

Abstract
The mechanism by which membrane-bound Bcl-2 inhibits the activation of cytoplasmic procaspases is unknown. Here we characterize an intracellular, membrane-associated form of procaspase-3 whose activation is controlled by Bcl-2. Heavy membranes isolated from control cells contained a spontaneously activatable caspase-3 zymogen. In contrast, in Bcl-2 overexpressing cells, although the caspase-3 zymogen was still associated with heavy membranes, its spontaneous activation was blocked. However, Bcl-2 expression had little effect on the levels of cytoplasmic caspase activity in unstimulated cells. Furthermore, the membrane-associated caspase-3 differed from cytosolic caspase-3 in its responsiveness to activation by exogenous cytochrome c. Our results demonstrate that intracellular membranes can generate active caspase-3 by a Bcl-2–inhibitable mechanism, and that control of caspase activation in membranes is distinct from that observed in the cytoplasm. These data suggest that Bcl-2 may control cytoplasmic events in part by blocking the activation of membrane-associated procaspases.
Keywords: apoptosis, Bcl-2, caspase, cytochrome c, programmed cell death
The caspases are a family of cysteine proteases that are essential effectors of the apoptotic process (Yuan et al., 1993; Alnemri et al., 1996; Cohen, 1997; Miller, 1997; Salvesen and Dixit, 1997). Caspases are synthesized as inactive zymogens, which are activated by proteolytic processing to yield large (∼18 kD) and small (∼12 kD) subunits that associate to form active enzymes (Thornberry et al., 1992; Nicholson et al., 1995; Stennicke and Salvesen, 1997). Diverse apoptotic stimuli cause the activation of specific caspases which then initiate a protease cascade by proteolytically processing additional caspases (Srinivasula et al., 1996; Yu et al., 1998). Once activated, these downstream caspases kill cells by cleaving specific molecular targets that are essential for cell viability or by activating proapoptotic factors (Liu et al., 1997; Salvesen and Dixit, 1997; Enari et al., 1998). Although caspases have been generally shown to be cytosolic proteins (Miller et al., 1993; Nicholson et al., 1995; Li et al., 1997b), immunochemical studies have suggested that in some instances, caspases might also be associated with the nucleus or plasma membrane (Singer et al., 1995; Krajewska et al., 1997; Krajewski et al., 1997; Posmantur et al., 1997). Recently published data has indicated an association of certain caspases with mitochondria and endoplasmic reticulum (Chandler et al., 1998; Mancini et al., 1998).
The Bcl-2 family constitutes another key set of regulators of the apoptotic pathway. These proteins can function to inhibit or induce apoptosis in a wide variety of cell systems (Oltvai and Korsmeyer, 1994; Reed, 1997). Bcl-2 family proteins contain one to four conserved domains, designated BH1-BH4, and most family members contain a COOH-terminal transmembrane anchor sequence which allows them to be associated with cellular membranes including the outer membrane of the mitochondria, the nuclear envelope and the endoplasmic reticulum (Krajewski et al., 1993; Lithgow et al., 1994; Yang et al., 1995; Reed, 1997). The over-expression of Bcl-2 has been shown to inhibit the activation of cytoplasmic caspases after apoptotic stimuli in several cell systems (Armstrong et al., 1996; Boulakia et al., 1996; Chinnaiyan et al., 1996; Srinivasan et al., 1996). However, it remains unclear how the membrane-bound Bcl-2 exerts control over the soluble cytoplasmic caspases.
Recent experiments have suggested several possible mechanisms for Bcl-2 family function. The Bcl-2 homologue Bcl-xL has been shown to be structurally similar to the diphtheria toxin channel-forming protein (Muchmore et al., 1996), and several Bcl-2 family members have been shown to form ion channels in vitro using reconstituted systems (Minn et al., 1997; Schendel et al., 1997; Schlesinger et al., 1997). These data have led to the hypothesis that Bcl-2 family members might function in cells as transmembrane channels (Vander Heiden et al., 1997). Other experiments demonstrated that Bcl-2 and Bcl-xL block the release of cytochrome c from mitochondria, preventing cytochrome c–mediated caspase activation (Kluck et al., 1997; Yang et al., 1997). This work suggested that Bcl-2 and Bcl-xL might act directly at the level of cytochrome c release. However, microinjection experiments have demonstrated that inhibition of apoptosis by Bcl-xL cannot be explained only by effects on cytochrome c compartmentalization (Li et al., 1997a). Yet other experiments have shown that CED-9, a Bcl-2 family member from the roundworm Caenorhabditis elegans (Horvitz et al., 1994), biochemically interacts with the adapter protein CED-4, blocking the CED-4–dependent activation of the caspase CED-3 (Chinnaiyan et al., 1997; Ottilie et al., 1997; Seshagiri and Miller, 1997; Spector et al., 1997; Wu et al., 1997). This work suggested that the mammalian Bcl-2 family members may similarly control apoptosis by directly affecting caspase activation mechanisms. Indeed, recent data indicates that Bcl-xL can bind to the mammalian CED-4 homologue Apaf-1, at least under some conditions (Hu et al., 1998; Pan et al., 1998).
Previous work has demonstrated that Bcl-2 inhibits the onset of apoptosis, but once apoptosis is initiated, Bcl-2 does not impede the process (McCarthy et al., 1997). This suggested that if Bcl-2 exerted direct control over caspases, it did not directly block the downstream caspases that effect cell killing, but rather, might affect regulatory mechanisms that trigger the downstream events. This prompted us to consider the existence of such triggering mechanisms in the Bcl-2–containing membrane compartments of the cell, and specifically, whether regulated caspases might be present there. This report describes the identification and characterization of membrane-derived caspase-3, the activation of which is suppressed by expression of Bcl-2.
Materials and Methods
Cell Lines and Cell Production
697 human lymphoblastoid cells stably infected with a retroviral expression construct containing bcl-2 cDNA or a control neomycin resistance gene (697-Bcl-2 and 697-neo cells1, respectively; obtained from Dr. John Reed, Burnham Institute; Miyashita and Reed, 1993) were used in these studies. The cells were maintained in mid-log phase growth in RPMI 1640 medium (Irvine Scientific) supplemented with 10% FBS (Hyclone), 0.2 mg/ml G-418 (GIBCO BRL) and 0.1 mg/ml penicillin/streptomycin (Irvine Scientific). Murine dopaminergic MN9D cells (obtained from Dr. A. Heller, University of Chicago) were grown in MEM (Irvine Scientific) supplemented with 10% FBS, 2 mM glutamine and 0.1 mg/ml penicillin/ streptomycin. Mouse brain cortical cells were prepared at E15 of gestation in Hank's buffered saline solution (Irvine Scientific) with 15 mM Hepes. The tissue was briefly dissociated with 0.1% trypsin and washed thoroughly with MEM supplemented with 10% FBS and 0.4 mg/ml DNase I (Sigma Chemical Co.), gently triturated and flash frozen. The human breast carcinoma cell line T47D was obtained from American Type Culture Collection and cultured as suggested by the manufacturer. MCF-7 cells stably transfected with an expression plasmid coding for procaspase-3 was kindly supplied by Dr. C. Froelich (Northwestern Healthcare Research Institute, Evanston, IL).
