

Abstract
Calreticulin is a ubiquitous Ca2+ binding protein, located in the endoplasmic reticulum lumen, which has been implicated in many diverse functions including: regulation of intracellular Ca2+ homeostasis, chaperone activity, steroid-mediated gene regulation, and cell adhesion. To understand the physiological function of calreticulin we used gene targeting to create a knockout mouse for calreticulin. Mice homozygous for the calreticulin gene disruption developed omphalocele (failure of absorption of the umbilical hernia) and showed a marked decrease in ventricular wall thickness and deep intertrabecular recesses in the ventricular walls. Transgenic mice expressing a green fluorescent protein reporter gene under the control of the calreticulin promoter were used to show that the calreticulin gene is highly activated in the cardiovascular system during the early stages of cardiac development. Calreticulin protein is also highly expressed in the developing heart, but it is only a minor component of the mature heart. Bradykinin-induced Ca2+ release by the InsP3-dependent pathway was inhibited in crt −/− cells, suggesting that calreticulin plays a role in Ca2+ homeostasis. Calreticulin-deficient cells also exhibited impaired nuclear import of nuclear factor of activated T cell (NF-AT3) transcription factor indicating that calreticulin plays a role in cardiac development as a component of the Ca2+/calcineurin/NF-AT/GATA-4 transcription pathway.
Keywords: cardiac development, NF-AT, calcineurin, endoplasmic reticulum, calcium binding protein, transcription
The ER performs an important role in controlling different intracellular processes including: protein synthesis, folding and modification, synthesis of membrane lipids, and regulation of Ca2+ storage and release. The ER lumen contains a characteristic set of resident proteins that are involved in many aspects of ER function, including Ca2+ binding and storage. One of the major Ca2+ binding chaperone proteins of the ER is calreticulin (Michalak et al., 1992). Calreticulin is an unusual lumenal ER protein. The protein contains both high affinity and high capacity Ca2+ binding sites (Ostwald and MacLennan, 1974; Baksh and Michalak, 1991) and has been implicated in the regulation of cytoplasmic Ca2+ homeostasis, even though it is located in the ER lumen (Liu et al., 1994; Bastianutto et al., 1995; Camacho and Lechleiter, 1995; Mery et al., 1996; Coppolino et al., 1997; Fasolato et al., 1998; John et al., 1998).
This regulatory function may be mediated by different Ca2+ binding sites in calreticulin (Baksh and Michalak, 1991; Krause and Michalak, 1997) and by the interaction of calreticulin with inositol 1,4,5-trisphosphate (InsP3)1 receptors and/or SERCA molecules (John et al., 1998). Other functions have been demonstrated for calreticulin including: modulation of gene expression (Burns et al., 1994; Dedhar et al., 1994; Michalak et al., 1996), chaperone activity (Nigam et al., 1994; Nauseef et al., 1995; Helenius et al., 1997), and regulation of cell adhesion (Coppolino et al., 1995, 1997; Opas et al., 1996). Calreticulin has also been reported to play a role in replication of Rubella virus RNA (Singh et al., 1994), in cytotoxic T-cell function/ activation (Burns et al., 1992; Dupuis et al., 1993; Andrin et al., 1998), in neutrophils (Stendahl et al., 1994), in sperm cell function (Nakamura et al., 1993), and in autoimmunity (Sontheimer et al., 1993).
The aim of this study was to investigate the physiological functions of calreticulin by generating a calreticulin-deficient mouse. The homozygous calreticulin knockout was an embryonic lethal because of defects in heart development and function. This was a surprising result because calreticulin is an ER lumenal protein and only a minor component of the mature heart (Fliegel et al., 1989a,b; Milner et al., 1991; Tharin et al., 1996). However, we generated transgenic mice expressing the green fluorescent protein (GFP) under the control of calreticulin promoter and showed that the calreticulin gene was activated during early stages of embryonic development. Furthermore, calreticulin-deficient cells have inhibited bradykinin-induced Ca2+ release from the ER and impaired nuclear import of the NF-AT3 transcription factor.
Materials and Methods
Construction of the Calreticulin Knockout Vector
The plasmid pNTK (containing the PGK neomycin cassette and PGK thymidine kinase) was used to generate the knockout vector. The calreticulin promoter was cloned previously from a mouse liver genomic library (Waser et al., 1997) and digested by SalI and HindIII restriction endonucleases. Afterwards, the 1.5-kb fragment was blunt ended and ligated into the pNTK plasmid to generate the pNTC1 plasmid. The plasmid pCM101 containing the full-length mouse calreticulin gene was cut with EcoRV. Furthermore, an 11-kb fragment, containing part of exon 4 and exons 5–9 and 3-kb of 3′ flanking region, was ligated with the NotI/SalI cut and blunted pNTC1 plasmid that resulted in the calreticulin gene knockout construct pNTC6 (see Fig. 1 A). In the pNTC6 plasmid the PGK NEO cassette replaced the first four exons of the calreticulin gene, thus removing the proposed transcription initiation site, the ATG start codon, and interrupting translation of the protein.
Figure 1.


Targeted disruption of the calreticulin gene. (A) Top shows the structure of the mouse calreticulin gene, indicating the ATG start codon and the restriction enzymes utilized (E, EcoRI; EV, EcoRV; H, HindIII; S, SalI). The arrows indicate the PCR primer sets (see Materials and Methods) used for recognition of the wild-type gene. The solid bar is the 5′ probe used for Southern blotting. Middle is the targeting vector designed to replace the first exons with the PGK NEO cassette. The targeted calreticulin allele is shown (bottom). The arrows indicate the PCR primers used for recognition of the mutant allele. (B) Southern blot of the EcoRI-digested genomic DNA isolated from wild-type (+/+), heterozygote (+/−), and homozygote (−/−) mice. The sizes of the wild-type (5.7 kb) and mutant allele (3.8 kb) are indicated. (C) Western blot analysis of proteins extracted from the biopsies of wild-type (+/+), heterozygote (+/−), and homozygote (−/−) mouse embryos. The blot was probed with the affinity-purified rabbit anticalreticulin antibody (Michalak et al., 1996).
Electroporation, Selection, and Screening of ES Cell Clones
J1 129/Sv embryonic stem (ES) cells (5 × 107) were electroporated with 100 μg of linearized pNTC6 vector using a Bio-Rad Gene Pulser at 400 V/cm and 25 μF. Cells were plated on mitomycin C–treated G-418–resistant mouse embryonic fibroblast feeder cells. Recombinant clones were selected with G418 (0.2 mg/ml) and gancyclovir (2 μM). 200 colonies were picked after 10 d in selection medium and expanded. 100 clones were screened for a homologous recombination event by PCR using the Expand Long Template PCR System (Boehringer Mannheim). The efficiency of homologous recombination was 1 in 30 clones. The heterozygote calreticulin knockout ES cells were identified by PCR using one 5′ primer (5′-GCTGGTCAAGTGTGATTCTCATGTTCCTGCCTG-3′) and two different 3′ primers (5′-CTCTGACCTTCACACTAGACACCCTTCATC-3′ for the wild-type gene and 5′-CTCTGACCTTCACACTAGACACCCTTCATC-3′ for the knockout allele) and confirmed by Southern blot analysis. These ES cells were microinjected into 3.5-d-old C57BL/6J blastocysts to generate chimeric mice. Chimeric males were analyzed for germline transmission by mating with C57BL/6J females, and the progeny were analyzed by PCR and Southern blot. For PCR amplification of the genomic DNA of knockout mice, two sets of primers were utilized (see Fig. 1 A). One resulted in detection of the wild-type gene (the primer sequences were: 5′-GAAGATCTAAACCAGTCAAAAGGACC-3′ and 5′-CTCCAGGTCCCCGTAAAATTTGCC-3′) and the second resulted in detection of the targeted knockout construct (the primer sequences were: 5′-CAGAGATCTCAGCAGCAAGGGC-3′ and 5′-CTCTGACC TTCACACTAGACACCCTTCATC-3′). EcoRI-digested genomic DNA was used for Southern blot detection of the calreticulin gene and for detection of the targeted mutant allele using the 5′ DNA probe indicated in Fig. 1 A.
