

Abstract
We show that specific mutations in the head of the thick filament molecule myosin heavy chain prevent a degenerative muscle syndrome resulting from the hdp2 mutation in the thin filament protein troponin I. One mutation deletes eight residues from the actin binding loop of myosin, while a second affects a residue at the base of this loop. Two other mutations affect amino acids near the site of nucleotide entry and exit in the motor domain. We document the degree of phenotypic rescue each suppressor permits and show that other point mutations in myosin, as well as null mutations, fail to suppress the hdp2 phenotype. We discuss mechanisms by which the hdp2 phenotypes are suppressed and conclude that the specific residues we identified in myosin are important in regulating thick and thin filament interactions. This in vivo approach to dissecting the contractile cycle defines novel molecular processes that may be difficult to uncover by biochemical and structural analysis. Our study illustrates how expression of genetic defects are dependent upon genetic background, and therefore could have implications for understanding gene interactions in human disease.
Keywords: Drosophila, muscle, myosin, myofibril, troponin I
Muscle contraction is the result of a series of protein–protein interactions and conformational changes that culminate in ATP-dependent movement of the myosin head of the thick filament when it is attached to actin of the thin filament. The action of the myosin head slides the thin filament relative to the thick filament, causing sarcomere shortening. Thin filaments are normally inhibited from interacting with thick filaments due to blockage of the myosin binding sites on actin by a strand of tropomyosin molecules, and possibly by the troponin I protein of the thin-filament based troponin complex. The inhibition is relieved by release of calcium ions from internal stores following neural activity. Ca2+ binds to troponin C protein, reconfiguring the troponin T–based interaction of the entire troponin complex with tropomyosin. The resulting movement of the tropomyosin strand from its inhibitory position permits the myosin crossbridge to bind to the thin filament. For a recent review, see Squire (1997).
There are numerous conformational rearrangements involved in thin-filament regulation of the crossbridge cycle (Farah and Reinach, 1995). Multiple Ca2+-induced changes in interaction among subunits of the troponin complex and between troponin and tropomyosin occur, although the details of the structural role of the troponin complex in this regulation are not known. Not only does tropomyosin shift during Ca2+ activation of the thin filament, but the actin monomer changes conformation (al-Khayat et al., 1995). Further, binding of the myosin head to the thin filament is a cooperative process that involves progressive tropomyosin movement (Vibert et al., 1997). The first myosin heads bind weakly to actin and interact with tropomyosin to push it further away from myosin binding sites on actin. This leads to a decreased duration of the ATP cycle, i.e., a fully on state (McKillop and Geeves, 1993; Metzger, 1995). Understanding the details of the contractile cycle is important for defining the mechanisms of human diseases, such as familial hypertrophic cardiomyopathy, where mutations in a number of sarcomeric contractile proteins can result in aberrant contractile properties and muscle hypertrophy (Watkins et al., 1995; Towbin, 1998).
Some success in mapping precise interaction sites of various contractile apparatus components has resulted from electron microscopy/image reconstruction, and from biochemical assays that assess interaction between intact proteins, proteolytic fragments, and expressed recombinant peptides. These studies are supplemented by determinations of atomic structure of contractile proteins that indicate the location of putative binding sites in particular conformational states. For instance, it has been shown recently that an NH2-terminal α-helical region of troponin I binds to troponin C at low Ca2+ conditions (Vassylyev et al., 1998). It is proposed that Ca2+ binding to troponin C releases this troponin I region and allows binding of an inhibitory region of troponin I, thereby allowing actomyosin interaction (Tripet et al., 1997; Vassylyev et al., 1998). It is important to note, however, that in vitro approaches represent a trade off between structural resolution and biological significance of derived conclusions. The inhibitory role of troponin I is a case in point. Inhibitory properties have been ascribed to the fragment between residues 104–115. However, this fragment's inhibiting efficiency is lower than the entire 1–116 fragment and this, in turn, is less inhibitory than the whole molecule (Tripet et al., 1997; Van Eyk et al., 1997).
An alternative method to assessing functional interactions of proteins during the contractile cycle involves genetic analysis, i.e., disrupting muscle function by mutating a particular contractile protein and searching for suppressor mutations that restore function. This is a particularly powerful approach in that interactions relevant to muscle function in vivo are clarified. In principle, suppressor mutations reveal sites of specific protein–protein interactions that are important to myofibril assembly and/or function. It is also possible that suppressor mutations work by less direct mechanisms, such as through interactions with an intermediary component of the contractile apparatus, or by a general change in protein function that compensates for the original mutation in a less specific way. The suppressor mutation approach has been applied most successfully to mapping muscle protein interactions in Caenorhabditis elegans (Greenwald and Horvitz, 1982; Moerman et al., 1982; Park and Horvitz, 1986; Gengyo-Ando and Kagawa, 1991).