Subcellular Fractionation
Frozen cell pellets containing ∼109 cells were thawed and resuspended in cold hypotonic buffer (10 mM Na-Hepes, 5 mM MgCl2, 42 mM KCl, pH 7.4) supplemented with 1 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 5 μg/ml aprotinin, 0.1 mM EDTA, 0.1 mM EGTA, and 5 mM DTT (Sigma Chemical Co.) to a density of ∼1.5 × 108 cells/ml. The samples were incubated on ice for 30 min at which time the cells were lysed using 30–40 strokes with a Dounce homogenizer. The sample was centrifuged twice for 10 min at 500 g, 4°C to separate the nuclei. The nuclear pellets were then washed twice in the same buffer supplemented with 1.6 M sucrose, yielding the nuclear fraction. The supernatant was then centrifuged at 14,000 g for 30 min at 4°C to pellet the heavy membranes. The heavy membranes were washed three times with 1.5 ml cold hypotonic buffer containing protease inhibitors and DTT. The washed membranes were resuspended in hypotonic buffer so that the total protein concentration was ∼2 mg/ml, yielding the heavy membrane fraction, that was either flash frozen or used immediately for enzymatic measurements without freezing. The 14,000 g supernatant was centrifuged at 100,000 g for 30 min at 4°C, yielding a supernatant (cytoplasmic fraction) and a pellet (light membrane fraction). Protein concentrations were measured using Protein Assay Kit II (Bio-Rad Laboratories) with bovine serum albumin as the calibration standard. In some experiments, cell pellets were lysed as above, but without a freezing step. To test effects of cytochrome c on caspase activity, some samples were treated with 10 μg/ml bovine cytochrome c (Sigma Chemical Co.) throughout the entire isolation procedure. In some experiments, mitochondrial fractions were prepared from lysed 697-neo and 697-Bcl-2 cells by the rat liver mitochondrial methods of Mancini and collaborators (Mancini et al., 1998) and used without freezing.
Western Immunoblotting
Subcellular fractions (50 μg protein per lane) were resolved by SDS-PAGE on 12% or 16% gels (Novex) and transferred to Immobilon PVDF membranes (Millipore). Membranes were blocked in PBS and 0.1% Tween 20 (PBST) + 0.4% casein (I-block, Tropix). Blots were incubated in 1 μg/ml primary antibody diluted in PBST/casein for 1 h. After three washes in PBST, blots were incubated for one hour in 1:15,000 dilutions of alkaline phosphatase conjugated goat anti–rabbit IgG or goat anti–mouse IgG (Tropix) in PBST/casein. Blots were then washed twice with PBST, twice in assay buffer (10 mM diethanolamine, pH 10.0, 1 mM MgCl2), and then incubated in 250 μM chemiluminescent substrate CSPD (Tropix) in assay buffer and exposed to Biomax film (Kodak) overnight. In some cases, after the secondary antibody incubations, the blots were washed with 10 mM Tris, pH 9.5, 1 mM MgCl2. The blots were then incubated for 30 min in 1.25 μg/ml DDAO phosphate (Amersham) dissolved in the Tris buffer. The blots were scanned using the STORM fluorescence imager (Molecular Dynamics). The antibodies used were against Bcl-2 (clone 7; Transduction Labs), caspase-3 (Srinivasan et al., 1998), cytochrome c (clone 7H8.2C12; PharMingen), cytochrome oxidase, subunit IV (clone 1A12-A12; Molecular Probes), D4-GDP dissociation inhibitor (D4-GDI; a kind gift of Dr. G. Bokoch, Scripps Research Institute, La Jolla, CA) and poly(ADP-ribose) polymerase (PARP) (clone C2-10; Enzyme Systems).
Immunocytochemistry
T47D human breast carcinoma cells, MCF7 human breast carcinoma cells transduced with a control vector or caspase-3 expression vector (Yang et al., 1998; MCF7/cont and MCF7/casp-3, respectively) were cultured on 8-chamber permanox slides (Nalge Nunc International Corporation). The MCF7/cont and MCF7/casp-3 cells were cultured in separate wells on the same 8-chamber slide. When the cells reached 40–50% confluence, they were fixed in ice-cold 10% formalin for 20 min, washed twice with PBS and immunostained immediately. For immunostaining, fixed cells were incubated for one hour at room temperature in blocking buffer (2% normal goat serum, 2% BSA, 0.2% nonfat milk powder, 0.4% Triton X-100 in PBS). Cells were then incubated with affinity-purified anti-caspase-3 rabbit polyclonal antibody CSP3 (Srinivasan et al., 1998) or purified rabbit IgG (PharMingen; 0.3–1.2 μg/ml), plus anti-cytochrome c mouse monoclonal antibody (clone 6H2.B4; PharMingen, 0.25 μg/ml) diluted in blocking buffer, for 1 h at room temperature. After three 5-min washes in wash buffer (PBS/0.1% Tween 20), cells were incubated for 1 h at room temperature with 0.8 μg/ml each of goat anti–rabbit IgG Alexa 488 conjugate and goat anti–mouse Alexa 594 conjugate (Molecular Probes). Finally, cells were washed three times, 5 min each, in wash buffer. The chamber divisions were removed and the cells were coverslipped under Citiflor mounting fluid (Ted Pella, Inc.). Immunstained cells were visualized by laser scanning confocal microscopy and conventional fluorescence microscopy; procaspase-3 and cytochrome c immunostaining were visualized with FITC and Texas red filters, respectively. The confocal images are single 0.4-μm optical sections.
Enzyme Activity and Inhibition Studies
Caspase activity was measured by mixing 50 μl of an enzyme-containing fraction and 200 μl of 25 μM acetyl-Asp-Glu-Val-Asp-aminomethylcoumarin(acDEVD-amc) substrate in ICE buffer (20 mM Hepes, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, 5 mM DTT, pH 7.5) in duplicate 96-well Cytoplate wells (Perseptive Biosystems). Product formation was monitored by the increase in fluorescence (ex = 360 nm, em = 460 nm) over 1–2 h at 30°C using the CytoFluor 4000 plate reader (Perseptive Biosystems). For kinetic studies, the substrate concentration was varied in the range 1–100 μM. For inhibition studies the enzyme was pretreated with 150 μl inhibitor for 30 min at room temperature before the addition of 50 μl of 50 μM substrate solution. Inhibitor IC50 values were determined using the equation:
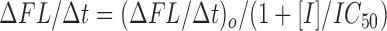 |
where ΔFL/Δt is the observed initial rate of fluorescence change at inhibitor concentration [I] and (ΔFL/Δt)o is the initial rate fluorescence change for the uninhibited enzyme.
Caspase Activation
Heavy membrane samples were diluted to 1 mg/ml in hypotonic buffer or in 0.25 M sucrose, 10 mM MOPS, 2 mM EDTA, pH 7.4 (Mancini et al., 1998) containing 5 mM DTT with or without 1% NP-40. Caspase activation was induced by adding either 60–160 ng/ml recombinant murine caspase-1 (in bacterial lysate), 2 μg/ml of purified human granzyme B (Enzyme Systems Products) or buffer, and incubating the samples for 60 min at 30°C or 37°C. After the activation period, the heavy membrane pellet was removed from the sample by centrifugation for 10 min at 14,000 g at 4°C. The acDEVD-amc cleaving activities in the resulting supernatants were corrected for the activity of the exogenous enzymes. To examine the time course of spontaneous activation of caspase activity from membranes, 50 μl of heavy membrane slurry containing 50–100 μg total protein was mixed with 200 μl hypotonic buffer containing 25 μM acDEVD-amc substrate and 6 mM DTT in 96-well Cytoplates and fluorescence was measured over time. At selected time points, aliquots were removed from some wells, centrifuged for 10 min at 14,000 g to remove the heavy membranes, and then the supernatant was added back into the 96-well plate to measure the soluble acDEVD-amc cleavage activity. In some experiments, subcellular fractions were treated with 1 μg/ml bovine cytochrome c (Sigma Chemical Co.) and 50 μM dATP (New England Biolabs) for 40 min at 30°C before measurement of caspase activity.