Generation of the GFP Reporter Gene Vector
The plasmid pS65T-C1 containing cDNA encoding GFP was purchased from CLONTECH Laboratories, Inc. The nucleotide sequence corresponding to the CMV promoter of this construct was removed using AseI and NheI restriction sites and the ends were blunted using Klenow polymerase. The CMV promoter was replaced with a 2.3-kb mouse calreticulin promoter (Waser et al., 1997), and cut with SmaI and StuI to generate blunt ends. The 3.58-kb vector containing the calreticulin promoter, GFP cDNA, and the SV-40 polyA site is referred to as the pCPGF construct. To generate transgenic mice, pCPGF was linearized with NaeI/SpeI (see Fig. 3 A), purified, and injected into the fertilized oocytes from the FVB/N mice (Taketo et al., 1991). Afterwards cells were transferred into pseudopregnant FVB/N mice. Genomic DNA was isolated from tail biopsies of each of the transgenic mouse litters and the presence of the GFP reporter targeting vector was detected by PCR using a 5′ primer in the CRT promoter region (5′-GATTCCTTCTGGGCAGTTCATAGTC-3′) and a 3′ primer in the GFP protein cDNA (5′-ATCTAATTCAACAAGAATTGGGACAA-3′). The locations of these primers are indicated in Fig. 3 A. Transgenic animals and calreticulin-deficient mice were generated in the Transgenic Facility (University of Alberta Health Sciences Laboratory Animal Services, Edmonton, Alberta, Canada).
Figure 3.
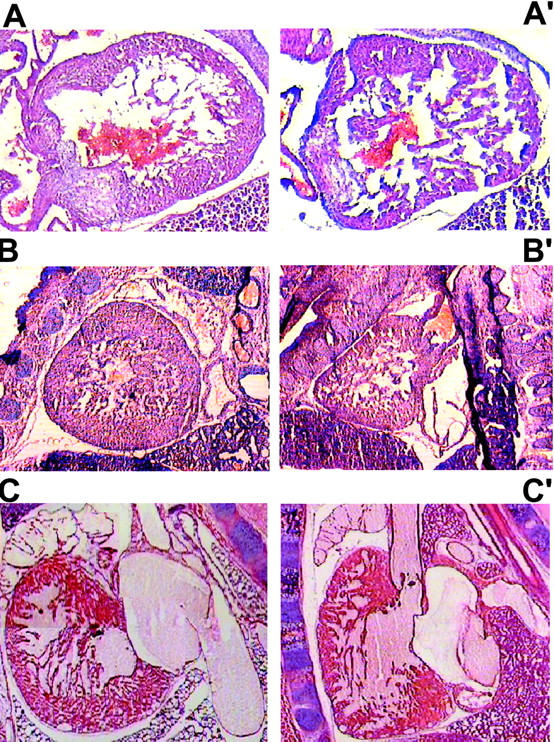
Histology of the hearts from crt +/− and crt −/− embryos. A, B, and C show histological analysis of the 12.5-, 14.5-, and 18-d-old crt +/− embryos, respectively. A′, B′, and C′ show histology of the 12.5-, 14.5-, and 18-d-old crt −/− embryos. crt −/− embryos show deep intertrabecular recesses and increased fenestration associated with the thinner ventricular wall. Sections were stained with hematoxylin and eosin as described in Materials and Methods.
Histological Analysis
Mouse embryos, at different gestational age, were dissected out of the uterus and fixed in 4% paraformaldehyde for 30–90 min. The embryos were embedded in 30% sucrose overnight at 4°C, washed once in PBS for 1 h, placed in 50% Tissue Tek OCT compound (mounting media) in PBS saline for 8 h at room temperature, and incubated in 100% OCT overnight at 4°C. The embryos were frozen in 2-methylbutane cooled in liquid nitrogen. Cryostat sections, 8–10 μm thick, were prepared in transverse and sagittal sections. The sections were either mounted in mounting media containing 0.2% DABCO (for fluorescence analysis) or processed for immunohistochemistry and standard hematoxylin and eosin staining. A confocal microscope (MRC600; Bio-Rad Laboratories) was used to obtain images at 10× or 60× and images were reconstructed using the Adobe Photoshop program. For better visualization, simulated phase-contrast images of each fluorescent image were generated using the Adobe Photoshop program.
Immunohistochemistry
Sections from wild-type and transgenic mouse embryos were stained with appropriate antibodies followed by staining using Vectastain Elite ABC kit and Vector DAB substrate kit (Vector Labs Inc.). Primary antibodies were polyclonal rabbit antibodies to GFP (1:150 dilution; CLONTECH Laboratories, Inc.) and affinity purified anticalreticulin (1:10 dilution) (CRT283 antibody described in Michalak et al., 1996). The sections were counterstained with hematoxylin.
Activation of NF-AT3 in Mouse Embryonic Fibroblasts
Embryos from two litters of heterozygous crosses were used to derive mouse embryonic fibroblasts. Embryos crt −/− and wild-type genotype were dissociated, washed, trypsinized for 30 min, and cultured in 6-well tissue culture plates. Cells were maintained in DME containing 20% FCS. For transfection experiments, plasmid DNA was purified by column chromatography (Qiagen Inc.). Cells were transfected transiently with NF-AT3 expression vector alone, or cotransfected with NF-AT3 and calreticulin expression vectors as described by Burns et al. (1994). After 1 d, cells were stimulated with 200 nM bradykinin (Waser et al., 1997) followed by indirect immunofluorescence with monoclonal anti–NF-AT (mAb 7A6, a gift of G.R. Crabtree, Stanford University) and polyclonal goat anticalreticulin antibodies (Milner et al., 1991). The secondary antibodies were: Texas red–conjugated sheep anti–mouse (diluted 1:50 in PBS) and FITC-conjugated donkey anti–goat (used at 1:50 dilution). For double labeling, all incubations were carried out sequentially. A confocal fluorescence microscope (MRC-600; Bio-Rad Laboratories) equipped with a krypton/argon laser light source was used.
Ca2+ Measurements
Wild-type and calreticulin-deficient mouse embryonic fibroblasts (1.5 × 106 cells/ml) were loaded with fura-2/AM (Molecular Probes, Inc.). Fluorescence measurements were carried out as described by Mery et al. (1996). Ca2+ release from internal stores was induced with either 1 μM thapsigargin (Sigma Chemical Co.), or 200 nM bradykinin (Sigma Chemical Co.). Changes in the cytoplasmic Ca2+ concentration were monitored in Ca2+-free media (Mery et al., 1996).
Miscellaneous
Proteins were separated by SDS-PAGE on 10% polyacrylamide gels as described by Laemmli (1970), transferred to nitrocellulose membranes, and stained by immunoblotting with affinity purified rabbit anticalreticulin antibody (Michalak et al., 1996).
Genomic DNA was isolated from mouse tissue as described by Ausubel et al. (1989) and digested with EcoRI. The DNA was separated by electrophoresis on 0.8% agarose gel and transferred to Hybond-N membranes (Amersham Pharmacia Biotech). The disruption of the calreticulin gene was characterized by Southern blotting (Ausubel et al., 1989).
Results
Calreticulin Knockout
Fig. 1 A summarizes the gene targeting strategy used to generate the calreticulin gene knockout mice. During the process of homologous recombination, the PGK NEO cassette was inserted into the calreticulin gene, replacing the first four exons, thus removing the initiator ATG and interrupting the expression of calreticulin protein (Fig. 1 A). PCR analysis of genomic DNA using the specific sets of primers depicted in Fig. 1 A helped to identify the genotype of the mice. Analysis of genomic DNA by Southern blotting (Fig. 1 B) showed two hybridizing bands in genomic DNA from crt +/− mice corresponding to the wild-type allele (5.7 kb) and the targeted knockout allele (3.8 kb), whereas Southern blotting of genomic DNA isolated from crt −/− mice (Fig. 1 B) showed only one DNA band of 3.8 kb corresponding to the size of the targeted knockout gene. Western blot analysis revealed that the interruption of a single allele in crt +/− mice resulted in a significant decrease in calreticulin protein level, whereas in the calreticulin gene knockout animals (crt −/−) there was no detectable expression of the protein (Fig. 1 C). crt +/− mice contained ∼50% lower level of calreticulin protein than wild-type mice, as estimated by densitometry. Identical results were obtained with three different anticalreticulin antibodies (not shown).
Phenotype of Calreticulin Knockout Mice
Chimeric male mice were crossed with wild-type females to generate first generation heterozygotes. The crt +/− mice had normal phenotype, being viable and fertile. Intercrossing of the crt +/− males with crt +/− females was carried out to generate homozygote (crt −/−) gene knockout mice. We were unable to obtain any viable crt −/− pups from this cross. Living crt −/− embryos were obtained at 18 d and earlier. In addition, a number of 12.5–16.5-d-old embryos were dead. Analysis of embryos at or after day 14.5 showed a deficit number of crt −/− embryos (15% instead of 25%), indicating that a significant fraction of crt −/− embryos died earlier. We concluded that the homozygote (crt −/−) gene knockout was embryonic lethal and that calreticulin is essential for survival.