Prado et al. (1995) described the isolation of suppressor mutations in Drosophila melanogaster for a particular point mutation of troponin I, the inhibitory subunit of the troponin complex. These suppressors prevent the heldup wings phenotype that arises from severe defects in the indirect flight muscles of the troponin I mutant. One suppressor is within the mutated troponin I protein itself (Prado et al., 1995). Four others are mapped to the second chromosome. Determination of mutant gene(s) that act to suppress troponin I defect, and definition of the precise location of mutations should reveal protein–protein interactions important to muscle function in vivo.
In this paper, we show that the four genetic suppressors of a Drosophila troponin I point mutation are within the myosin heavy chain (MHC)1. We determine molecular alterations in the myosin molecule and map these on the three-dimensional structure of globular head in an effort to understand the molecular basis of suppression. We show that observed suppression is allele-specific, i.e., it is dependent on a specific mutated residue in troponin I and particular sites within MHC. We elucidate the degree of phenotypic suppression observed in indirect flight muscles of adult flies using light and electron microscopy, and demonstrate that different myosin suppressor alleles suppress the troponin defects to different degrees. Finally, we discuss the possibility that our work reveals an interaction between MHC and troponin I, a prospect not previously proposed based on structural or biochemical studies.
Materials and Methods
Isolation, Mapping, and Sequencing of Suppressor Mutations
Isolation of dominant suppressors of heldup2 was described in Prado et al. (1995). In brief, adult hdp2 males were mutagenized with ethyl-methane sulfonate (EMS) according to standard procedures, and crossed to females of the genotype C(1)M3/Y;Sco/CyO or C(1)M3/Y;TM1/TM3. Male offspring with near normal wing position, instead of the expected heldup wings, were crossed individually to balancer stocks to identify the chromosome containing the suppressor. Stocks with a series of recessive markers were used to determine the map position of each suppressor on a particular chromosome based upon recombination between markers. Each isolated suppressor should be designated as Su(hdp2)D followed by an identification number. For brevity, they are referred to as D mutations in the text. As per standard practice, gene abbreviations are designated in italics and proteins are in capital Roman type.
We obtained recessive–lethal, homozygous suppressor strain embryos for DNA amplification and sequencing by using a second chromosome balancer line (CyO y+) marked with the yellow+ gene (y+; Mardahl et al., 1993) in combination with an X chromosome marked with the y and w (white eye) mutations. To this end, hdp2;D mutation/CyO males were mated with y w;CyO y+/Bc Elp females. Male offspring of genotype y w;D mutation/CyO y+ were backcrossed to y w;CyO y+/Bc Elp females. Resulting males and females of the y w;D mutation/CyO y+ genotype were mated to produce a stable stock. Embryos with dark mouth hooks carry one or two copies of the second chromosome marked with CyO y+, while homozygotes for the D suppressor mutation display yellow mouth hooks.
Genomic DNA was extracted from homozygous embryos of each suppressor mutant according to the method of Jowett (1986). 60 embryos were frozen in an Eppendorf tube and stored at −80°C for at least 1 h. 40 μl of single fly homogenization buffer (10 mM Tris-HCl, pH 7.5, 60 mM NaCl, 50 mM EDTA, 150 μM spermine, 150 μM spermidine) were added and the samples were ground with a plastic pestle. 40 μl of single fly lysis buffer (1.25% [wt/vol] SDS, 300 mM Tris-HCl, pH 8, 100 mM EDTA, 5% [wt/vol] sucrose, 0.75% freshly added diethyl pyrocarbonate) were added. The mixture was incubated for 30 min at 60°C. The sample was cooled to room temperature and 12 μl of 8 M potassium acetate was added. After cooling on ice for 45 min, debris was pelleted by 1 min centrifugation in a microfuge. Supernatant was removed to a fresh tube and 200 μl of 100% ethanol was added. DNA was precipitated at room temperature for 10 min and pelleted in a microfuge for 10 min. The sample was washed with 80% ethanol and vacuum dried. The pellet was resuspended in 60 μl TE (10 mM Tris-HCl, pH 8, 1 mM EDTA).