Production of Recombinant Caspase-1 and Caspase-3 Proteins
BL21 (DE3) cells harboring a plasmid containing the cloned human caspase-3 cDNA (Fernandes-Alnemri et al., 1994; provided by Dr. E. Alnemri, Thomas Jefferson University, Philadelphia, PA) ligated into the BamHI/XhoI sites of pET21b (Novagen) were grown in one liter LB medium containing 0.1 mg/ml ampicillin at 37°C. When the culture density reached A600 = 1, IPTG (Sigma Chemical Co.) was added to a concentration of 1 mM and the culture was incubated at 25°C for 3 h. The cells were harvested by centrifugation at 2,000 g for 15 min at 4°C. The cells were lysed using one freeze-thaw cycle in 100 ml binding buffer (20 mM Tris-HCl, 500 mM NaCl, 5 mM imidazole, 0.1% Triton X-100) with 0.1 mg/ml lysozyme. Cell debris was removed from the sample by centrifugation at 20,000 g, for 30 min at 4°C. The lysed cells were treated just before centrifugation with 0.5 mM MgCl2 and 2 μg/ml DNase I (Sigma Chemical Co.) to reduce viscosity. The supernatant was filtered through a 0.45-μm syringe filter and loaded onto a 1 ml Ni2+-charged HiTrap chelating column (Amersham Pharmacia) at a 1 ml/min flow rate. The column was washed at 1 ml/min with 10 ml binding buffer followed by 10 ml binding buffer containing 60 mM imidazole. The caspase-3 protein was eluted from the column using a 30-ml linear gradient of imidazole (60–500 mM).
Recombinant murine caspase-1 was expressed using BL21 (DE3) pLys S cells harboring pET3ap30mICEFLAG plasmid (a generous gift of Drs. H.R. Horvitz and Ding Xue, Massachusetts Institute of Technology) which contains the p30 caspase-1 cDNA inserted into the NdeI/BamHI sites of the pET3a expression vector (Novagen). A 3-liter culture was grown at 37°C in induction medium (20 g/liter tryptone, 10 g/liter yeast extract, 6 g/liter NaCl, 3 g/liter Na2HPO4, 1 g/liter KH2PO4, 1 mM MgCl2, 0.1 mM CaCl2, pH 7.4) containing 0.1 mg/ml ampicillin and 0.025 mg/ml chloramphenicol. When the culture reached a density of A600 = 1.0, IPTG was added to 1 mM and the culture was shaken at 25°C for 3 h. The cells were collected by centrifugation at 2,000 g for 15 min at 4°C and resuspended in 100 ml cold buffer containing 25 mM Tris-HCl, pH 8.0, 25 mM KCl, 0.1% Triton X-100, and 0.1 mg/ml lysozyme (InovaTech). The cells were lysed using one freeze/thaw cycle and the lysate was clarified by treating the sample with 2 μg/ml DNase I, 0.5 mM MgCl2 for 60 min and then centrifuging at 20,000 g for 30 min at 4°C to remove cell debris.
Results
Characterization of Subcellular Fractions from 697 Cells
Subcellular fractions were prepared from 697 cells stably infected with retroviral constructs expressing either bcl-2 cDNA or a neomycin resistance gene (697-Bcl-2 and 697-neo cells, respectively; Miyashita and Reed, 1993). Nuclear, heavy membrane, light membrane, and cytosolic fractions were isolated from these cells, and were characterized by Western blot analysis with antibodies specific for proteins with distinct known subcellular distributions. Antibodies used were directed against cytochrome oxidase, specific for mitochondrial inner membrane (Tzagoloff, 1982), PARP, specific for nuclei (Berger, 1985), D4-GDP dissociation inhibitor (D4-GDI), specific for cytoplasm (Na et al., 1996) and Bcl-2. As shown in Fig. 1, the mitochondrial marker was found almost exclusively in the heavy membrane fraction, the nuclear marker only in the nuclear fraction, and the cytoplasmic marker only in the cytoplasmic fraction. Thus, the fractionation methods used generated fractions with the expected subcellular distribution of marker proteins. Importantly, we could not detect cytoplasmic contamination of the nuclear and membrane fractions, and detected only minimal mitochondrial contamination of nuclear fractions (the diffuse D4-GDI reactive band in the nuclear fraction shown in Fig. 1 is nonspecific). Western analysis of fractions from 697-neo cells with an antibody to human Bcl-2 (Fig. 1) demonstrated strong reactivity in nuclear and heavy membrane fractions, weaker reactivity in the light membrane fraction, and undetectable signal in cytoplasm, in accord with previous results (Krajewski et al., 1993; Yang et al., 1995; Lithgow et al., 1994). Similar analysis of fractions from 697-Bcl-2 cells showed significant overexpression.
Figure 1.

Immunoblots of subcellular fractions from 697-neo and 697-Bcl-2 cells using antibodies to PARP, cytochrome oxidase (subunit IV), D4-GDI and Bcl-2. The immunoblots were visualized on film by chemiluminescence, except the cytochrome oxidase immunoblot which was visualized by chemifluorescence. Nuc, nuclear fraction; HM, heavy membrane fraction; LM, light membrane fraction; S100, cytosolic fraction. Arrows indicate the specific immunoreactive band. The diffuse signal present in the nuclear fractions probed with the D4-GDI antibody represents a non-specific background band.
Subcellular Distribution of acDEVD-amc Cleavage Activity in 697 Cells
Preliminary experiments indicated that caspase activity was associated with membranes derived from unstimulated cells. To determine the subcellular distribution of such caspases, we quantitated the caspase activity in the subcellular fractions from 697-neo cells by incubating them with the substrate acDEVD-amc, and measuring the increase in fluorescence over the subsequent 2 h. acDEVD-amc is a useful substrate for all caspases characterized to date, with the exception of caspase-2 (Talanian et al., 1997; data not shown). While most of the acDEVD-amc cleavage activity (∼75%) was in the cytoplasmic fraction, a substantial amount of the cleavage activity was found in the nuclear, heavy membrane and light membrane fractions (Fig. 2, a and c). The major acDEVD-amc cleaving activity in each fraction was indeed caspase activity since it was potently blocked by specific caspase inhibitors (Table I, column 1, and data not shown).
Figure 2.

acDEVD-amc cleavage activity in subcellular fractions of 697 cells. 697 cells transfected with neo control or Bcl-2 expression vectors were fractionated and the caspase activity of each subcellular fraction was assayed using acDEVD-amc as substrate. The observed cleavage activity values in the histogram are normalized for constant number of cells (A and B) or mg protein (C and D). The values listed for each column in A and B indicate the percent of total cleavage activity present in each fraction. The error bars indicate the range of observed values for two independent 697 cell preparations.
Table I.
Heavy Membrane (HM)-derived Caspases from Various Cell Types and Recombinant Human Caspase-3: Inhibition by Peptide Aldehydes
| IC50 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inhibitor | 697-neo HM (spontaneous activity) | 697-neo HM (caspase-1 treated) | 697-Bcl-2 HM (caspase-1 treated) | Cortical cell HM (caspase-1 treated) | MN9D HM (caspase-1 treated) | r-caspase-3 (His)6 | ||||||
| DEVD-ald | 2.3 | 2.8 | 1.3 | 1.0 | 0.72 | 1.0 | ||||||
| DFLD-ald | 3.4 | 4.5 | 3.6 | 2.3 | 2.5 | 1.5 | ||||||
| YVAD-ald | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 | ||||||
Bcl-2 Suppresses Membrane-derived Caspase Activity
Next, we examined the effect of Bcl-2 on the caspase activities in the various subcellular fractions. When subcellular fractions derived from 697-Bcl-2 cells were prepared and incubated with acDEVD-amc substrate, substantially reduced caspase activity was observed in the nuclear and heavy membrane fractions compared with 697-neo cells (Fig. 2 b). This Bcl-2 effect was evident when the caspase activity was measured on a per cell basis or per mg protein and resulted in an 80–90% reduction in caspase activity in these fractions (Fig. 2, b and d). The effect of Bcl-2 expression on caspase activity in these fractions was specific, since little if any suppression was seen in the activities observed in the cytoplasmic or light membrane fractions (Fig. 2, a–d). These observations suggested that the membrane-associated caspase activity was not simply derived from a small percentage of apoptotic cells in the 697-neo cultures whose numbers were suppressed in the 697-Bcl-2 cultures. If that were the case, we would also have expected to see major differences in caspase activities between cytoplasmic fractions derived from 697-neo vs. 697-Bcl-2 cells. Indeed, control experiments demonstrated that when 697-neo cells were induced to undergo apoptosis by staurosporine treatment, the major increase in caspase activity was found in the cytoplasm (data not shown). The ability of Bcl-2 to suppress membrane-associated caspase activity was not limited to the 697 lymphoblastoid cells, since similar effects were observed in Jurkat T cells and FL5.12 cells (data not shown). Since our data, as well as other published studies, have demonstrated that Bcl-2 protein is found predominantly in nuclear envelope and heavy membrane fractions (Fig. 1; Krajewski et al., 1993; Yang et al., 1995), our results were compatible with the possibility that Bcl-2 might act locally to regulate this membrane-derived caspase activity. In an effort to begin analyzing such mechanisms, we further characterized this membrane-derived, Bcl-2–suppressible caspase activity and focused our efforts on the heavy membrane fraction.