Accordingly, serial timed matings were set up to follow embryonic development in an attempt to find the cause of death of the crt −/− mice. Fig. 2, A and B, shows photographs of 18-d-old crt +/− (left) and crt −/− (right) mouse embryos. At this level of analysis the most visible difference between the crt +/− and crt −/− embryos was the failure of absorption of the umbilical hernia (omphalocele) in the crt −/− embryos (Fig. 2, A and B, arrow). Histological analysis of crt −/− mice confirmed the omphalocele and the presence of midgut in the umbilical hernia (Fig. 2 B). The other significant defect was observed in morphology of the hearts of the crt −/− mice (Fig. 2 B). Fig. 2 B shows that there was a marked decrease in the thickness of the ventricular wall in crt −/− as compared to crt +/− mice. Histological analysis of 12.5-d-old crt −/− embryo also revealed defects in morphology of the heart (Fig. 2 C), indicating that calreticulin is essential for cardiac development at relatively early stages of cardiogenesis. No other gross morphological changes were detected in crt −/− mice (Fig. 2). Higher magnification analysis of hearts from 12.5-, 14.5-, and 18-d-old embryos revealed deep intertrabecular recesses and increased fenestration that were associated with the thinner ventricular wall (Fig. 3). However, no significant changes in the histology of the atrial wall was observed (Figs. 2 and 3). At high magnification, pictures of the ventricular wall of 18-d-old crt −/− embryo further demonstrate all of the following: increased fenestration, thinner ventricular wall, and the impaired growth of the compact layer of the ventricles as compared to the crt +/− embryos (Fig. 4).
Figure 2.



Phenotypes of heterozygote (crt +/−) and homozygote (crt −/−) mouse embryos. (A) Phenotypes of 18-d-old crt +/− and crt −/− mouse embryos are shown. The arrow indicates the umbilical hernia. Leg and tail biopsies were removed for better visualization of the defect and were used for DNA and protein analysis. (B) Low magnification histology of 18-d-old crt +/− and crt −/− mouse embryos is shown. The arrow indicates the umbilical hernia. (C) Phenotypes of wild-type (wt) and homozygote (crt −/−) 12.5-d-old mouse embryos. Sagittal sections of embryos were stained with eosin and hematoxylin.
Figure 4.
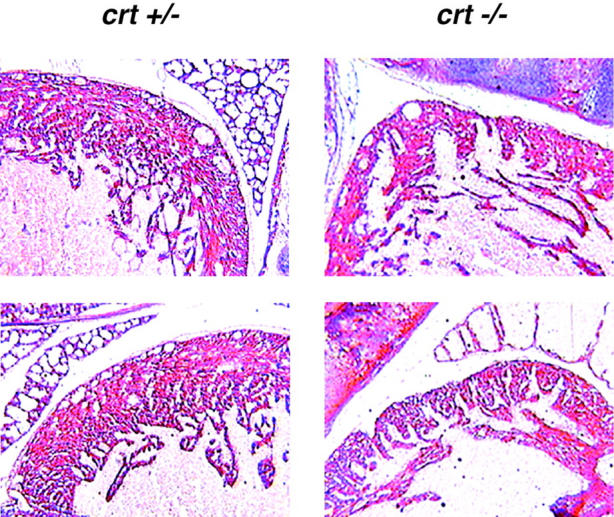
Histological analysis of the heart of crt +/− and crt −/− 18-d-old embryos. Sections were stained with hematoxylin and eosin as described in Materials and Methods. The upper panel shows the apex of the heart in crt +/− and crt −/− embryos. The lower panel shows ventricular walls in crt +/− and crt −/− animals.
Developmental Activation of the Calreticulin Promoter
The mature heart contains a very low level of calreticulin (Fliegel et al., 1989a,b; Milner et al., 1991; Tharin et al., 1996) but histological analysis of calreticulin-deficient mice suggests an essential role for the protein in cardiac development. To answer the question of whether the calreticulin gene is activated during cardiac development we studied the activation of its promoter in transgenic mice expressing the GFP reporter gene under control of the calreticulin promoter. Fig. 5 A illustrates the strategy used to construct the reporter transgene. The calreticulin promoter (2.3 kb) was introduced upstream of the GFP reporter gene. Genomic DNA from the transgenic mice was analyzed by PCR for the presence of the GFP transgene (Fig. 5 B). Three separate transgenic founder mice (L9, L13, and L17) were generated and all three gave identical results. Activation of the calreticulin promoter was monitored by detection of the fluorescent signal obtained from calreticulin promoter-driven expression of the GFP reporter gene. The first generation (F1) male transgenic mice were used in serial timed matings with wild-type FVB/N females. Embryos were harvested at different gestation times, fixed, and frozen. Every embryo was tested for the presence of the GFP transgene by PCR, followed by analysis of sections prepared from the transgenic and wild-type littermates.
Figure 5.

Construction and analysis of the GFP reporter gene vector. (A) 2.3-kb calreticulin promoter (Waser et al., 1997) was subcloned upstream of the cDNA encoding GFP. The arrows show the locations of the primer set used for PCR-driven detection of integration of the transgene into the mice genome. (B) PCR analysis of the genomic DNA of transgenic mice: lane M, molecular size ladder; lane 1, positive control purified plasmid DNA used as a template; lane 2, wild-type mouse; and lanes 3 and 4, GFP-transgenic mice.
Fig. 6, A and B, shows sagittal sections of a 9.5-d-old transgenic mouse embryo. The highest fluorescent signal, indicative of high expression of GFP, was found in the cardiovascular system including: ventricular walls, atrial walls, aortic sac, sinus venosus, dorsal aorta, and some of the smaller arteries (e.g., branchial arch arteries). This indicates that the highest activity of the calreticulin promoter occurs in these tissues at day 9.5 of embryonic development. The optic vesicle and the lining of brain ventricles also exhibited high calreticulin promoter activity (Fig. 6, A and B). At 10.5 d of embryonic development the calreticulin promoter remained highly active in cardiovascular and brain tissue, but its activity was also detected in the midgut and in intersomitic vessels (Fig. 6, C and D). In older embryos (13.5-d-old) high activity of the calreticulin promoter was maintained in the heart and arteries (Fig. 6 E). In addition, activation of the calreticulin promoter was also seen in the liver, midgut, and the umbilical hernia.
Figure 6.
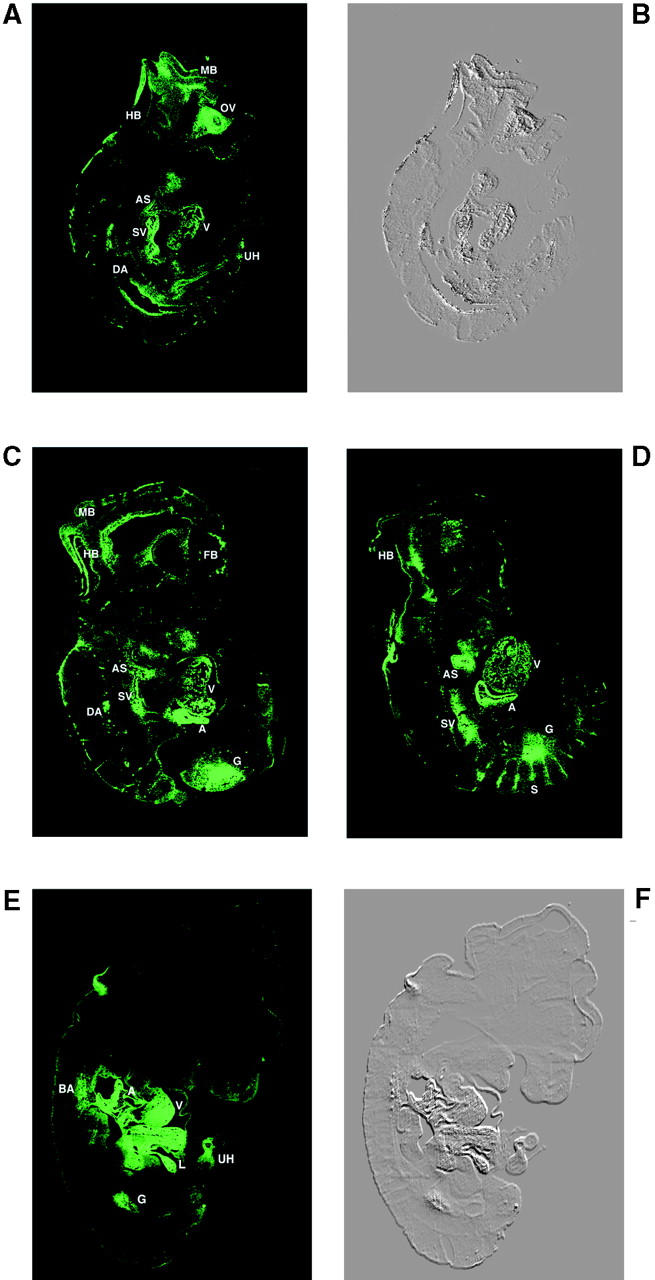
Developmental activation of calreticulin promoter. (A and B) Sagittal section of a 9.5-d-old mouse embryo expressing the GFP reporter protein under the control of the calreticulin promoter. (B) A phase-contrast of the image seen in A. (C and D) Sagittal sections of 10.5-d-old mouse embryos from GFP transgenic mice. (C) The first section shows high expression of GFP in the atria (A) and ventricles (V). Fluorescent signals are seen mainly in the cardiovascular and the nervous systems. (D) Additional sections of the embryo shown in C indicate high fluorescence in the intersomitic vessels (S). (E and F) Analysis of 13.5-d-old transgenic mouse embryo. (E) Confocal image showing the fluorescent signal in the cardiovascular system, liver, gut, and umbilical hernia. (F) A phase-contrast image of E. Tissues expressing GFP show a highly fluorescent signal: A, atria; AS, aortic sac; BA, branchial arteries and branches from the pulmonary arteries and arches of the aorta; DA, dorsal aorta; FB, fore brain; G, gut; HB, hind brain; L, liver; MB, mid brain; MG, midgut; OV, optic vesicle; SV, sinus venosus; S, somite; UH, umbilical hernia; and V, ventricles.