Genomic DNA from each mutant was used in PCR to generate 11 fragments that cover the entire coding region, plus flanking introns of the Mhc gene. The following oligonucleotide primers were used for amplification (sequences given for noncoding strand in a 5′ to 3′ orientation): 1, ATGCCGAAGCCAGTCGCAAAT (position 1924), GGAATTCGATACGGATGAATTTACC (position 4141); 2, TAAGCTTGAAGACCGATGAGGCC (position 3948), ATAGCCGTCACTACATAGAGC (position 5941); 3, TTATGTTCTTCTTGCTAAACC (position 6456), ATCTGACTAAAATCCTCAGA (position 8185); 4, GATACACTGCAGCACTAT (position 8367), TGATCGGAGGCCTTGGGGAAC (position 10131); 5, GTTCCCCAAGGCCTCCGATCA (position 10131), GTGTGGGGATTCAATTGAAAG (position 11087); 6, GGAATCAAAAACGAACTCTAC (position 11206), CTAATTGTGGAAGGAGC (position 11818); 7, GTTAAGATCAACTGTAACTAA (position 12206), AGACCCAGGCTGGTCTCGTT (position 14095); 8, CTTCAGCCCGAATCGACCGCC (position 15455), TCAGATCTCTCTATCTCGAT (position 16958); 9, TTGAAGGATCTACAGTTTACA (position 16959), GGGTGACAGACGCTGCTTGGT (position 18365); 10, GTCCCAGGTGTCTCAGCTGT (position 18045), GGCGGGCGGCATCGACCATAG (position 19512); and 11, TGCGTCGTGAGAACAAGAACC (position 18653), TATTACTCTCTTGTTTT (position 20368). Each PCR sample contained 5 μl of genomic DNA, 20 μl of 10× PCR buffer (Promega Corp.), 20 μl of 5 μM solutions of each dNTP (80 μl total), 16 μl of 25 mM MgCl2, 100 pmol each of two primers, 0.8 μl of Taq polymerase (Promega Corp.), and was brought to a total volume of 200 μl with distilled H2O. Paraffin oil was placed on top of the sample to prevent evaporation, and DNA was amplified in an Ericomp thermocycler as follows: one cycle at 95°C for 1 min, 45°C for 2 min, 72°C for 40 min; 28 cycles at 95°C for 1 min, 45°C for 2 min, 72°C for 6 min; and one cycle at 95°C for 1 min, 45°C for 2 min, 72°C for 15 min. Paraffin oil was then removed and DNA was chloroform extracted and precipitated.
PCR products were cloned before sequencing. Amplified products were separated by agarose gel electrophoresis, isolated using GeneClean (Bio 101), and blunt ends were created with the Klenow fragment of Escherichia coli DNA polymerase I (Sambrook et al., 1989). Each fragment was cloned into the EcoRV site of pKS plasmid (Stratagene) and DNA sequencing was performed using a Sequenase kit (United States Biochemicals) or on an automated DNA sequencer (Applied Biosystems).
Reverse Transcription and Amplification of Mhc mRNA
First strand synthesis of cDNA was performed using 1 μg of total RNA (isolated as described in Hess and Bernstein, 1991), 100 pmol of 3′ primer (TGATCGGAGGCCTTGGGGAAC, position 10131), 1.4 μl of 5× first strand buffer (250 mM Tris, pH 8.5, 375 mM KCl, 5 mM MgCl2, 50 mM dithiothreitol), brought to a total volume of 7 μl with distilled H2O. The mixture was placed in boiling water for 30 s, then allowed to cool to 37°C. 1 μl of Inhibitase (1 U/μl; Promega Corp.) was then added along with 0.5 μl of each dNTP at 10 mM. Then 0.6 μl of 5× first strand buffer was added plus 0.5 μl of distilled H2O. The reaction was started by addition of 1.0 μl of M-MLv reverse transcriptase (100 U/μl; GIBCO BRL) and the sample was incubated at 37°C for 1.5 h. The reaction was terminated on ice by adding 20 μl of 0.3 M NaOH/0.03 M EDTA. RNA was hydrolyzed at 60°C for 1 h. The solution was neutralized by adding 3.4 μl of 3 M sodium acetate, pH 5.2, and cDNA was precipitated with 2.5 vol of 100% ethanol. After centrifugation in the microfuge for 15 min at 4°C, the DNA pellet was washed with 80% ethanol and vacuum dried. The sample was resuspended in 10 μl distilled H2O. Half the sample was amplified using the 3′ primer at position 10131 and 5′ primer GGCTGGTGCTGATATTGAGA (position 4182), as described for genomic DNA above.
In Situ Hybridization
Slides were cleaned by thorough washing with liquid hand soap, then treated with subbing solution (0.5% gelatin, 0.05% chrome alum). Slides were dried overnight in a dust-free environment. Tissue was prepared by embedding whole flies (with wings removed) in OCT compound and freezing on dry ice. Frozen tissue sections (8–16 μm) were taken using a microtome. These were placed onto treated slides and allowed to dry. Tissue was fixed with 4% paraformaldehyde for 20 min and then washed three times in 1× PBT (1.3 M NaCl, 0.07 M Na2HPO4, 0.03 M NaH2PO4, 1% Tween 20). Sections were then treated with 50 μg/ml proteinase K in PBT for 3 min. This was followed by treatment with 2 mg/ml glycine in PBT for 1 min (repeated once). Slides were washed in PBS (1.3 M NaCl, 0.07 M Na2HPO4, 0.03 M NaH2PO4) for 1 min and placed in 4% paraformaldehyde for 20 min. This was followed by two washes with PBS for 5 min each. The samples were dehydrated in 30% ethanol, 50% ethanol, 70% ethanol, 80% ethanol, 95% ethanol, 100% ethanol (5 min each), and placed under the vacuum for 40 min.
Transcription of digoxigenin-labeled probes was according to the procedure provided in Genius 3 Kit (Boehringer Mannheim). Antisense probes from each copy of exon 7 were prepared from the following fragments that had been cloned into a plasmid containing a T3 or T7 RNA polymerase binding site: exon 7a, XbaI (4568) to HindIII (4940); exon 7b, HindIII (4940) to HindIII (5300); exon 7c, Hind III (5300) to EcoRV (5900); exon 7d, EcoRV (5900) to EcoRI (6600). 1 μg of RNA probe was added to 25 μl of 10 mg/ml tRNA and brought to a total volume of 100 μl with distilled H2O. The probe was denatured by heating at 75°C for 10 min.