Membrane-derived Caspase Activity Reflects Spontaneous Activation and Membrane Release
It was possible that the membrane associated caspase activity was due either to an active membrane-bound enzyme, or alternatively, to the spontaneous activation and release of a soluble active enzyme. We therefore designed a set of experiments to distinguish between these two possibilities. First, to freshly prepared heavy membranes derived from 697-neo cells (neo-membranes), we immediately added hypotonic buffer and acDEVD-amc substrate at room temperature, and measured the emergence of amc fluorescence over a 90-min period (Fig. 3 a). The data demonstrate that there is little detectable fluorescence change over the first 15 min of incubation, but after this lag period, the rate of amc production increases markedly (Fig. 3 a). These results indicated that the freshly prepared membranes did not contain active caspase, but that activation occurred spontaneously during the incubation period. To assess whether this newly activated caspase was soluble or membrane bound, membranes were incubated for different periods of time, after which the samples were centrifuged and the resulting supernatants were assayed for caspase activity with acDEVD-amc substrate. These data demonstrated that very little caspase activity was present in the supernatant initially, but that soluble caspase activity appeared thereafter (Fig. 3 b). Quantitative analysis of these data demonstrated that for each supernatant, fluorescence increased linearly, indicating that once released from the membranes, no further activation occurred. Furthermore, the slopes of these curves (Fig. 3 b) approximate the instantaneous slopes of the corresponding time points in the progress curve for the heavy membrane slurry (Fig. 3 a). Therefore, all of the caspase-3 activity can be accounted for in the supernatant fraction, indicating that all active enzyme had been released from the membranes. In contrast to the neo-membranes, membranes derived from the 697-Bcl-2 cells (Bcl-2-membranes) failed to generate significant acDEVD-amc cleaving activity (Fig. 3 a).
Figure 3.


Spontaneous activation of membrane-associated procaspase-3. (A) Spontaneous activation of caspase activity in heavy membranes from 697-neo and Bcl-2 cells. The acDEVD-amc cleavage activity of was measured by adding 20 μg of freshly prepared membranes into hypotonic buffer containing 20 μM acDEVD-amc (final concentration). The evolution of amc product was measured by the change in fluorescence (ex, 360 nm; em, 460 nm) at room temperature. (B) Generation of soluble caspase activity from neo-membranes. Neo-membranes were added to hypotonic buffer containing acDEVD-amc. At the indicated time points from 0 to 90 min (right hand box), the sample was centrifuged for 10 min at 14,000 g at 10°C to remove the membranes. The acDEVD-amc cleavage activity of the supernatant was measured and is plotted as the increase in fluorescence over the subsequent 30-min period after centrifugation for each time point.
Procaspase-3 Is Present in Heavy Membranes from Both 697-neo and 697-Bcl-2 Cells
The lack of acDEVD-amc cleaving activity in the Bcl-2-membranes could be due either to the absence of activatable procaspase or suppression of procaspase activation. To distinguish between these alternatives, we first performed Western blot analysis on the membrane fractions with antibodies specific for caspase-3, since the measured acDEVD-amc cleavage activity is in fact due to caspase-3 (see below). The results (Fig. 4 a) demonstrate the presence of a caspase-3 reactive band that is of similar intensity in both the neo-membranes and Bcl-2-membranes, and that is approximately the size expected for the procaspase zymogen. Interestingly, the electrophoretic mobility of the membrane-derived bands was slightly slower than that of cytoplasmic procaspase-3. To further demonstrate the presence of procaspase-3 in both neo- and Bcl-2-membranes, we attempted to activate these fractions by treatment with exogenous caspase-1, since procaspases can be activated by proteolytic cleavage at aspartic acid residues between their large and small subunits (Srinivasula et al., 1996; Stennicke and Salvesen, 1997; Salvesen and Dixit, 1997). As we have shown above, membranes derived from Bcl-2 cells showed almost no caspase activity when measured under our standard conditions. However, treatment of the Bcl-2-membranes with caspase-1 caused a robust induction of enzymatic activity (Fig. 4 b). The neo-membranes were also activated by exogenous caspase-1. But importantly, after activation, the resulting caspase activities from the Bcl-2- and neo-membranes were always similar, within a factor of two (Fig. 4 b). Together with the procaspase-3 immunoblot data, this supports the conclusion that comparable levels of procaspase-3 are present in neo- and Bcl-2-membranes.
Figure 4.


Neo-membranes and Bcl-2-membranes contain similar amounts of procaspase-3. (A) Immunoblot of heavy membrane and cytosolic fractions from 697-neo and 697-Bcl-2 cells using an affinity-purified rabbit polyclonal antibody to caspase-3. The arrowheads indicate the migration of protein size markers (Rainbow Markers; Novex); the arrow indicates procaspase-3. HM, heavy membrane fractions; S100, cytosolic fraction. Note: The immunoreactive procaspase-3 band in heavy membrane fractions migrates more slowly than the cytosolic form of the protein. (B) Activation of membrane-associated acDEVD-amc cleavage activity by exogenous caspase-1. Heavy membrane fractions (containing 50 μg total protein) from 697-Bcl-2 and 697-neo cells were resuspended and treated with murine caspase-1 or buffer for 1 h at room temperature. After centrifugation, the acDEVD-amc cleavage activity of the resulting supernatant was measured. The acDEVD-amc cleavage activity of caspase-1–treated samples was corrected for exogenous caspase-1 activity by subtracting the fluorescence of control samples containing only caspase-1 from the observed fluorescence. The error bars represent the standard deviation of the observed values in three independent experiments.
Caspase-1-treatment of membranes not only activated the endogenous caspase activity, but also released it from the membranes, since the activity remained in the supernatant when the membranes were removed by centrifugation (Fig. 4 b). This induction and release were due to the proteolytic activity of caspase-1, since the caspase-1 activation could be completely blocked by 200 nM acYVAD-aldehyde which inhibits caspase-1, but not the membrane caspase, at this concentration (data not shown). Our results indicate that both neo- and Bcl-2–expressing cells contain similar amounts of a membrane-associated inactive procaspase that can be activated by caspase-1. However, without exogenous caspase treatment, only membranes derived from the neo-expressing cells demonstrated spontaneous caspase activation.
To further document the presence of procaspase-3 in heavy membrane fractions we performed immunocytochemical studies, using adherent cell lines for ease of experimentation. First, we demonstrated that our affinity-purified antibody CSP3, generated against recombinant caspase-3 and used in our Western blots (Fig. 4 a; Srinivasan et al., 1998), was specific when used as an immunocytochemical reagent. Our staining results showed that the antibody did not react with MCF7/cont breast carcinoma cells (Fig. 5 f) which lack procaspase-3 due to a genetic deletion (Li et al., 1997a; Janicke et al., 1998). However, the antibody showed intense staining when reacted with MCF7/casp-3 cells overexpressing recombinant procaspase-3 (Fig. 5 d). To analyze the distribution of endogenous procaspase-3 in an untransfected cell line we used T47D breast carcinoma cells. The caspase-3 antibody demonstrated both diffuse and punctate staining (Fig. 5 a). Much of the punctate staining colocalized with mitochondria, as visualized by anti–cytochrome c antibody (Fig. 5, b and c). Nonspecific purified rabbit IgG did not stain these cells (Fig. 5 h). These results confirm that procaspase-3 immunoreactivity associates with heavy membrane elements in cells, as was also shown using other cell types (Mancini et al., 1998).