A high level of activation of the calreticulin promoter continued in the cardiovascular system and umbilical hernia in the 14.5-d-old mouse embryo (Fig. 7 A). The expression of GFP reporter protein was localized exclusively to the cytosol of the myocytes of atria and ventricles (Fig. 7 B). There was no fluorescent signal in the thoracic wall and blood cells, indicating differential expression of GFP in these tissues. At late stages of development (18-d-old embryos) a relatively low fluorescent signal, indicative of a lower expression of GFP, was found in the heart (Fig. 7 C). A negligible level of fluorescence was found in the heart of 3-wk-old transgenic mice (Fig. 7 E). This indicates that the activity of the calreticulin promoter is downregulated at late stages of development and after birth. These findings are in agreement with earlier observations that mature hearts express a low level of calreticulin (Fliegel et al., 1989a,b; Milner et al., 1991; Tharin et al., 1996).
Figure 7.
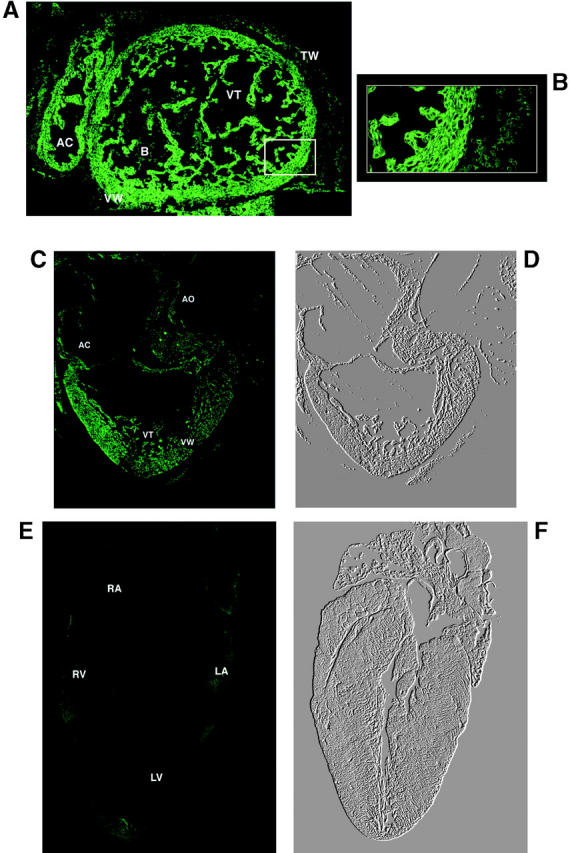
Confocal image of the hearts at different stages of development. (A and B) Cross-sections of the heart from 14.5-d-old embryo. GFP expression is localized to both atrial and ventricular walls but the fluorescent signal is also seen in the atrial and ventricular trabeculae. (B) The highest fluorescent signal found in the cardiomyocytes is shown. (C) GFP expression in the heart of 18-d-old embryo. (D) A phase-contrast image of C. (E) A negligible level of expression of GFP in the heart of 3-wk-old mouse is shown. (F) A phase-contrast image of E. Symbols: AC, atrial chamber; B, blood cells; TW, thoracic wall; VT, ventricular trabeculae; and VW, ventricular wall.
To confirm the expression of GFP protein was under the control of the calreticulin promoter in the cardiovascular system, we carried out immunohistological analysis of GFP transgenic embryos. Fig. 8 shows immunohistological staining of sagittal section of hearts from transgenic mice with antibodies to GFP. In agreement with observed fluorescent signals of GFP (Figs. 6 and 7) the protein was highly expressed in the embryonic heart (Fig. 8, C and D). There was no staining of the wild-type embryonic heart with anti-GFP antibodies (Fig. 8, A and B). To determine if activation of the calreticulin promoter correlated with expression of calreticulin protein we carried out immunohistological analysis of mouse embryos with anticalreticulin antibodies. Fig. 9 shows low (A–D) and high (A′–D′) magnification of immunohistological staining of 9.5- (A, A′), 13.5- (B, B′), and 18- (C, C′) d-old embryos and mature (D) hearts. Calreticulin protein was highly expressed in myocytes during early stages of embryonic development (Fig. 9, A′, B′, and C′). The highest expression of calreticulin was observed in the 13.5-d-old embryonic heart (Fig. 9 B′). In agreement with our earlier biochemical studies (Fliegel et al., 1989a,b; Milner et al., 1991; Tharin et al., 1996), virtually no staining for calreticulin was detected in the mature heart (Fig. 9 D′). This is in full agreement with the levels of GFP expressed in GFP transgenic mice (Figs. 6 and 7).
Figure 8.
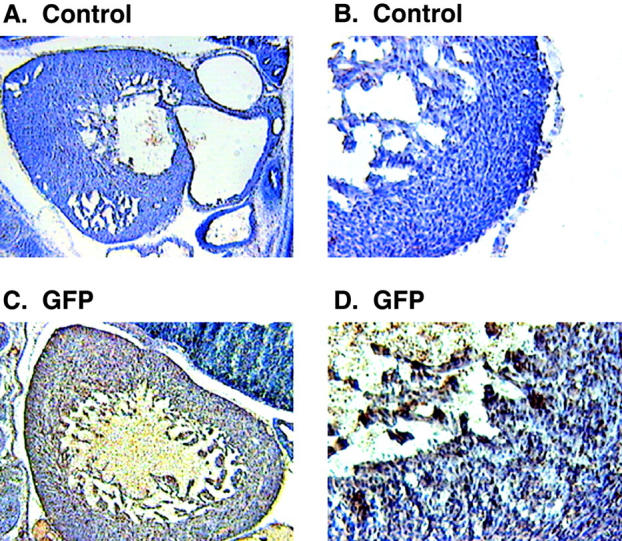
Immunohistochemical analysis of the GFP transgenic mice. Sagittal sections of 13.5-d-old mouse embryos were stained with specific antibodies followed by DAB (brown color). The sections were counter-stained with hematoxylin to visualize the nuclei. Low (A) and high magnification (B) images from cryostat sections of a wild-type mouse embryo stained with anti-GFP antibody. Low (C) and high (D) magnification images from GFP transgenic mouse showing the GFP positive staining in the ventricular myocytes.
Figure 9.