Hybridization was carried out by adding the denatured probe to 400 μl of hybridization buffer (50% formamide, 10% dextran sulfate, 0.3 M NaCl, 10 mM Tris-HCl, pH 8, 1 mM EDTA, 0.1% Tween 20, 50 μg/ml heparin, 1× Denhardt's solution). 100 μl of the probe in hybridization solution were placed onto each slide. Slides were covered with a plastic sealer (HybriWell, Research Products International) and placed in a sealed box. Hybridization was allowed to proceed for at least 18 h at 56°C.
After hybridization, slides were washed with 4× SSC (twice for 10 min each). This was followed by RNase A treatment (20 μg/ml in 0.5 M NaCl, 10 mM Tris, pH 7.5, 1 mM EDTA) to remove single-stranded probe for 30 min at 37°C. Slides were washed in PBT for 5 min (repeated once), and then incubated with antibody conjugate at a ratio of 1:500 in PBT plus 5% normal goat serum for 120 min. Unbound antibody was washed off with buffer 3 (100 mM Tris, pH 9.53, 100 mM NaCl, 50 mM MgCl2) for 5 min. This was repeated. Color reaction buffer was prepared by adding 20 ml of buffer 3 to 100 μl of NBT and 75 μl of X phosphate. This reaction was allowed to proceed for at least 1 h and as long as overnight. The reaction was stopped by rinsing in H2O.
Protein Analysis
One-dimensional SDS-PAGE was performed by the method of Laemmli (1970). Upper thoraces from 10 flies were dissected, homogenized in 100 μl sample buffer and boiled. Samples (10 μl) were loaded on gels containing 9.5% acrylamide. After staining in Coomassie blue, scanning was performed using a Molecular Dynamics densitometer. MHC levels were normalized to actin levels within the same lane to account for differences in protein loading levels.
Flight Testing
Flight testing was performed using the method of Drummond et al. (1991) on young (2-d-old flies).
Microscopy
For transmission electron microscopy, flies were dissected according to the protocol of Peckham et al. (1990). Once the heads, wings, and abdomens were removed, thoraces were fixed overnight at 4°C in 4% paraformaldehyde, 1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.2. The dorsolongitudinal muscles (DLMs) were dissected from the thoraces, washed several times in buffer, and postfixed in 2% OsO4 in buffer for 45 min at 4°C in the dark. After dehydration, DLMs were embedded in Araldite resin. Silver sections (60–70 nm) were cut on a Reichert Ultracut E ultramicrotome, collected on Formvar-coated grids, and counterstained with uranyl acetate (10 min) and lead citrate (10 min). Micrographs were obtained using a JEOL 1200 EX electron microscope. Morphological analysis at the light microscope level was carried out on paraffin-embedded samples stained with Toluidine blue (Prado et al., 1995).
Results
Phenotypic Analysis of Dorsolongitudinal Muscles in Normal Flies and heldup2 Troponin I Mutants
The DLMs are composed of six fibers (a–f) attached to the anterior and posterior sides of the thorax (Fig. 1 A). The DLM fibers, like the opposing dorsoventral indirect flight muscles, are termed fibrillar muscles. This is because each fiber contains several hundred myofibrils that can be easily teased apart. Individual fibrils are subdivided by transverse bands of electron dense material, the Z bands, that define the unit of contraction, the sarcomere (Fig. 1 B). In a transverse view, the circular fibril contains a crystalline-like array of thick and thin filaments that is arranged in a 1:6 hexagonal pattern (Fig. 1 C). In the normal strain used here, Canton-S (CS), ∼1,000 thick and 2,000 thin filaments accumulate in each fibril. These numbers are fairly constant within a muscle showing only a 5% variability in DLM muscle (a) of our CS stock. Note, however, that other normal strains may exhibit up to 1,500 thick filaments per fibril.
Figure 1.
