Milstein C. Preparation of monoclonal antibodies: strategies and procedures. Methods Enzymol. 1981;73:3–46. doi: 10.1016/0076-6879(81)73054-4. [DOI] [PubMed] [Google Scholar]
- Gerace L, Blum A, Blobel G. Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction. Interphase and mitotic distribution. J Cell Biol. 1978;79:546–566. doi: 10.1083/jcb.79.2.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MW, Allen TD. High resolution scanning electron microscopy of the nuclear envelope: demonstration of a new, regular, fibrous lattice attached to the baskets of the nucleoplasmic face of the nuclear pores. J Cell Biol. 1992;119:1429–1440. doi: 10.1083/jcb.119.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MW, Allen TD. The nuclear pore complex and lamina: three-dimensional structures and interactions determined by field emission in-lens scanning electron microscopy. J Mol Biol. 1996;257:848–865. doi: 10.1006/jmbi.1996.0206. [DOI] [PubMed] [Google Scholar]
- Grandi P, Emig S, Weise C, Hucho F, Pohl T, Hurt EC. A novel nuclear pore protein Nup82p which specifically binds to a fraction of Nsp1p. J Cell Biol. 1995;130:1263–1273. doi: 10.1083/jcb.130.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco A, Pierotti MA, Bongarzone I, Pagliardini S, Lanzi C, Della G, Porta TRK-T1 is a novel oncogene formed by the fusion of TPR and TRK genes in human papillary thyroid carcinomas. Oncogene. 1992;7:237–242. [PubMed] [Google Scholar]
- Heath CV, Copeland CS, Amberg DC, Del Priore V, Snyder M, Cole CN. Nuclear pore complex clustering and nuclear accumulation of poly(A)+ RNA associated with mutation of the Saccharomyces cerevisiaeRAT2/NUP120 gene. J Cell Biol. 1995;131:1677–1697. doi: 10.1083/jcb.131.6.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins D, Thompson J, Gibson T. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw JE, Carragher BO, Milligan RA. Architecture and design of the nuclear pore complex. Cell. 1992;69:1133–1141. doi: 10.1016/0092-8674(92)90635-p. [DOI] [PubMed] [Google Scholar]
- Kartenbeck J, Zentgraf H, Scheer U, Franke WW. The nuclear envelope in freeze-etching. Ergeb Anat Entwicklungsgesch. 1971;45:3–55. doi: 10.1007/978-3-662-10390-6. [DOI] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AE. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. . J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV, Dyos SL, Kershaw D, Finch JT. A spacer protein in the Saccharomyces cerevisiaespindle poly body whose transcript is cell cycle-regulated. J Cell Biol. 1993;123:1175–1184. doi: 10.1083/jcb.123.5.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolling R, Nguyen T, Chen EY, Botstein D. A new yeast gene with a myosin-like heptad repeat structure. Mol Gen Genet. 1993;237:359–369. doi: 10.1007/BF00279439. [DOI] [PubMed] [Google Scholar]
- Kraemer DM, Strambio-de-Castillia C, Blobel G, Rout MP. The essential yeast nucleoporin NUP159 is located on the cytoplasmic side of the nuclear pore complex and serves in karyopherin-mediated binding of transport substrate. J Biol Chem. 1995;270:19017–19021. doi: 10.1074/jbc.270.32.19017. [DOI] [PubMed] [Google Scholar]
- Lawrence JB, Singer RH, Marselle LM. Highly localized tracks of specific transcripts within interphase nuclei visualized by in situ hybridization. Cell. 1989;57:493–502. doi: 10.1016/0092-8674(89)90924-0. [DOI] [PubMed] [Google Scholar]
- Lawrence JB, Carter KC, Xing X. Probing functional organization within the nucleus: is genome structure integrated with RNA metabolism? . Cold Spring Harb Symp Quant Biol. 1993;58:807–818. doi: 10.1101/sqb.1993.058.01.088. [DOI] [PubMed] [Google Scholar]
- Li O, Heath CV, Amberg DC, Dockendorff TC, Copeland CS, Snyder M, Cole CN. Mutation or deletion of the Saccharomyces cerevisiaeRAT3/NUP133 gene causes temperature-dependent nuclear accumulation of poly(A)+ RNA and constitutive clustering of nuclear pore complexes. Mol Biol Cell. 1995;6:401–417. doi: 10.1091/mbc.6.4.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas AD, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Meier UT, Blobel G. Nopp140 shuttles on tracks between nucleolus and cytoplasm. Cell. 1992;70:127–138. doi: 10.1016/0092-8674(92)90539-o. [DOI] [PubMed] [Google Scholar]