Figure 5.

Procaspase-3 immunoreactvity partially colocalized with mitochondria. T47D cells (a–c, h, and i) were double-labeled with antibodies to procaspase-3 and cytochrome c and visualized by confocal microscopy (0.4-μm optical sections). These cells show both diffuse and punctate staining with anti–procaspase-3 antibody (a). The punctate staining largely colocalizes with anti–cytochrome c staining (b, anti–cytochrome c; c, merged image from a and b). This staining is specific as shown by the lack of staining with control rabbit IgG (h), whereas anti–cytochrome c demonstrated the presence of several cells (i). The specificity of the procaspase-3 antibody is further demonstrated by the lack of staining of MCF7/cont cells that lack procaspase-3 (f), although anti–cytochrome c staining demonstrated the presence of several cells (g). However, MCF7/casp-3 cells that overexpress procaspase-3 show intense staining with anti–procaspase-3 (d) as well as with anti–cytochrome c (e). d–g are conventional fluorescence microscope images. Scale markings are in microns.
Enzymological Characterization of the Induced and Spontaneous Caspase Activities
We further characterized the membrane-derived caspase activities by measuring the inhibition of acDEVD-amc cleavage by several peptide aldehyde inhibitors (Table I). The IC50 values for the inhibition of acDEVD-amc activity derived from activated Bcl-2 membranes are quite similar to those for the inhibition of the activity derived from neo-membranes, suggesting that caspase-1 activates the same procaspase in both membrane preparations. Furthermore, these IC50 values are similar to those for the spontaneously activated acDEVD-amc activity derived from neo-membranes, suggesting that the spontaneous and caspase-1–induced activities derive from the same caspase. In all cases, the inhibition data fit well to a simple competitive inhibition curve as described in the Materials and Methods, suggesting that each acDEVD-amc activity arose from a single caspase rather than a mixture of enzymes. The observed IC50 values for the membrane associated caspases are very similar to those for purified fully processed recombinant human caspase-3. Kinetic measurements also indicate that Km values for hydrolysis of acDEVD-amc by the membrane-derived caspases (10 μM) are similar to that observed with fully processed caspase-3 (Nicholson et al., 1995). NH2-terminal microsequence analysis of activated, affinity-purified heavy membrane caspase confirms that this enzyme is indeed human caspase-3 (manuscript in preparation).
To determine if the presence of membrane-associated caspase activity is a general property of mammalian cells, we measured the acDEVD-amc cleavage activity in heavy membranes from two other cell sources: mouse E15 primary brain cortical cells and the mouse dopaminergic MN9D cell line (Choi et al., 1992). Heavy membrane fractions were prepared using identical procedures to those used for the 697 cells and were activated with caspase-1. These fractions contained a membrane-associated caspase activity with similar cleavage activities per mg protein as observed in 697 cells (data not shown) and that was blocked by caspase inhibitors with a similar potency to that observed with fractions derived from 697 cells or with recombinant caspase-3 (Table I). We conclude that the existence of membrane-derived caspase activity is not specific to 697 cells, but appears to be a more general phenomenon.
Addition of Exogenous Cytochrome c Does Not Activate Membrane-associated Procaspase-3
Several recent reports have shown that the release of cytochrome c from mitochondria can cause the activation of cytoplasmic caspase-3 (Liu et al., 1996; Li et al., 1997a). Other reports have demonstrated that cytochrome c is released from mitochondria after apoptotic insults and that Bcl-2 can inhibit that release (Kluck et al., 1997; Yang et al., 1997). Thus it was possible that the difference we observed between caspase activities in heavy membranes from Bcl-2– and neo-expressing cells simply reflected inhibition by Bcl-2 of cytochrome c release during preparation of the heavy membrane fractions or during subsequent incubation of these fractions. To investigate this possibility, we performed cell fractionation in the presence of exogenous cytochrome c and measured whether this influenced caspase activation. If the Bcl-2-membranes had low caspase activity because of a Bcl-2 effect on cytochrome c sequestration, then the addition of exogenous cytochrome c during membrane fractionation should increase the caspase activity derived from those membranes to the levels seen in membranes from neo-cells. Accordingly, during the fractionation procedure for heavy membranes from neo- and Bcl-2–expressing cells, we added 10 μg/ml cytochrome c to the cell lysate immediately after homogenization, and 10 μg/ml to the buffers used to suspend and wash the heavy membranes. This concentration of cytochrome c was chosen since it represents the estimated total amount of cytochrome c present in the starting cell pellets (Li et al., 1997a). Finally, these membranes were resuspended in 1 μg/ml cytochrome c plus 50 μM dATP, incubated, and then assayed for acDEVD-amc cleaving activity (Fig. 6 a). This activity was compared with that from our usual membrane preparations prepared without cytochrome c, and incubated without cytochrome c or dATP. The data demonstrate that inclusion of cytochrome c during membrane fractionation and incubation has no effect on membrane-derived caspase activity; the activity in the membranes derived from Bcl-2–expressing cells remained low compared with the activity in the neo-membranes, and furthermore, there was also no effect of cytochrome c on the caspase activity derived from the neo-membranes (Fig. 6 a). Although the cytochrome c treatments did not activate the membrane-associated caspase, the enzyme could still be activated by subsequent treatment with exogenous caspase-1 (data not shown). The lack of a cytochrome c effect on the activation of the membrane caspase was not due to an inactive preparation of cytochrome c, since the acDEVD-amc cleavage activity of the cytoplasmic fractions from both neo and Bcl-2 cells were strongly activated by inclusion of cytochrome c during fractionation and assay (Fig. 6 b). We conclude that Bcl-2 expression suppresses the activation of the membrane-associated procaspase-3, but that this effect is not overcome by addition of exogenous cytochrome c. Furthermore, Bcl-2 overexpression did not affect the ability of cytochrome c to activate caspase-3 in cytoplasmic fractions.
Figure 6.


Membrane-associated caspase activation is not stimulated by exogenous cytochrome c. Subcellular fractions were prepared from 697-neo and 697-Bcl-2 cells. After the cells were lysed by Dounce homogenization, the sample was split into two tubes. One tube was processed using standard buffers, while bovine cytochrome c was added to the other (10 μg/ml final concentration), and cytochrome c was maintained in that sample throughout membrane isolation including the heavy membrane pellet wash steps. Aliquots of the cytochrome c–treated heavy membranes and cytoplasmic fractions were then incubated with hypotonic buffer containing 50 μM dATP/1 μg/ml cytochrome c for 40 min at 30°C, while the membranes and cytoplasmic samples that had not been treated with cytochrome c were incubated only with buffer. Each sample was then centrifuged and acDEVD-amc cleavage activity in the supernatant was measured. The graphs represent data from one out of three equivalent experiments. (A) Heavy membrane–derived caspase activities. (B) Cytoplasmic caspase activities.
Release of Membrane-associated Caspase Activity Is Not Due to Simple Leakage from Organelles
A recent report described the presence of procaspase-3 in the intermembrane space within mitochondria (Mancini et al., 1998). Thus, it was possible that this material could account for the activatable caspase activity that we measured in our mitochondria-containing heavy membrane fractions. Furthermore, it was possible that the spontaneous activity that we measured in membrane fractions from 697-neo cells was due to leakage of active caspase from mitochondria, and that mitochondria isolated from 697-Bcl-2 cells were simply less leaky (Yang et al., 1997). However, several experiments suggested that the activity we measured was not due to leakage from mitochondria, and that the activity is distinct from that described by Mancini et al. (1998).