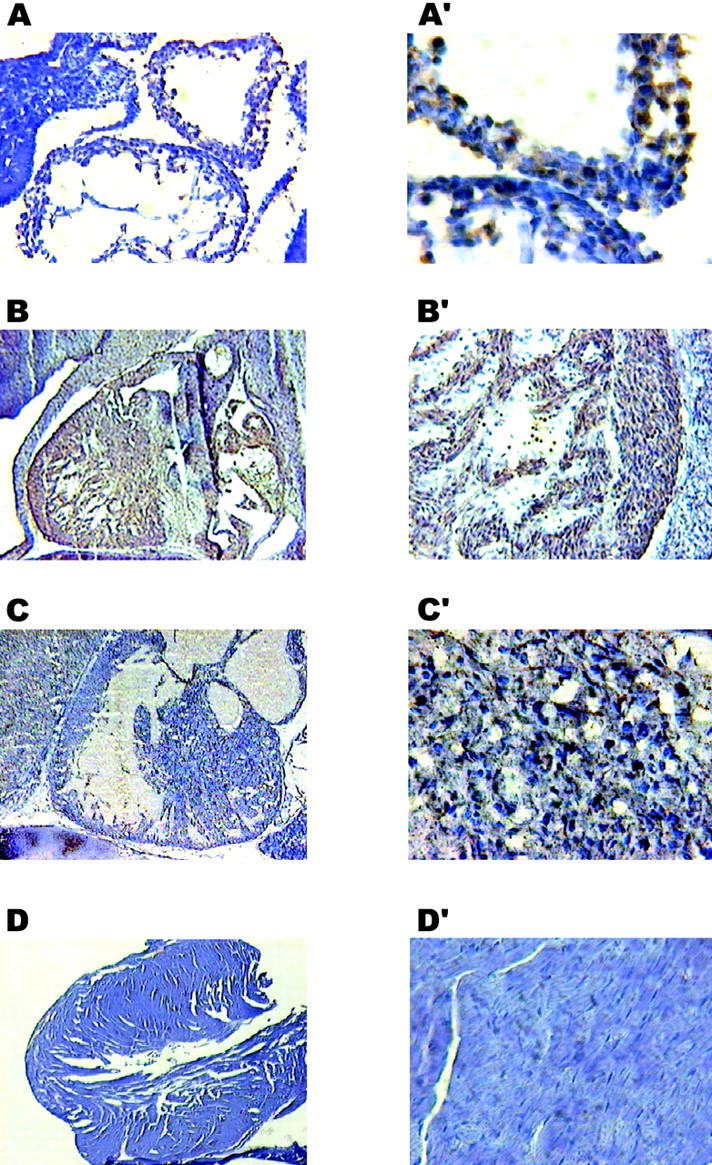
Immunohistochemical analysis of the transgenic mice with anticalreticulin antibodies. Sagittal sections of 9.5- (A), 13.5- (B), and 18- (C) d-old embryos and mature (D) mouse hearts were stained with anticalreticulin antibodies followed by DAB (brown color). High magnification picture (A′–D′) shows that calreticulin protein was highly expressed in myocytes during early stages of embryonic development (A′, B′, and C′). No significant staining for calreticulin was detected in the mature heart (D′). The sections were counterstained with hematoxylin.
Nuclear Translocation of NF-AT3 Is Impaired in Calreticulin Knockout Cells
Recent observations indicate that the NF-AT3 transcription factor plays an important role in cardiac hypertrophy and development (de la Pompa et al., 1998; Molkentin et al., 1998; Ranger et al., 1998). The activation of NF-AT is controlled by calcineurin, a Ca2+ calmodulin-dependent phosphatase (Timmerman et al., 1996). Dephosphorylation of NF-AT by activated calcineurin triggers its nuclear translocation (Rao et al., 1997). To determine whether the role of calreticulin in cardiac development might be associated with NF-AT nuclear translocation, we isolated mouse embryonic fibroblasts from crt −/− and wild-type embryos and tested them for nuclear import of NF-AT3 transcription factor. Cells were stimulated with bradykinin to induce depletion of the intracellular Ca2+ stores (Hashii et al., 1993; Waser et al., 1997) and activation of calcineurin (Timmerman et al., 1996). Fig. 10 A shows that wild-type cells expressed calreticulin and the protein is localized to an ER-like network. As expected, there was no expression of calreticulin in crt −/− cells (Fig. 10 B). In wild-type cells stimulated with bradykinin, NF-AT3 was efficiently translocated to the nucleus (Fig. 10 A′). However, nuclear import of NF-AT3 in crt −/− cells was impaired and the majority of the transcription factor was found in the cytoplasm (Fig. 10 B′) indicating that NF-AT3 transcription factor was not efficiently translocated into the nucleus. Nuclear translocation of NF-AT3 was reestablished in the crt −/− cells transiently transfected with calreticulin expression vector (Fig. 10, C and C′).
Figure 10.

Inhibition of nuclear import of NF-AT3 transcription factor in crt −/− cells. Mouse embryonic fibroblasts isolated from 14.5-d-old crt +/− (row A) and crt −/− (rows B and C) embryos were transiently transfected with either NF-AT3 (rows A and B) or NF-AT3 and calreticulin (row C) expression vectors. A, B, and C show localization of calreticulin, while A′, B′, and C′ show localization of NF-AT3 in the same cells. Cells were stimulated with 200 nM bradykinin. A′ shows that in the stimulated crt +/− cells NF-AT3 translocates to the nucleus. In crt −/− cells the majority of NF-AT3 is found in the cytoplasm (B′). In crt −/− cells transfected with calreticulin expression vector, NF-AT3 is again able to translocate the nucleus after stimulation (C′). A″, B″, and C″ are phase-contrast images of the same cells. Bar, 25 μm.
InsP3-dependent Ca2+ Release Is Inhibited in Calreticulin-deficient Cells
It is well established that Ca2+ release by the InsP3-dependent pathway is required to activate calcineurin, leading to nuclear import of NF-AT transcription factor (Timmerman et al., 1996; Dolmetsch et al., 1997; Kehlenbach et al., 1998). To assess whether calreticulin-deficient cells exhibit any alteration in Ca2+ homeostasis, we used the fluorescent Ca2+ indicator fura-2. Wild-type and crt −/− mouse embryonic fibroblasts were stimulated with either thapsigargin, an inhibitor of SERCA type Ca2+ pumps (Thastrup et al., 1990), or with bradykinin, a potent activator of the InsP3-dependent Ca2+ release channel located in the ER (Hashii et al., 1993). When cells were stimulated with thapsigargin, the peak amplitude and the duration of enhanced cytoplasmic Ca2+ concentration were comparable in wild-type and in crt −/− mouse embryonic fibroblasts (Fig. 11 A). Next we investigated the effects of bradykinin, a receptor agonist known to activate phospholipase C and InsP3-dependent Ca2+ release from the ER (Hashii et al., 1993). Fig. 11 B (solid line) shows that bradykinin caused a rapid and transient increase in the cytoplasmic Ca2+ concentration in wild-type cells, but not in crt −/− mouse embryonic fibroblasts (Fig. 10 B, dotted line). Ca2+ release by InsP3-dependent pathway was clearly impaired in calreticulin-deficient cells.
Figure 11.

Bradykinin-induced elevation of cytoplasmic Ca2+ concentration is inhibited in crt −/− cells. Wild-type and crt −/− mouse embryonic fibroblasts (MEF) were loaded with the fluorescent Ca2+ indicator fura-2. Cells were stimulated with 1 μM thapsigargin, a Ca2+-ATPase inhibitor (A), or with 200 nM bradykinin (B). Solid line shows wild-type mouse embryonic fibroblasts and the dotted line shows calreticulin (CRT)-deficient mouse embryonic fibroblasts.
Discussion
In this study we demonstrated that disruption of the calreticulin gene results in embryonic lethality. Analysis of the crt +/− and crt −/− embryos showed defects in cardiac morphology. The crt −/− mice show a marked decrease in ventricular wall thickness and deep intertrabecular recesses in the ventricular walls. Investigation of the activation pattern of the calreticulin gene during embryonic development revealed that at early stages of embryogenesis the highest level of activation of the calreticulin gene was in the cardiovascular system: ventricular walls, atrial walls, aortic sac, sinus venosus, dorsal aorta, and some of the smaller arteries. In later stages of embryogenesis a high activity of the promoter in the cardiovascular system was maintained, but activation of the gene was also seen in the brain, midgut, intersomitic vessels, and, finally, in the liver and the umbilical hernia. Analysis of InsP3-dependent Ca2+ release and of nuclear import of NF-AT3 transcription factor in cells isolated from crt −/− and wild-type mice revealed that calreticulin is a component of the Ca2+ regulatory system in these cells. Therefore, calreticulin influences the Ca2+ and calcineurin-dependent pathway in developing heart.