- Meier UT, Blobel G. NAP57, a mammalian nucleolar protein with a putative homolog in yeast and bacteria. J Cell Biol. 1994;127:1505–1514. doi: 10.1083/jcb.127.6.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Caceres JF, Spector DL. The dynamics of a pre-mRNA splicing factor in living cells. Nature. 1997;387:523–527. doi: 10.1038/387523a0. [DOI] [PubMed] [Google Scholar]
- Mitchell PJ, Cooper CS. The human tprgene encodes a protein of 2094 amino acids that has extensive coiled-coil regions and an acidic C-terminal domain. Oncogene. 1992;7:1319–1332. [PubMed] [Google Scholar]
- Moir RD, Spann TP, Goldman RD. The dynamic properties and possible functions of nuclear lamins. Int Rev Cytol. 1995;162:B141–B182. doi: 10.1016/s0074-7696(08)62616-9. [DOI] [PubMed] [Google Scholar]
- Monneron A, Bernhard W. Fine structural organization of the interphase nucleus in some mammalian cells. J Ultrastruct Res. 1969;27:266–288. doi: 10.1016/s0022-5320(69)80017-1. [DOI] [PubMed] [Google Scholar]
- Mortimer RK, Johnson JR. Genealogy of principal strains of the yeast genetic stock center. Genetics. 1986;113:35–43. doi: 10.1093/genetics/113.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedenthal RK, Riles L, Johnston M, Hegemann JH. Green fluorescent protein as a marker for gene expression and subcellular localization in budding yeast. Yeast. 1996;12:773–786. doi: 10.1002/(SICI)1097-0061(19960630)12:8%3C773::AID-YEA972%3E3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Olson MV, Dutchik JE, Graham MY, Brodeur GM, Helms C, Frank M, MacCollin M, Scheinman R, Frank T. Random-clone strategy for genomic restriction mapping in yeast. Proc Natl Acad Sci USA. 1986;83:7826–7830. doi: 10.1073/pnas.83.20.7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Dean M, Cooper CS, Schmidt M, O'Brien SJ, Blair DG, Vande GF, Woude Mechanism of metoncogene activation. Cell. 1986;45:895–904. doi: 10.1016/0092-8674(86)90564-7. [DOI] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson T. Thinking about a nuclear matrix. J Mol Biol. 1998;277:147–159. doi: 10.1006/jmbi.1997.1618. [DOI] [PubMed] [Google Scholar]
- Pemberton LF, Rout MP, Blobel G. Disruption of the nucleoporin gene NUP133 results in clustering of nuclear pore complexes. Proc Natl Acad Sci USA. 1995;92:1187–1191. doi: 10.1073/pnas.92.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss D, Mulholland J, Kaiser CA, Orlean P, Albright C, Rose MD, Robbins PW, Botstein D. Structure of the yeast endoplasmic reticulum: localization of ER proteins using immunofluorescence and immunoelectron microscopy. Yeast. 1991;7:891–911. doi: 10.1002/yea.320070902. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Mills AD, Dilworth SM, Laskey RA, Dingwall C. Nuclear protein migration involves two steps: rapid binding at the nuclear envelope followed by slower translocation through nuclear pores. Cell. 1988;52:655–664. doi: 10.1016/0092-8674(88)90403-5. [DOI] [PubMed] [Google Scholar]
- Ris H. The three dimensional structure of the nuclear pore complexes as seen by high voltage electron microscopy and high resolution low voltage scanning electron microscopy. EMSA Bull. 1991;21:54–56. [Google Scholar]
- Ris H. High-resolution field-emission scanning electron microscopy of nuclear pore complex. Scanning. 1997;19:368–375. doi: 10.1002/sca.4950190504. [DOI] [PubMed] [Google Scholar]
- Ris H, Malecki M. High-resolution field emission scanning electron microscope imaging of internal cell structures after epon extraction from sections: a new approach to correlative ultrastructural and immunocytochemical studies. J Struct Biol. 1993;111:148–157. doi: 10.1006/jsbi.1993.1045. [DOI] [PubMed] [Google Scholar]
- Rothstein R. Targeting, disruption, replacement and allele rescue: integrative transformation in yeast. Methods Enzymol. 1990;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Rout MP, Kilmartin JV. Components of the yeast spindle and spindle pole body. J Cell Biol. 1990;111:1913–1927. doi: 10.1083/jcb.111.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Blobel G. Isolation of the nuclear pore complex. J Cell Biol. 1993;123:771–783. doi: 10.1083/jcb.123.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout, M.P., and J.V. Kilmartin. 1994. Preparation of yeast spindle pole bodies. In Cell Biology: A Laboratory Handbook. Vol. 1. J.E. Celis, editor. Academic Press, London. 605–612.