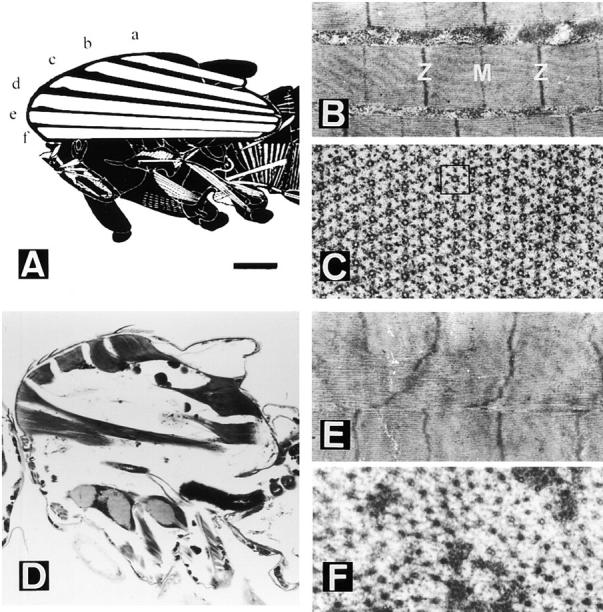
Normal and heldup2 mutant dorsolongitudinal muscles (DLM). (A) Diagram showing the six (a–f) DLM fibers in a sagittal view. Anterior is to the left, and dorsal is up. (B) Detail of a fibril showing a sarcomere between Z bands and including an M line. (C) Transverse view of a fibril showing the 1:6 array of thick to thin filaments. The square box has one thick filament in the center and six surrounding thin filaments in a hexagonal pattern. (D) Sagittal section of a hdp2 male. Note the remnants of the six DLMs near their attachment sites. (E) Hypercontracted sarcomeres in which the M line is no longer detected. (F) Cross section of a mutant muscle in which only thick filaments are visible. Bar: (A) 250 μm; (B and E) 750 nm; (C and F) 120 nm; (D) 240 μm.
In the troponin I mutant heldup2, the six DLMs appear torn apart from the center (Fig. 1 D). In the remaining muscle material, near the attachment sites, the sarcomere length is 40% reduced and the thick–thin filament pattern is destroyed mostly due to the collapse of thin filaments (Fig. 1, E and F). It appears as if the mutant muscles were clamped in a state of hypercontraction. The mutation hdp2 is a single amino acid change, Ala55Val, affecting all known isoforms of troponin I (Beall and Fyrberg, 1991; our unpublished data). This corresponds to residue 25 in rabbit skeletal muscle troponin I (see Vassylyev et al., 1998).
D Suppressors on Chromosome II are Mhc Mutations
To identify molecular interactions between muscle proteins and troponin I, we screened for mutations that suppress the heldup wing position of the troponin I hdp2 mutation and isolated four D mutations that map to chromosome II (Prado et al., 1995). We employed meiotic recombination to discern their locations on the second chromosome, and found they map between markers rd and pr. Further, we localized recessive lethality associated with mutations D41, 45, and 62 to the interval uncovered by Df(2)H20. This deficiency removes polytene chromosome regions 36A8–36A9;36F1 and contains the myosin heavy chain (Mhc) gene.
To determine whether the suppressor mutations are Mhc alleles, we performed genetic complementation tests with known Mhc alleles (for details on these alleles, see Lindsley and Zimm, 1992). We crossed each of the D-suppressor mutants to a null mutant (Mhc1), a hypomorphic mutant (Mhc2), and several point mutants (Mhc5, Mhc6, Mhc8). Mhc1, Mhc2, and Mhc8 are recessive lethal alleles, while Mhc5 and Mhc6 are viable as homozygotes. Our results show that the D-suppressor mutants are likely to be Mhc alleles, since none of the suppressors produced progeny over Mhc null or hypomorphic alleles, except for D1 which occasionally was viable in combination with Mhc1. The suppressors produced viable progeny in combination with the various point mutations, except that D1 is lethal in combination with Mhc5, D41 is lethal with Mhc8, and D62 produces very few viable adults in combination with Mhc8. These data demonstrate interaction, and likely allelism, between the D-suppressor mutants and Mhc.
Since Mhc null alleles are recessive lethal (O'Donnell and Bernstein, 1988), as are three of the four D-series suppressor mutants, it is important to determine whether the latter exert their suppression effect through failure to accumulate MHC. We determined whether MHC protein accumulates in the suppressor strains by crossing each to Mhc10 and measuring MHC levels in upper thoraces of heterozygotes. Mhc10 adults fail to accumulate MHC in the jump and indirect flight muscles due to a mutation in an alternative exon specifically used in these muscle types (O'Donnell et al., 1989). Each of the D/Mhc10 heterozygotes accumulate more MHC than Mhc10/Mhc10 adults, but less than +/Mhc10 individuals (Table I). This indicates that suppressor mutations produce stable MHC protein. While the suppressor mutants accumulate only ∼65–85% as much MHC as flies carrying one copy of wild-type Mhc gene, it is clear that suppressor alleles are not null mutations for Mhc. It is also noteworthy that Mhc missense mutations that cause flight muscle dysfunction typically result in less than wild-type levels of MHC accumulation (Mogami et al., 1986; Kronert et al., 1995).
Table I.