In the adult, calreticulin is expressed mainly in nonmuscle and smooth muscle cells, and is only a minor component of the skeletal muscle and cardiac sarcoplasmic reticulum (Fliegel et al., 1989a,b; Milner et al., 1991; Tharin et al., 1996). However, data presented in Figs. 6–9 show that calreticulin is highly expressed in the cardiovascular system during early embryogenesis. Thus, it was not surprising to find that crt −/− mice have defects in cardiac development and function. The only other gross morphological change in crt −/− embryos was the failure to absorb the umbilical hernia. It is unlikely that embryonic lethality of calreticulin-deficient mice occurs because of failure to absorb the umbilical hernia since this pathology is not embryonic lethal in humans (Byrne, 1991). Embryonic lethality of crt −/− mice most likely resulted from a lesion in cardiac development.
Numerous functions have been postulated for calreticulin including: modulation of steroid-mediated gene expression (Burns et al., 1994; Dedhar et al., 1994; Michalak et al., 1996), chaperone activity (Helenius et al., 1997), regulation of cell adhesion (Coppolino et al., 1995, 1997; Opas et al., 1996), and regulation of Ca2+ homeostasis (Liu et al., 1994; Bastianutto et al., 1995; Camacho and Lechleiter, 1995; Mery et al., 1996; Coppolino et al., 1997; Fasolato et al., 1998; John et al., 1998). A loss of any of these potential functions of calreticulin could lead to a lesion in cardiac development. Because there were no obvious histological abnormalities in any other tissues during embryogenesis, it is unlikely that this is directly due to chaperone function of calreticulin, or its role in the regulation of cell adhesion.
Cardiac development is an extremely complex process under strict transcriptional control (Rossant, 1996; Fishman and Chien, 1997; Sucov, 1998; Creazzo et al., 1998). The first morphogenetic event in mouse cardiac development is the formation of a primordial heart tube by cells that segregate from the splanchnic mesoderm in 7-d-old embryos (DeRuiter et al., 1992). Between 8 and 9.5 d, the primitive heart tube begins to exhibit rhythmic contractions and undergoes asymmetrical elongation, creating an S-shape loop (DeRuiter et al., 1992). Later in embryogenesis, the growing heart progresses through several additional morphological stages including all of the following: the formation of endocardial cushion tissue, septation, trabeculation, compaction (expansion of the ventricular wall), and envelopment of the epicardial mantle (DeRuiter et al., 1992; Mikawa and Fischman, 1996). Based on analogies to skeletal myogenesis, it is presumed that cardiomyocyte lineage commitment is directed by a group of tissue-specific transcription factors (Olson and Srivastova, 1996).
However, the factors essential for cardiomyocyte differentiation in vertebrates remain poorly characterized (Olson and Srivastova, 1996). How can calreticulin, an ER lumenal protein, affect cardiac development? In this work we have shown that nuclear import of NF-AT3 transcription factor is impaired in crt −/− cells indicating that calreticulin plays a role in the Ca2+/calcineurin/NF-AT/GATA-4 transcription pathway (Molkentin et al., 1998). Cardiomyogenesis depends on activation of the GATA family of transcription factors (Grepin et al., 1994; Evans, 1997). GATA-4 null mice display a severe defect in the formation of the cardiac tube, which is required for the migration and folding of the precardiogenic splanchnic mesoderm (Kuo et al., 1997; Molkentin et al., 1997). Furthermore, inhibition of GATA-4 expression affects terminal cardiomyocyte differentiation (Grepin et al., 1994). During cardiomyogenesis NF-AT, in the nucleus, forms active heterodimer complexes with GATA-4 and significantly enhances its transcriptional activity (Molkentin et al., 1998). The ability of NF-AT to translocate to the nucleus depends on the activation of calcineurin phosphatase activity and efficient dephosphorylation of the transcription factor (Rao et al., 1997). Therefore, it is not surprising that NF-AT–deficient mice are not viable and the major defect in these animals relates to cardiac development (de la Pompa et al., 1998; Ranger et al., 1998).
Activation of calcineurin and nuclear import of NF-AT requires Ca2+ release by the InsP3-dependent pathway (Timmerman et al., 1996; Dolmetsch et al., 1997). In this study we show that calreticulin-deficient mouse embryonic fibroblasts do not have a measurable InsP3-dependent Ca2+ release when stimulated with bradykinin. This is in agreement with a previous report in which the Ca2+ response to bradykinin was diminished as a result of antisense oligodeoxynucleotide downregulation of calreticulin expression (Liu et al., 1994). Calreticulin is a Ca2+-binding protein that affects Ca2+ storage in the ER lumen (Bastianutto et al., 1995; Mery et al., 1996; Fasolato et al., 1998). It may regulate function of the ER Ca2+-release channel (InsP3 receptor), Ca2+-ATPase (SERCA2b) (Camacho and Lechleiter, 1995; John et al., 1998), and store-operated Ca2+ influx (Mery et al., 1996). Camacho and Lechleiter (1995) proposed that calreticulin may be responsible for modulation of the Ca2+ release function of the InsP3 receptor and/or of the Ca2+ transport function of SERCA. Recently, John et al. (1998) reported that calreticulin may interact with SERCA2b, resulting in a lower transport capacity by the Ca2+-ATPase. Here we show that loss of calreticulin reduces Ca2+ release through the InsP3 receptor. Despite inhibition of the InsP3-dependent Ca2+ release pathway, cardiomyocytes cultured from 14.5-d-old crt −/− embryos undergo spontaneous contraction indicating that their excitation-contraction coupling is functional in these cells (Mesaeli, N., and M. Michalak, unpublished observations). This implies that Ca2+ handling by the ER, but not by the cardiac sarcoplasmic reticulum, is affected in calreticulin knockout cardiomyocytes. ER and sarcoplasmic reticulum membranes may form separate functional entities in muscle cells (Tharin et al., 1996).
An intriguing finding of this work is that calreticulin-deficient mice develop omphalocele (umbilical hernia). At the early stages of embryogenesis the small intestine develops outside the body to form a physiological umbilical hernia. However, in 15.5-d-old embryos the intestine enters the body cavity and the body wall closes (only the umbilical cord and its contents remain in this region) (Kaufman, 1992). Failure of the intestine to return to the coelom results in an omphalocele (Byrne, 1991).
Recent studies indicate that umbilical hernia is associated with the function of a cyclin-dependent kinase inhibitory protein p57KIP2, a regulator of cell proliferation (Eggenschwiler et al., 1997; Zhang et al., 1997). IGF-II receptor mutation and overexpression of IGF-II also results in omphalocele (Eggenschwiler et al., 1997). Disruption of the MARCKS gene that encodes a substrate of protein kinase C also leads to an omphalocele (Stumpo et al., 1995; Chen et al., 1996). Protein kinase C is activated by Ca2+, suggesting that Ca2+ release from the ER may play an important role in the closure of umbilical hernia. GATA-4 transcription factor is also expressed during gut development by playing a role in the regulation of intestine epithelial cell differentiation (Gao et al., 1998). It is conceivable that a similar Ca2+/calcineurin/NF-AT/GATA-4 transcription pathway may be activated during cardiac development and closure of umbilical hernia. This may explain why NF-AT3–deficient mice also show signs of abdominal necrosis (de la Pompa et al., 1998).
In Fig. 12 we present a model in which calreticulin is linked to NF-AT function and cardiac development. Ca2+ release through the InsP3-dependent pathway is required to activate calcineurin and to maintain NF-AT transcription factor in the nucleus (Timmerman et al., 1996; Dolmetsch et al., 1997). We propose that calreticulin regulates calcineurin activity indirectly by affecting Ca2+ release from the ER and the ability of NF-AT to translocate to the nucleus (Fig. 12). This model is supported by our demonstration that InsP3-dependent Ca2+ release is inhibited in calreticulin-deficient cells, concomitant with the inhibition of the NF-AT nuclear import.
Figure 12.

A model for the proposed role of calreticulin in the regulation of cardiac development. Myogenic signal from extracellular space activates the production of IP3 that results in the release of Ca2+ from ER under the regulation of calreticulin (CRT). Increased intracellular Ca2+ binds to calmodulin (CaM) and activates calcineurin (CaN). CaN dephosphorylates NF-AT that translocates to the nucleus. In the nucleus NF-AT forms complexes with the GATA-4 transcription factors leading to activation of transcription of genes essential for cardiac development (Molkentin et al., 1998).