- Rout MP, Wente SR. Pores for thought: nuclear pore complex proteins. Trends Cell Biol. 1994;4:357–365. doi: 10.1016/0962-8924(94)90085-x. [DOI] [PubMed] [Google Scholar]
- Rout, M.P., and C. Strambio-de-Castillia. 1998. Isolation of yeast nuclear pore complexes and nuclear envelopes. In Cell Biology: A Laboratory Handbook. Vol. 2. J.E. Celis, editor. Academic Press, London. 143–151.
- Rout MP, Blobel G, Aitchison JD. A distinct nuclear import pathway used by ribosomal proteins. Cell. 1997;89:715–725. doi: 10.1016/s0092-8674(00)80254-8. [DOI] [PubMed] [Google Scholar]
- Senior A, Gerace L. Integral membrane proteins specific to the inner nuclear membrane and associated with the nuclear lamina. J Cell Biol. 1988;107:2029–2036. doi: 10.1083/jcb.107.6.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Tugendreich S, Forbes D. Major binding sites for the nuclear import receptor are the internal nucleoporin nup153 and the adjacent nuclear filament protein Tpr. J Cell Biol. 1998;141:31–49. doi: 10.1083/jcb.141.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., G.R. Fink, and J.B. Hicks. 1986. Methods in Yeast Genetics. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- Shulga N, Roberts P, Gu Z, Spitz L, Tabb MM, Nomura M, Goldfarb DS. In vivo nuclear transport kinetics in Saccharomyces cerevisiae: a role for heat shock protein 70 during targeting and translocation. J Cell Biol. 1996;135:329–339. doi: 10.1083/jcb.135.2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simos G, Tekotte H, Grosjean H, Segref A, Sharma K, Tollervey D, Hurt EC. Nuclear pore proteins are involved in the biogenesis of functional tRNA. EMBO (Eur Mol Biol Organ) J. 1996;15:2270–2284. [PMC free article] [PubMed] [Google Scholar]
- Singer RH, Green MR. Compartmentalization of eukaryotic gene expression: causes and effects. Cell. 1997;91:291–294. doi: 10.1016/s0092-8674(00)80411-0. [DOI] [PubMed] [Google Scholar]
- Siniossoglou S, Wimmer C, Rieger M, Doye V, Tekotte H, Weise C, Emig S, Segref A, Hurt EC. A novel complex of nucleoporins, which includes Sec13p and a Sec13p homolog, is essential for normal nuclear pores. Cell. 1996;84:265–275. doi: 10.1016/s0092-8674(00)80981-2. [DOI] [PubMed] [Google Scholar]
- Smith V, Chou KN, Lashakari D, Botstein D, Brown PO. Functional analysis of the genes of yeast chromosome V by genetic footprinting. Science. 1996;274:2069–2074. doi: 10.1126/science.274.5295.2069. [DOI] [PubMed] [Google Scholar]
- Soman NR, Correa P, Ruiz BA, Wogan GN. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc Natl Acad Sci USA. 1991;88:4892–4896. doi: 10.1073/pnas.88.11.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell. 1997;90:1041–1050. doi: 10.1016/s0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- Strambio-de-Castillia C, Blobel G, Rout MP. Isolation and characterization of nuclear envelopes from the yeast Saccharomyces. . J Cell Biol. 1995;131:19–31. doi: 10.1083/jcb.131.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher JW, Shaw JM, Dickson WJ. Marginal fitness contributions of nonessential genes in yeast. Proc Natl Acad Sci USA. 1998;95:253–257. doi: 10.1073/pnas.95.