First we tested whether the addition of 1% NP-40 to neo-membranes affected the level of either spontaneous activity or the activity induced by caspase-1 or granzyme B. We reasoned that if procaspase and/or active caspase was sequestered within organelles, then enhanced activity would be measured in the presence of NP-40. Treatment with 1% NP-40 was sufficient to release almost all of the cytochrome c present in heavy membrane preparations (data not shown). Furthermore, it was shown by Mancini and colleagues that treatment of their mitochondrial preparations with 1% NP-40 allowed granzyme B to cleave procaspase-3 whereas no cleavage was observed in the absence of detergent (Mancini et al., 1998). However, our results demonstrate that 1% NP-40 had little effect either on spontaneous activity or the activity induced by treatment with caspase-1 or granzyme B (Fig. 7 a). Next, to analyze whether membrane preparations from 697-Bcl-2 cells may have low spontaneous activity due to enhanced sequestration of a caspase, we added acDEVD-amc to Bcl-2- and neo-membrane preparations, incubated them in buffer alone or buffer plus 1% NP-40, and measured the appearance of fluorescence (Fig. 7 b). The results indicate that 1% NP-40 had only a minor effect on the magnitude or rate of fluorescence increase. Preparations derived from 697-Bcl-2 cells had low activity regardless of whether 1% NP-40 was present, demonstrating that this low level of activity was not due to sequestration of an active caspase. Finally, we prepared mitochondrial fractions from 697-neo and 697-Bcl-2 cells using the methods described by Mancini et al. (1998) to more directly assess the relationship between our results and their published data. As shown in Fig. 7 c, fractions from both 697-neo and 697-Bcl-2 made by these methods have granzyme B–activatable caspase activity in the absence of NP-40. However, in the presence of 1% NP-40, granzyme B treatment yielded enhanced caspase activity (Fig. 7 c). Thus, under these conditions, granzyme B generates caspase activity in both NP-40–independent and –dependent manners.
Figure 7.



Effects of permeabilizing detergent NP-40 on membrane caspase activity. (A) NP-40 has minimal effects on spontaneous and induced caspase activities in neo-membranes. 160 μl of neo-membranes were diluted with 180 μl hypotonic buffer and treated with 40 μl 10% NP-40 detergent or dH2O (final vol = 380 μl). The diluted membranes were activated by the addition of 20 μl granzyme B or caspase-1 lysate or buffer, and incubated for 60 min at 30°C. After activation, the heavy membranes were removed by centrifugation and the acDEVD-amc cleaving activity of each sample was measured by adding 50 μl of each supernatant to 200 μl of 25 μM acDEVD-amc substrate in ICE buffer. (B) NP-40 has minimal effects on spontaneous caspase activation in Bcl-2- and neo-membranes. The effect of NP-40 on the progress curve for heavy membrane catalyzed acDEVD-amc hydrolysis was measured by adding 50 μl freshly prepared neo- or Bcl-2-membranes to 200 μl of 25 μM acDEVD-amc in hypotonic buffer pH 7.5 (containing 4 mM DTT) with or without 1% NP-40 detergent. Fluorescence was measured as in Fig. 3 a. (C) NP-40– dependent and –independent activation of procaspase-3 by granzyme B in fractions enriched in intact mitochondria. Mitochondrial fractions were prepared from lysed 697-neo and 697-Bcl-2 cells by the methods of Mancini et al. (1998) using isotonic buffers (see Materials and Methods). Diluted membranes, with or without 1% NP-40, were activated by the addition of granzyme B or buffer for 60 min, centrifuged, and assayed for acDEVD-amc cleavage activity as described in A.
Discussion
The present work was motivated by genetic and biochemical studies that suggested that the Bcl-2 homologue CED-9 functions by regulating the activity of the caspase CED-3 through protein-protein interactions (Horvitz et al., 1994; Chinnaiyan et al., 1997; Ottilie et al., 1997; Seshagiri and Miller, 1997; Spector et al., 1997; Wu et al., 1997). Given that Bcl-2 and the related death-inhibiting protein Bcl-xL are both localized to intracellular membranes (Krajewski et al., 1993; González-Garcia et al., 1994), we reasoned that these molecules may act locally to regulate a membrane-associated caspase. As an initial step in investigating this hypothesis, we first demonstrated that intracellular membrane fractions do in fact contain an activatable procaspase, which when characterized, was shown to be caspase-3. Immunocytochemical evidence further supported the conclusion that heavy membrane components contain procaspase-3 (Fig. 5). Quantitatively, the amount of membrane-derived caspase activity is relatively small compared with that in the cytoplasmic fraction; the total acDEVD-amc cleavage activity in the heavy membrane fraction was generally ∼5–10% of the total cytoplasmic activity in unstimulated cells (Fig. 2). In cells stimulated to undergo apoptosis, there are increases in caspase activity in both membrane and cytoplasmic fractions, but the percentage that is membrane-associated remains low (data not shown). However, our results suggest that the pool of membrane-associated caspase is uniquely regulated by Bcl-2. Heavy membrane and nuclear fractions derived from 697-Bcl-2 cells demonstrated only low levels of spontaneous activation of caspase activity compared with similar fractions derived from control 697-neo cells (Figs. 2, 3, and 7 b). The heavy membranes from the Bcl-2–expressing cells did, however, contain appreciable amounts of procaspase-3, measured in two ways: directly by Western analysis (Fig. 4 a), and indirectly, by measurement of acDEVD-amc cleaving activity after activation by exogenous caspase-1 (Fig. 4 b). This demonstrated that Bcl-2 was not affecting the ability of procaspase-3 to associate with membranes, but rather, it exerted specific control over enzyme activation. The Western blot analysis demonstrated that the membrane-associated procaspase-3 had an electrophoretic mobility distinct from that of cytoplasmic procaspase-3. However, we do not yet know the biochemical basis for this observation.
There are many possible mechanisms by which procaspase-3 could become associated with heavy membranes. It has recently been shown that soluble procaspase-3 is present within the mitochondrial intermembrane space (Mancini et al., 1998), and thus it was possible that this material represented the Bcl-2–regulated activity that we observed in our heavy membrane fractions. However, whereas the procaspase sequestered in the intermitochondrial space is protected from activation by exogenous granzyme B (Mancini et al., 1998), we observed that heavy membrane caspase activity in our preparations is readily activated by exogenous caspase-1 (Fig. 4 b) or granzyme B (Fig. 7). In addition, during the course of membrane-associated procaspase activation, active enzyme is continuously released into the medium (Fig. 3) in accord with the idea that a bound, inactive procaspase is converted to a soluble, active enzyme. Addition of the membrane-permeabilizing detergent NP-40 to our standard heavy membrane fractions had only a minimal effect on either spontaneous or caspase-1 activated caspase activity (Fig. 7, a and b), suggesting that these activities were not sequestered within organelles. Thus, we favor the hypothesis that the Bcl-2–regulated procaspase-3 is physically associated with membranes and not simply soluble within a sequestered compartment, mitochondrial or other. However, we did observe that addition of NP-40 enhanced the granzyme B–activated caspase activity when fractions were prepared using methods designed to isolate intact mitochondria (Fig. 7 c). Thus, it is possible that mitochondria may contain two pools of procaspase, one that is accessible to activators only with detergent, and one accessible without detergent.