Genetic and Molecular Properties of the D-series Suppressors and Other Mhc Alleles
| Mhc genotype | Lethal over Mhc null | Protein* | Wild-type sequence | Mutant sequence‡ | ||||
|---|---|---|---|---|---|---|---|---|
| D1 | no | 73 | Asp | Gly (aa 625) | ||||
| GAT | GGT | |||||||
| D41 | yes | 65 | AspAspAlaGlu | AspGluStop---- | ||||
| GATGATGCTGAG | GATGAGTAGCTGAG | |||||||
| [changes aa 328, creates stop codon, 5′ splice site (underlined)] | ||||||||
| D45 | yes | 85 | Ala | Thr (aa 261) | ||||
| GCT | ACT | |||||||
| D62 | yes | 77 | GlyGlyArgGlyLysLysGlyGlyGlyPhe | GlyPhe | ||||
| GGAGGTCGTGGCAAGAAGGGCGGTGGCTTC | GGCTTC | |||||||
| (deletion in D62 underlined) | (deletion of aa 638-645) | |||||||
| Mhc5 | no | 88 | Gly | Asp (aa 200) | ||||
| GGT | GAT | |||||||
| Mhc6§ | no | 90-100 | Arg | His (MHC rod) | ||||
| CGC | CAC | |||||||
| Mhc8 | yes | 79‖ | Tyr | His (aa 832) | ||||
| TAC | CAC |
Protein levels of MHC standardized to actin levels and expressed as a percentage of wild-type MHC accumulation.
Chicken amino acid sequences given to allow localization on the chicken myosin S1 structure.
Data from Kronert et al. (1995).
Protein accumulation data from Mogami et al. (1986).
To demonstrate that each suppressor mutation resides within Mhc, and to determine their molecular lesions, we cloned and sequenced the Mhc gene from homozygous embryos of each strain. We found that each suppressor strain has a discrete region of the Mhc coding sequence altered, and all mutations affect the head domain (S1 fragment) of the myosin molecule. We mapped encoded aberrations onto the three-dimensional map of chicken myosin head (Fig. 2). Amino acid identity between Drosophila and chicken myosin is high, and the atomic resolution crystal structure of the chicken molecule (Rayment et al., 1993b) serves as an excellent model for visualizing Drosophila alternative coding regions and mutations (Bernstein and Milligan, 1997).
Figure 2.
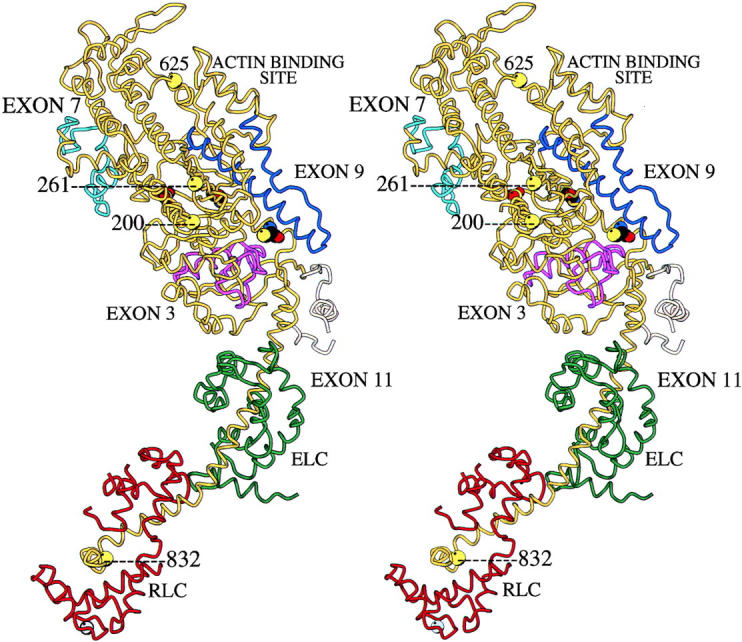
Locations of mutations on the three-dimensional structure of the myosin head. The figure depicts a stereo-pair image of the atomic resolution structure of chicken myosin S1 (Rayment et al., 1993b) with regions encoded by Drosophila alternative exons and Drosophila Mhc mutations superimposed. The backbone of the heavy chain is yellow, the essential light chain is green, and the regulatory light chain is red. The location of the β-phosphate group of ATP is shown as a red sphere, with the reactive thiols depicted as tricolored residues. The Drosophila MHC regions encoded by alternative exons are purple (exon 3), light blue (exon 7), dark blue (exon 9), and tan (exon 11; Bernstein and Milligan, 1997). Two hdp2 suppressor mutations are at the actin binding loop. Mutation D1 affects residue 625 at the base of the loop while mutation D62 is an 8 amino acid deletion in the loop itself. The loop is not visible in the structure due to its flexible nature, but its ends are located behind residue 625 and at the terminus of a long helical structure just to its right. Mutation D45 is at residue 261, on the surface of the molecule. This residue is just to the right of the two free ends of the molecule representing the flexible loop at the lip of the nucleotide binding pocket. The loop itself is not visible in the structure. Mutation D41 affects the lip of the nucleotide pocket by causing the substitution of exon 7 alternative exons (light blue). Residue 200, which is mutated in Mhc5, is also in this general vicinity of the molecule and acts to enhance the effects of hdp2. A mutation in the regulatory light chain binding domain at residue 832 (Mhc8) enhances the effects of the hdp2 allele to a lesser extent. Graphic produced in collaboration with Dr. Ronald Milligan (The Scripps Research Institute) using the Molscript program.