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B, Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- Unwin PN, Milligan RA. A large particle associated with the perimeter of the nuclear pore complex. J Cell Biol. 1982;93:63–75. doi: 10.1083/jcb.93.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Blobel G. A temperature-sensitive NUP116 null mutant forms a nuclear envelope seal over the yeast nuclear pore complex thereby blocking nucleocytoplasmic traffic. J Cell Biol. 1993;123:275–284. doi: 10.1083/jcb.123.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Blobel G. NUP145 encodes a novel yeast glycine-leucine-phenylalanine-glycine (GLFG) nucleoporin required for nuclear envelope structure. J Cell Biol. 1994;125:955–969. doi: 10.1083/jcb.125.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Rout MP, Blobel G. A new family of yeast nuclear pore complex proteins. J Cell Biol. 1992;119:705–723. doi: 10.1083/jcb.119.4.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winey M, Yarar D, Giddings TH, Jr, Mastronarde DN. Nuclear pore complex number and distribution throughout the Saccharomyces cerevisiaecell cycle by three-dimensional reconstruction from electron micrographs of nuclear envelopes. Mol Biol Cell. 1997;8:2119–2132. doi: 10.1091/mbc.8.11.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KD, Shields DC. Molecular evidence of an ancient duplication event of the entire yeast genome. Nature. 1997;387:708–713. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- Worman HJ, Yuan J, Blobel G, Georgatos SD. A lamin B receptor in the nuclear envelope. Proc Natl Acad Sci USA. 1988;85:8531–8534. doi: 10.1073/pnas.85.22.8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman HJ, Evans CD, Blobel G. The lamin B receptor of the nuclear envelope inner membrane: a polytopic protein with eight potential transmembrane domains. J Cell Biol. 1990;111:1535–1542. doi: 10.1083/jcb.111.4.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak RW, Rout MP, Aitchison JD. Karyopherins and kissing cousins. Trends Cell Biol. 1998;8:184–188. doi: 10.1016/s0962-8924(98)01248-3. [DOI] [PubMed] [Google Scholar]
- Wray BE, Sealock R. Ultrastructural immunocytochemistry of particulate fractions using polyvinyl chloride microculture wells. J Histochem Cytochem. 1984;32:1117–1120. doi: 10.1177/32.10.6481151. [DOI] [PubMed] [Google Scholar]
- Xing Y, Johnson CV, Dobner PR, Lawrence JB. Higher level organization of individual gene transcription and RNA splicing. Science. 1993;259:1326–1330. doi: 10.1126/science.8446901. [DOI] [PubMed] [Google Scholar]
- Yang Q, Rout MP, Akey CW. Three-dimensional architecture of the isolated yeast nuclear pore complex: functional and evolutionary implications. Mol Cell. 1998;1:223–234. doi: 10.1016/s1097-2765(00)80023-4. [DOI] [PubMed] [Google Scholar]
- Zachar Z, Kramer J, Mims IP, Bingham PM. Evidence for channeled diffusion of pre-mRNAs during nuclear RNA transport in metazoans. J Cell Biol. 1993;121:729–742. doi: 10.1083/jcb.121.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimowska G, Aris JP, Paddy MR. A DrosophilaTpr protein homolog is localized both in the extrachromosomal channel network and to nuclear pore complexes. J Cell Sci. 1997;110:927–944. doi: 10.1242/jcs.110.8.927. [DOI] [PubMed] [Google Scholar]