Caspase activity rises dramatically in the cytoplasm of cells induced to undergo apoptosis and the generation of this activity is blocked by various agents, including Bcl-2, that inhibit apoptosis (Armstrong et al., 1996, 1997; Chinnaiyan et al., 1996; Srinivasan et al., 1996). Thus, we considered whether the inhibition of caspase activation by Bcl-2 in heavy membranes was an indirect consequence of general apoptotic inhibition. For example, the caspase activity in neo-membranes could simply reflect the existence of more apoptotic cells in unstimulated 697-neo cultures vs. 697-Bcl-2 cultures. However, two observations argue against this interpretation. First, the inhibition of caspase activity by Bcl-2 in unstimulated cultures was only seen in heavy membrane and nuclear fractions. However, in 697 cells induced to undergo apoptosis by staurosporine treatment, or in Jurkat cells stimulated with anti-Fas antibody, the largest increase in caspase activity is seen in the cytoplasmic fraction, and this is blocked by Bcl-2 (data not shown). The absence of an effect of Bcl-2 on the cytoplasmic caspase activity in unstimulated cells suggests that the difference between activity in the neo-membranes and Bcl-2-membranes is not simply a passive consequence of more apoptotic cells in the 697-neo cultures. Note, however, that although the number of apoptotic cells in resting 697-neo cultures is low (<5%), this represents generally two to three times more apoptotic cells than in 697-Bcl-2 cultures, as measured by Hoechst dye or annexin V staining (data not shown). Second, the activity that we measured in neo-membranes does not reflect procaspases that were activated during prior apoptotic events since these membranes do not contain pre-existing active caspase; rather, the activity is generated during subsequent incubation (Fig. 3). However, it is possible that the ability to autoactivate heavy membrane caspase is a specific property of a subpopulation of pre-apoptotic cells. A recent report suggests that caspases activated in the cytoplasm during apoptosis can subsequently become membrane bound (Chandler et al., 1998) but this phenomenon does not appear to account for our observation of an activatable procaspase in membranes derived from unstimulated cells.
Another striking difference between membrane bound and cytoplasmic caspase-3 relates to activation by cytochrome c. The membrane-associated procaspase showed no activation in the presence of exogenous cytochrome c, even when the cytochrome c was present throughout the membrane isolation procedure and in the caspase assay buffers. It is unlikely that the failure of cytochrome c to activate this caspase was due to an inability of the cytochrome c to obtain access to the procaspase, since activation was readily effected by exogenous granzyme B. It was possible that cytochrome c failed to enhance caspase activation in neo-membranes because endogenous cytochrome c was saturating the activation mechanism. However, this is also unlikely because NP-40 treatment released endogenous cytochrome c but did not inhibit caspase activation (Fig. 7, a and b). In contrast, cytoplasmic procaspase-3 could be robustly activated by cytochrome c when added either during fractionation, during assay, or both (Fig. 6 and data not shown). Note also that cytochrome c effectively activated cytoplasmic caspases regardless of whether the cytoplasm was derived from neo- or Bcl-2–expressing cells, in accord with published data indicating that Bcl-2 functions upstream of cytochrome c–induced caspase activation (Kluck et al., 1997; Duckett et al., 1998). Thus, our results imply the existence of two distinct procaspase-3 activation mechanisms: a Bcl-2–regulated pathway specific to membranes and insensitive to exogenous cytochrome c; and a cytoplasmic activation pathway, directly activated by cytochrome c, but which is not directly Bcl-2–regulated. However, the activation of the cytoplasmic procaspase-3 is indirectly controlled by Bcl-2, since Bcl-2 blocks all downstream caspase events associated with apoptotic stimuli (Armstrong et al., 1996; Chinnaiyan et al., 1996; Boulakia et al., 1996).
The fact that the heavy membrane procaspase-3 from control cells becomes active spontaneously implies that it undergoes proteolytic cleavage by some protease also present in the membranes. It is possible that this cleavage occurs through autoactivation without the intervention of a separate activating protease, but this may be unlikely; procaspase-3 in the cytoplasm does not self-activate, but requires a first cleavage by caspase-9 before a second autocatalytic step (Martin et al., 1996; Li et al., 1997b). Thus, analogous to the caspase-9/procaspase-3 activating mechanism described in the cytoplasm, we speculate that the heavy membrane and nuclear fractions also contain an activating caspase capable of cleaving the membrane-associated procaspase-3. This membrane-associated activating caspase could be a form of caspase-9, or perhaps another caspase, that is regulated by Bcl-2.
The specific effect of Bcl-2 on the activation of the membrane-associated caspase suggests that this caspase may play an important role in controlling apoptosis. We speculate that the membrane-associated caspase might function as a specific trigger to promote downstream apoptotic activity leading ultimately to cytoplasmic caspase activation. If so, this would place Bcl-2 at a critical control point, regulating the trigger, and thereby inhibiting diverse apoptotic events in cells.
Acknowledgments
We would like to thank Drs. E. Alnemri, C. Froelich, H.R. Horvitz, J. Reed, and D. Xue for cDNA clones and cell lines, E. Monosov for help with confocal microscopy, and Lisa Trout and Chris Knowles for invaluable assistance in the preparation of this manuscript.
Abbreviations used in this paper
- 697-neo cells
697 cells stably infected with a retrovirus expressing the neomycin resistance gene
- 697-Bcl-2 cells
697 cells stably infected with a retrovirus expressing human bcl-2 cDNA
- acDEVD-amc
acetyl-Asp-Glu-Val-Asp-aminomethylcoumarin
- acYVAD-ald
acetyl-Tyr-Val-Ala-Asp-aldehyde
- Bcl-2-membranes
heavy membranes prepared from 697-Bcl-2 cells
- neo-membranes
heavy membranes prepared from 697-neo cells
- PARP
poly(ADP-ribose) polymerase
Footnotes
Address correspondence to Lawrence C. Fritz, IDUN Pharmaceuticals, 11085 N. Torrey Pines Road, Suite 300, La Jolla, CA 92037. Tel.: (619) 623-1330. Fax: (619) 625-2677. E-mail: lfritz@idun.com
References
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Armstrong R, Aja T, Xiang J, Gaur S, Krebs J, Hoang K, Bai X, Korsmeyer S, Karanewsky D, Fritz L, Tomaselli K. Fas-induced activation of the cell death related protease CPP32 is inhibited by Bcl-2 and by ICE family protease inhibitors. J Biol Chem. 1996;271:16850–16855. doi: 10.1074/jbc.271.28.16850. [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, Tomaselli KJ. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci. 1997;15:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. [PubMed] [Google Scholar]
- Boulakia CA, Chen G, Ng FWH, Teodoro JG, Branton PE, Nicholson DW, Poirier GG, Shore GC. Bcl-2 and adenovirus E1B 19 kDA protein prevent E1A-induced processing of CPP32 and cleavage of poly(ADP-ribose) polymerase. Oncogene. 1996;12:29–36. [PubMed] [Google Scholar]
- Chandler JM, Cohen GM, MacFarlane M. Different subcellular distribution of caspase-3 and caspase-7 after Fas-induced apoptosis in mouse liver. J Biol Chem. 1998;273:10815–10818. doi: 10.1074/jbc.273.18.10815. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Orth K, O'Rourke K, Duan H, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway: Bcl-2 and Bcl-XLfunction upstream of the ced-3-like apoptotic proteases. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science. 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- Choi HK, Won L, Roback JD, Wainer BH, Heller A. Specific modulation of dopamine expression in neuronal hybrid cells by primary cells from different brain regions. Neurobiology. 1992;89:8943–8947. doi: 10.1073/pnas.89.19.8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckett CS, Li F, Wang Y, Tomaselli KJ, Thompson CB, Armstrong RC. Human IAP-like protein regulates programmed cell death downstream of Bcl-xLand cytochrome c. Mol Cell Biol. 1998;18:608–615. doi: 10.1128/mcb.18.1.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis eleganscell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- González-Garcia M, Pérez-Ballestero R, Ding L, Duan L, Boise LH, Thompson CB, Núñez G. Bcl-xLis the major Bcl-X mRNA form expressed during murine development and its product localizes to mitochondria. Development. 1994;120:3033–3042. doi: 10.1242/dev.120.10.3033. [DOI] [PubMed] [Google Scholar]