The role of the InsP3 receptor and the InsP3-dependent pathway in cardiac development is further supported by studies on another regulator of the InsP3 receptor and Ca2+ release, the immunophilin protein FKBP12 (FK506 binding protein) (Brillantes et al., 1994; Cameron et al., 1995). Knockout of the FKBP12 gene is also embryonic lethal and the animals show cardiac defects (Shou et al., 1998). FKBP12 could be considered as a cytosolic regulator of the ER Ca2+ release channel, whereas calreticulin could be a lumenal regulator of Ca2+ homeostasis. The calreticulin/Ca2+/calcineurin/NF-AT/GATA-4 regulatory pathway is critical for the developing heart (Molkentin et al., 1998), but it is likely to be switched off in the adult myocardium and many components are absent or present at very low levels (NF-AT, GATA-4, and calreticulin) in the mature cardiomyocytes (de la Pompa et al., 1998; Fliegel et al., 1989a,b; Molkentin et al., 1998, and the references therein). In mature organisms NF-AT and calreticulin may play a more important role in the immune response (Burns et al., 1992; Dupuis et al., 1993; Rao et al., 1997; Andrin et al., 1998). Our results demonstrate an essential role for calreticulin during embryogenesis. Although calreticulin is not a transcription factor, this work shows that the protein is a regulator of Ca2+ homeostasis and of transcriptional pathways involved in cardiac development.
Acknowledgments
We thank Mathilde Waser for help with construction of the vector pNTC6. We thank K. Bagnal and T. Krukoff (Department of Cell Biology and Anatomy, University of Alberta) and J.C. Russell (Department of Surgery, University of Alberta) for invaluable help with histological analysis. We thank P. D'Obrenan for help with artwork. We thank G.R. Crabtree (Stanford University) for the anti–NF-AT mAb (7A6) and T. Hoey (Tularik Inc., San Francisco, CA) for the NF-AT cDNA. We thank L. Agellon and R.C. Bleackley (University of Alberta) for critically reading the manuscript and helpful discussions. The technical assistance of Karolina Michalak and Antoinette Monod is greatly appreciated.
This work was supported by grants to M. Michalak from the Medical Research Council of Canada (MRCC), and the Heart and Stroke Foundation of Alberta, to M. Opas from the MRCC and the Heart and Stroke Foundation of Ontario (HSFO), to K.-H. Krause from the Swiss National Foundation (3100-04 589.95/1), and to D.H. MacLennan from the MRCC and the HSFO. N. Mesaeli was a postdoctoral fellow of the Heart and Stroke Foundation of Canada. K. Nakamura is a postdoctoral fellow of the Alberta Heritage Foundation for Medical Research (AHFMR). M. Michalak is a MRCC Senior Scientist and an AHFMR Medical Scientist.
Abbreviations used in this paper
- ES cells
embryonic stem cells
- GFP
green fluorescent protein
- InsP3
inositol 1,4,5-trisphosphate
- NF-AT
nuclear factor of activated T cell
Footnotes
Address correspondence to Dr. Marek Michalak, MRC Group in Molecular Biology of Membranes, Department of Biochemistry, 3-56 Medical Sciences Building, University of Alberta, Edmonton, Alberta, Canada T6G 2H7. Tel.: 780-492-2256. Fax: 780-492-0886. E-mail: marek.michalak @ualberta.ca
References
- Andrin C, Pinkoski MJ, Burns K, Atkinson EA, Krahenbuhl O, Hudig D, Fraser SA, Winkler U, Tschopp J, Opas M, et al. Interaction between a Ca2+binding protein calreticulin and perforin, a component of the T-cell granules. Biochemistry. 1998;37:10386–10394. doi: 10.1021/bi980595z. [DOI] [PubMed] [Google Scholar]
- Ausubel, F.M., R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhe. 1989. Current Protocols in Molecular Biology. Vol. 1–3. John Wiley & Sons, Inc., New York.
- Baksh S, Michalak M. Expression of calreticulin in Escherichia coli and identification of its Ca2+binding domains. J Biol Chem. 1991;266:21458–21465. [PubMed] [Google Scholar]
- Bastianutto C, Clementi E, Codazzi F, Podini P, De Giorgi F, Rizzuto R, Meldolesi J, Pozzan T. Overexpression of calreticulin increases the Ca2+ capacity of rapidly exchanging Ca2+stores and reveals aspects of their lumenal microenvironment and function. J Cell Biol. 1995;130:847–855. doi: 10.1083/jcb.130.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Lander M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Burns K, Helgason CD, Bleackley RC, Michalak M. Calreticulin in T-lymphocytes. Identification of calreticulin in T-lymphocytes and demonstration that activation of T-cells correlates with increased levels of calreticulin mRNA and protein. J Biol Chem. 1992;267:19039–19042. [PubMed] [Google Scholar]
- Burns K, Duggan B, Atkinson AE, Famulski KS, Nemer M, Bleackley RC, Michalak M. Modulation of gene expression by calreticulin binding to the glucocorticoid receptor. Nature. 1994;367:476–480. doi: 10.1038/367476a0. [DOI] [PubMed] [Google Scholar]
- Byrne, W.J. 1991. Disorders of the umbilical cord, abdominal wall, urachus, and omphalomesenteric duct. In Diseases of the Newborn. H.W. Taeusch, R.A. Ballard, and M.E. Avery, editors. W.B. Saunders Co. Philadelphia. 694–701.
- Camacho P, Lechleiter JD. Calreticulin inhibits repetitive intracellular Ca2+waves. Cell. 1995;82:765–771. doi: 10.1016/0092-8674(95)90473-5. [DOI] [PubMed] [Google Scholar]
- Cameron AM, Steiner JP, Sabatini DM, Kaplin AI, Walensky LD, Snyder SH. Immunophilin FK506 binding protein associated with inositol 1,4,5-trisphosphate receptor modulates calcium flux. Proc Natl Acad Sci USA. 1995;92:1784–1788. doi: 10.1073/pnas.92.5.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chang S, Duncan SA, Okano HJ, Fishell G, Aderem A. Disruption of the MARCKS gene prevents cranial neural tube closure and results in anencephaly. Proc Natl Acad Sci USA. 1996;93:6275–6279. doi: 10.1073/pnas.93.13.6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppolino M, Leung-Hagesteijn CY, Dedhar S, Wilkins J. Inducible interaction of integrin α2β1with calreticulin. J Biol Chem. 1995;270:23132–23138. doi: 10.1074/jbc.270.39.23132. [DOI] [PubMed] [Google Scholar]
- Coppolino M, Woodside MJ, Demaurex N, Grinstein S, St-Arnaud R, Dedhar S. Calreticulin is essential for integrin-mediated calcium signaling and cell adhesion. Nature. 1997;386:843–847. doi: 10.1038/386843a0. [DOI] [PubMed] [Google Scholar]
- Creazzo TL, Godt RE, Leatherbury L, Conway SJ, Kirby ML. Role of cardiac neural crest cells in cardiovascular development. Annu Rev Physiol. 1998;60:267–286. doi: 10.1146/annurev.physiol.60.1.267. [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Marengere L, Langille BL, Crabtree GR, et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–185. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- Dedhar S, Rennie PS, Shago M, Hagesteijn CY, Yang H, Filmus J, Hawley RG, Bruchovsky N, Cheng H, Matusik RJ, et al. Inhibition of nuclear hormone receptor activity by calreticulin. Nature. 1994;367:480–483. doi: 10.1038/367480a0. [DOI] [PubMed] [Google Scholar]
- DeRuiter MC, Poelmann RE, VanderPlas-de Vries I, Mentink MM, Gittenberger-de Groot AC. The development of the myocardium and endocardium in mouse embryos. Fusion of two heart tubes? . Anat Embryol. 1992;185:461–473. doi: 10.1007/BF00174084. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- Dupuis M, Schaerer E, Krause K-H, Tschopp J. The calcium binding protein calreticulin is a major constituent of lytic granules in cytolytic T lymphocytes. J Exp Med. 1993;177:1–7. doi: 10.1084/jem.177.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler J, Ludwig T, Fisher P, Leighton PA, Tilghman SM, Efstratiadis A. Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith-Wiedemann and Simpson-Golabi-Behmel syndromes. Genes Dev. 1997;11:3128–3142. doi: 10.1101/gad.11.23.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans T. Regulation of cardiac gene expression by GATA-4/5/6. Trends Card Med. 1997;7:75–83. doi: 10.1016/S1050-1738(97)00010-8. [DOI] [PubMed] [Google Scholar]
- Fasolato C, Pizzo P, Pozzan T. Delayed activation of the store-operated calcium current induced by calreticulin overexpression in RBL-1 cells. Mol Biol Cell. 1998;9:1513–1522. doi: 10.1091/mbc.9.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman MC, Chien KR. Fashioning the vertebrate heart: earliest embryonic decisions. Development. 1997;124:2099–2117. doi: 10.1242/dev.124.11.2099. [DOI] [PubMed] [Google Scholar]
- Fliegel L, Burns K, MacLennan DH, Reithmeier RAF, Michalak M. Molecular cloning of the high affinity calcium binding protein (calreticulin) of skeletal muscle sarcoplasmic reticulum. J Biol Chem. 1989a;264:21522–21528. [PubMed] [Google Scholar]
- Fliegel L, Burns K, Opas M, Michalak M. The high affinity calcium binding protein of skeletal muscle sarcoplasmic reticulum. Biochim Biophys Acta. 1989b;982:1–8. doi: 10.1016/0005-2736(89)90166-1. [DOI] [PubMed] [Google Scholar]
- Gao X, Sedgwick T, Shi Y-B, Evans T. Distinct functions are implicated for the GATA-4, -5, and -6 transcription factors in the regulation of intestine epithelial cell differentiation. Mol Cell Biol. 1998;18:2901–2911. doi: 10.1128/mcb.18.5.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grepin C, Dagnino L, Robitaille L, Haberstroh L, Antakly T, Nemer M. A hormone-encoding gene identifies a pathway for cardiac but not skeletal muscle gene transcription. Mol Cell Biol. 1994;14:3115–3129. doi: 10.1128/mcb.14.5.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashii M, Nozawa Y, Higashida H. Bradykinin-induced cytosolic Ca2+ oscillations and inositol tetrakisphosphate-induced Ca2+ influx in voltage-clamped ras-transformed NIH/3T3 fibroblasts. J Biol Chem. 1993;268:19403–19410. [PubMed] [Google Scholar]
- Helenius A, Trombetta ES, Hebert DN, Simons JF. Calnexin, calreticulin and the folding of glycoproteins. Trends Cell Biol. 1997;7:193–200. doi: 10.1016/S0962-8924(97)01032-5. [DOI] [PubMed] [Google Scholar]
- John LM, Lechleiter JD, Camacho P. Differential modulation of SERCA2 isoforms by calreticulin. J Cell Biol. 1998;142:963–973. doi: 10.1083/jcb.142.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, M.H. 1992. The atlas of mouse development. Academic Press, London.