D1 is a point mutation in exon 10 (A→ G), changing amino acid 625 (chicken MHC numbering system) from Asp to Gly (Table I). This mutation affects an amino acid at the base of the second loop of the molecule (Fig. 2). This loop is involved in actin binding (Mornet et al., 1981; Sutoh, 1982; Rayment et al., 1993a,b; Uyeda et al., 1994; Rovner et al., 1995). If the mutation affects the mobility of the loop, it could dampen acto-myosin interaction.
Mutation D62 also affects exon 10, and is a 24-bp in-frame deletion starting at amino acid 638 (Table I). Like D1, this mutation affects the loop that binds actin. It removes eight amino acids within the loop and clearly would be expected to affect actomyosin interaction. The loop, which runs from residue 627 to 646, is not visible in Fig. 2 due to its flexible nature (Rayment et al., 1993b).
Mutation D45 is a point mutation in exon 5 (G→ A), changing amino acid 261 from Ala to Thr (Table I). This amino acid is in the general vicinity of ATP entry and the ATP binding site (Fig. 2). However, it is on the surface of the molecule, away from direct interactions with the nucleotide. It is located very close to loop 1 of the molecule (residues 204–216), which is not visible in the structure. This loop is important for regulating nucleotide entry and exit from the ATP binding pocket (Murphy and Spudich, 1998; Sweeney et al., 1998).
Mutation D41 is a 2-bp insertion into exon 7a, interrupting amino acid codon 328. It places this alternative exon out of frame and inserts a stop codon (Table I). The mutation also produces a potential 5′ splice junction, GTAGCT. This could disrupt alternative splicing. To study this, we used RT-PCR to amplify the exon 7 region in adult upper thoraces from this mutant. Since this mutation is recessive lethal, the thoraces were taken from D41/Mhc10 organisms (note that Mhc10 RNA fails to accumulate in fly thoraces due to a splicing defect; Collier et al., 1990). We cloned the PCR products from D41/ Mhc10 heterozygotes and analyzed a number of clones by DNA sequencing or restriction enzyme digestion. Normally exon 7d is used in indirect flight muscles (Hastings and Emerson, 1991), which make up the bulk of the thorax. We found this to be the case in all 17 clones analyzed from wild-type thoraces. However, we observed an extreme reduction in exon 7d usage, replaced by in-frame inclusion of exons 7b or 7c, in clones of Mhc PCR products from thoraces of D41/Mhc10 organisms (1 exon 7b, 13 exon 7c, and 4 exon 7d). Thus, the insertion of a splice junction in exon 7a appears to disrupt the alternative splicing process.
We next used in situ hybridization to investigate the possibility of tissue-specific alternative splicing disruption in thoracic musculature of D41 adults. Alternative exon-specific probes were prepared and hybridized to sections of young adults, either wild-type or D41/Mhc10 mutant. The hybridization results clearly showed that exon 7d accumulates in indirect flight muscles of wild-type, but is below detectable levels in D41 indirect flight muscles (Fig. 3). High levels of exon 7c accumulate in D41 indirect flight muscles, but no trace of this exon is detected in wild-type indirect flight muscle transcripts. Thus, the unusual effect of the mutation is to disrupt the alternative splicing apparatus through the introduction of a 5′ splice site, resulting in use of a different alternative exon than is normally employed in indirect flight muscles. Exon 7 encodes a region at the lip of the nucleotide binding pocket (light blue in Fig. 2). It is possible that using the wrong version of this alternative exon disrupts MHC function by changing nucleotide affinity and disrupting the ATPase cycle.
Figure 3.
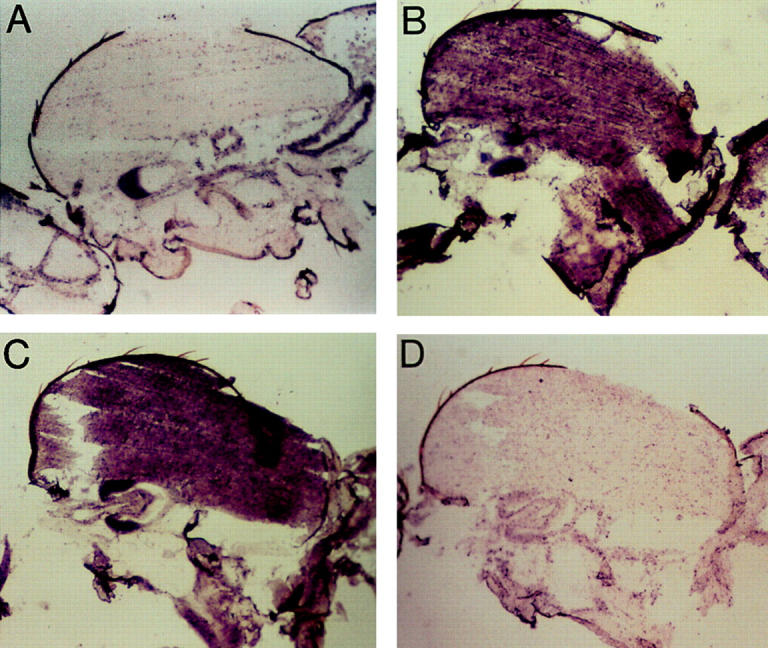
In situ hybridization analysis of Mhc mRNA accumulation in wild-type and D41 Mhc mutant. D41 mutation creates a splice junction-like sequence resulting in misregulation of alternative splicing of the exon 7 series. Probes specific to alternative versions of exon 7 show that exon 7d is used in wild-type indirect flight muscles, but replaced with exon 7c in D41. Panels are brightfield micrographs of parasagittal sections of thoraces (anterior is left, dorsal is up). (A) Wild-type male probed with exon 7c, showing no hybridization to the indirect flight muscles that comprise the bulk of the upper thoracic region. (B) Wild-type male probed with exon 7d, showing strong hybridization to the indirect flight muscles. (C) hdp2/Y;D41/Mhc10 males probed with exon 7c, showing strong hybridization to the indirect flight muscles. (D) hdp2/Y;D41/Mhc10 males probed with exon 7d showing failure of the indirect flight muscles to hybridize with this probe.