- Horvitz HR, Shaham S, Hengartner MO. The genetics of programmed cell death in the nematode Caenorhabditis elegans. . Cold Spring Harbor Symp Quant Biol. 1994;111:377–385. doi: 10.1101/sqb.1994.059.01.042. [DOI] [PubMed] [Google Scholar]
- Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Bcl-xLinteracts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA. 1998;95:4386–4391. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Bossey-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Wang H-G, Krajewski S, Zapata JM, Shabaik A, Gascoyne R, Reed JC. Immunohistochemical analysis of in vivopatterns of expression of CPP32 (caspase-3), a cell death protease. Cancer Res. 1997;57:1605–1613. [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigations of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- Krajewski S, Gascoyne RD, Zapata JM, Krajewska M, Kitada S, Chhanabhai M, Horsman D, Berean K, Piro LD, Fugier-Vivier I, et al. Immunolocalization of the ICE/Ced-3-family protease, CPP32 (caspase-3), in non-Hodgkin's lymphomas, lymphocytic leukemias, and reactive lymph nodes. Blood. 1997;89:3817–3825. [PubMed] [Google Scholar]
- Li F, Srinivasan A, Wang Y, Armstrong RC, Tomaselli KJ, Fritz LC. Cell-specific induction of apoptosis by microinjection of cytochrome c: Bcl-xLhas activity independent of cytochrome c release. J Biol Chem. 1997a;272:30299–30305. doi: 10.1074/jbc.272.48.30299. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997b;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lithgow T, van Driel R, Bertram JF, Strasser A. The protein product of the oncogene bcl-2 is a component of the nuclear envelope, the endoplasmic reticulum, and the outer mitochondrial membrane. Cell Growth Differ. 1994;3:411–417. [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Liu X, Zou H, Staughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Mancini M, Nicholson DW, Roy S, Thornberry NA, Peterson EP, Casciola-Rosen LA, Rosen A. The caspase-3 precursor has a cytosolic and mitochondrial distribution: implications for apoptotic signaling. J Cell Biol. 1998;140:1485–1495. doi: 10.1083/jcb.140.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Amarante-Mendes GP, Shi L, Chuang T-H, Casiano CA, O'Brien GA, Fitzgerald P, Tan EM, Bokoch GM, Greenberg AH, Green DR. The cytotoxic cell protease granzyme B initiates apoptosis in a cell-free system by proteolytic processing and activation of the ICE/CED-3 family protease, CPP32, via a novel two-step mechanism. EMBO (Eur Mol Biol Organ) J. 1996;15:2407–2416. [PMC free article] [PubMed] [Google Scholar]
- McCarthy NJ, Whyte MK, Gilbert CS, Evan GI. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–217. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DK. The role of the caspase family of cysteine proteases in apoptosis. Semin Immunol. 1997;9:35–49. doi: 10.1006/smim.1996.0058. [DOI] [PubMed] [Google Scholar]
- Miller DK, Ayala JM, Egger LA, Raju SM. Purification and characterization of active human interleukin-1β-converting enzyme from THP.1 monocytic cells. J Biol Chem. 1993;268:18062–18069. [PubMed] [Google Scholar]
- Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-xLforms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood. 1993;81:151–157. [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Changs BS, Thompson CB, Wong S, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- Na S, Chuang TH, Cunningham A, Turi TG, Hanke JH, Bokoch GM, Danley DE. D4-GDI, a substrate of CPP32, is proteolyzed during Fas-induced apoptosis. J Biol Chem. 1996;271:11209–11213. doi: 10.1074/jbc.271.19.11209. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Korsmeyer SJ. Checkpoints of dueling dimers foil death wishes. Cell. 1994;79:189–192. doi: 10.1016/0092-8674(94)90188-0. [DOI] [PubMed] [Google Scholar]
- Ottilie S, Wang Y, Banks S, Chang J, Vigna NJ, Weeks S, Armstrong RC, Fritz LC, Oltersdorf T. Mutational analysis of the interacting cell death regulators CED-9 and CED-4. Cell Death Differ. 1997;4:526–533. doi: 10.1038/sj.cdd.4400288. [DOI] [PubMed] [Google Scholar]
- Pan G, O'Rourke K, Dixit VM. Caspase-9, Bcl-xL, and Apaf-1 form a ternary complex. J Biol Chem. 1998;273:5841–5845. doi: 10.1074/jbc.273.10.5841. [DOI] [PubMed] [Google Scholar]
- Posmantur R, McGinnis K, Nadimpalli R, Gilbertsen RB, Wang KK. Characterization of CPP32-like protease activity following apoptotic challenge in SH-SY5Y neuroblastoma cells. J Neurochem. 1997;68:2328–2337. doi: 10.1046/j.1471-4159.1997.68062328.x. [DOI] [PubMed] [Google Scholar]
- Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC. Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA. 1997;94:5113–5118. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger PH, Gross A, Yin X-M, Yamamoto K, Saito M, Waksman G, Korsmeyer SJ. Comparison of the ion channel characteristics of proapoptotic Bax and antiapoptotic Bcl-2. Proc Natl Acad Sci USA. 1997;94:11357–11362. doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshagiri S, Miller LK. Caenorhabditis elegansCED-4 stimulates CED-3 processing and CED-3-induced apoptosis. Curr Biol. 1997;7:455–460. doi: 10.1016/s0960-9822(06)00216-8. [DOI] [PubMed] [Google Scholar]
- Singer II, Scott S, Chin J, Bayne EK, Limjuco G, Weidner J, Miller KD, Chapman K, Kostura MJ. The interleukin-1 beta-converting enzyme (ICE) is localized on the external cell surface membranes and in the cytoplasmic ground substance of human monocytes by immuno-electron microscopy. J Exp Med. 1995;182:1447–1459. doi: 10.1084/jem.182.5.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector MS, Desnoyers S, Hoeppner DJ, Hengartner MO. Interaction between the C. eleganscell-death regulators CED-9 and CED-4. Nature. 1997;385:653–656. doi: 10.1038/385653a0. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Foster LM, Testa MP, Ord T, Keane RW, Bredesen DE, Kayalar C. Bcl-2 expression in neural cells blocks activation of ICE/CED-3 family proteases during apoptosis. J Neurosci. 1996;16:5654–5660. doi: 10.1523/JNEUROSCI.16-18-05654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan A, Roth KA, Sayers RO, Shindler KS, Wong AM, Fritz LC, Tomaselli KJ. In situimmunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Litwack G, Alnemri ES. Molecular ordering of the Fas-apoptotic pathway: the Fas/ APO-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases. Proc Natl Acad Sci USA. 1996;93:14486–14491. doi: 10.1073/pnas.93.25.14486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennicke HR, Salvesen GS. Biochemical characteristics of caspases-3, -6, -7, and -8. J Biol Chem. 1997;272:25719–25723. doi: 10.1074/jbc.272.41.25719. [DOI] [PubMed] [Google Scholar]
- Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HD, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;396:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Tzagoloff, A. 1982. Mitochondria. Plenum Press, New York. 342 pp.
- Vander Heiden, M.G., N.S. Chandel, E.K. Williamson, P.T. Schumacker, and C.B. Thompson. Bcl-xLregulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Wu D, Wallen HD, Nunez G. Interaction and regulation of subcellular localization of CED-4 by CED-9. Science. 1997;275:1126–1129. doi: 10.1126/science.275.5303.1126. [DOI] [PubMed] [Google Scholar]
- Yang T, Kozopas K, Craig R. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J Cell Biol. 1995;128:1173–1184. doi: 10.1083/jcb.128.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yang X, Stennicke HR, Wang B, Green DR, Janicke RU, Srinivasan A, Seth P, Salvesen GS, Froelich C. Granzyme B mimics apical caspases. J Biol Chem. 1998;273:34278–34283. doi: 10.1074/jbc.273.51.34278. [DOI] [PubMed] [Google Scholar]
- Yu R, Mandlekar S, Harvey KJ, Ucker DS, Kong A-NT. Chemopreventive isothiocyanates induce apoptosis and caspase-3-like protease activity. Cancer Res. 1998;58:402–408. [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. eleganscell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]