- Kehlenbach RH, Dickmanns A, Gerace L. Nucleocytoplasmic shuttling factor including Ran and CRM1 mediate nuclear export of NFAT in vitro. J Cell Biol. 1998;141:863–874. doi: 10.1083/jcb.141.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause K-H, Michalak M. Calreticulin. Cell. 1997;88:439–443. doi: 10.1016/s0092-8674(00)81884-x. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:8145–8149. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4 . Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Liu N, Fine RE, Simons E, Johnson RJ. Decreasing calreticulin expression lowers the Ca2+response to bradykinin and increases sensitivity to ionomycin in NG-108-15 cells. J Biol Chem. 1994;269:28635–28639. [PubMed] [Google Scholar]
- Mery L, Mesaeli N, Michalak M, Opas M, Lew DP, Krause KH. Overexpression of calreticulin increases intracellular Ca2+-storage and decreases store-dependent Ca2+influx. J Biol Chem. 1996;271:9332–9339. doi: 10.1074/jbc.271.16.9332. [DOI] [PubMed] [Google Scholar]
- Michalak M, Milner RE, Burns K, Opas M. Calreticulin. Biochem J. 1992;285:681–692. doi: 10.1042/bj2850681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak M, Burns K, Andrin C, Mesaeli N, Jass GH, Busaan JL, Opas M. Endoplasmic reticulum form of calreticulin modulates glucocorticoid-sensitive gene expression. J Biol Chem. 1996;271:29436–29445. doi: 10.1074/jbc.271.46.29436. [DOI] [PubMed] [Google Scholar]
- Mikawa T, Fischman DA. The polyclonal origin of myocyte lineages. Annu Rev Physiol. 1996;58:509–521. doi: 10.1146/annurev.ph.58.030196.002453. [DOI] [PubMed] [Google Scholar]
- Milner RE, Baksh S, Shemanko C, Vance JE, Carpenter M, Smillie L, Opas M, Michalak M. Calreticulin, and not calsequestrin, is the major calcium binding protein of smooth muscle sarcoplasmic reticulum and liver endoplasmic reticulum. J Biol Chem. 1991;266:7155–7156. [PubMed] [Google Scholar]
- Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA-4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Moriya M, Baba T, Michikawa Y, Yamanobe T, Arai K, Okinaga S, Kobayashi T. An endoplasmic reticulum protein, calreticulin, is transported into the acrosome of rat sperm. Exp Cell Res. 1993;205:101–110. doi: 10.1006/excr.1993.1063. [DOI] [PubMed] [Google Scholar]
- Nauseef WM, McCormick SJ, Clark RA. Calreticulin functions as a molecular chaperone in the biosynthesis of myeloperoxidase. J Biol Chem. 1995;270:4741–4747. doi: 10.1074/jbc.270.9.4741. [DOI] [PubMed] [Google Scholar]
- Nigam SK, Goldberg AL, Ho S, Rhode MF, Bush KT, Sherman MY. A set of endoplasmic reticulum proteins possessing properties of molecular chaperones includes Ca2+-binding proteins and numbers of the thioredoxin superfamily. J Biol Chem. 1994;269:1744–1749. [PubMed] [Google Scholar]
- Olson EN, Srivastova D. Molecular pathways controlling heart development. Science. 1996;272:617–676. doi: 10.1126/science.272.5262.671. [DOI] [PubMed] [Google Scholar]
- Opas M, Szewczenko-Pawlikowski M, Jass GK, Mesaeli N, Michalak M. Calreticulin modulates cell adhesiveness via regulation of vinculin expression. J Cell Biol. 1996;135:1913–1923. doi: 10.1083/jcb.135.6.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostwald TJ, MacLennan DH. Isolation of a high affinity calcium binding protein from sarcoplasmic reticulum. J Biol Chem. 1974;249:974–979. [PubMed] [Google Scholar]
- Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NF-AT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Rossant J. Mouse mutants and cardiac development. Circ Res. 1996;78:349–353. doi: 10.1161/01.res.78.3.349. [DOI] [PubMed] [Google Scholar]
- Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- Singh NK, Atreya CD, Nakhasi HL. Identification of calreticulin as a rubella virus RNA binding protein. Proc Natl Acad Sci USA. 1994;91:12770–12774. doi: 10.1073/pnas.91.26.12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer RD, Lieu T-S, Capra JD. Calreticulin: the diverse functional repertoire of a new human autoantigen. Immunologist. 1993;1:155–160. [Google Scholar]
- Stendahl O, Krause KH, Kirschner J, Jerström P, Theler JM, Clark RA, Carpentier JL, Lew DP. Redistribution of intracellular Ca2+stores during phagocytosis in human neutrophils. Science. 1994;265:1439–1441. doi: 10.1126/science.8073285. [DOI] [PubMed] [Google Scholar]
- Stumpo DJ, Bock CB, Tuttle JS, Blackshear PJ. MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proc Natl Acad Sci USA. 1995;92:944–948. doi: 10.1073/pnas.92.4.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucov HM. Molecular insights into cardiac development. Annu Rev Physiol. 1998;60:287–308. doi: 10.1146/annurev.physiol.60.1.287. [DOI] [PubMed] [Google Scholar]
- Taketo M, Schroeder AC, Mobraaten LE, Gunning KB, Hanten G, Fox RR, Roderick TH, Stewart CL, Lilly F, Hansen CT, et al. FVB/N: an inbred mouse strain preferable for transgenic analyses. Proc Natl Acad Sci USA. 1991;88:2065–2069. doi: 10.1073/pnas.88.6.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharin S, Hamel PA, Conway EM, Michalak M, Opas M. Regulation of calcium storage proteins, calreticulin and calsequestrin, during differentiation of the myogenic cell line L6. J Cell Physiol. 1996;166:547–560. doi: 10.1002/(SICI)1097-4652(199603)166:3<547::AID-JCP9>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR. Rapid shuttling of NF-AT in discrimination of Ca2+signals and immunosuppression. Nature. 1996;383:837–840. doi: 10.1038/383837a0. [DOI] [PubMed] [Google Scholar]
- Waser M, Mesaeli N, Spencer C, Michalak M. Regulation of calreticulin gene expression by calcium. J Cell Biol. 1997;138:547–557. doi: 10.1083/jcb.138.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Liegeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ. Altered cell differentiation and proliferation in mice lacking p57KIP2indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387:151–158. doi: 10.1038/387151a0. [DOI] [PubMed] [Google Scholar]