We also studied use of the aberrant version of exon 7a in the D41 mutant. In wild-type embryos, alternative exon 7a is abundantly expressed in body wall muscles (Zhang and Bernstein, manuscript in preparation). Our RT-PCR analysis of RNA from wild-type embryos confirmed that this is the major exon 7 version used at this stage (16 clones examined) and showed that exon 7a is incorporated in all reverse transcribed mRNAs studied from homozygous D41 embryos (14 clones). The normal splice junction is used in the mutant. This would result in premature termination of translation due to the stop codon described above, and explains the recessive lethality of the mutation. The suppressive effect of the D41 mutation upon the hdp2 phenotype, however, appears to result from misexpression of exon 7c in the indirect flight muscles.
Functional and Structural Effects of D Suppressors
We examined the degree of rescue of hdp2 phenotypes by each suppressor mutation that maps within the head domain of MHC. While the suppressed wing position phenotype is evident in all hdp2;D/+ males, none can jump or fly under standard criteria (Prado et al., 1995). We analyzed the structural effects of the suppressors in hdp2;D/+ males at light and electron microscopic levels (Fig. 4). In general, the organization of the six DLMs is restored with similar efficiency by the four D mutations. However, the e and f muscles, their posterior region in particular, are still very sensitive to contraction, and appear grossly abnormal at 3–5 d (Fig. 4, A, E, I, and M).
Figure 4.

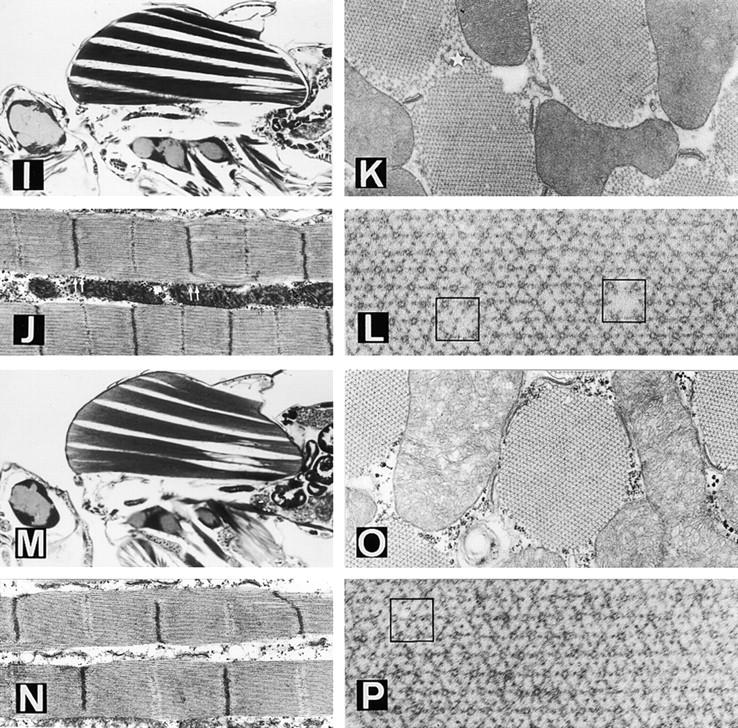
Suppression of the troponin I mutation hdp2 by the D-series Mhc mutations. D1 (A–D) and D62 (E–H) affect the actin-binding loop of MHC. (A) Sagittal, slightly tilted, view of a hdp2;D1/+ male. Note the almost normal appearance of DLM fibers a–d but the collapse of muscles e and f (arrow- head). (B) Detail of two fibrils. Note the restoration of the M line in the sarcomeres. (C) Cross section of a suppressed muscle. m = mitochondrion. (D) Detail of the thick–thin filament array. Some structural failures such as the absence of a thick filament (arrow- head) or an additional thin filament (arrow) can be identified. (E) Sagittal view of a hdp2;D62/+ male. Note the persistence of gross structural defects on muscles e and f near the posterior attachment site. (F) Detail of fibrils showing virtually normal sarcomeres. (G) Cross section showing a near normal fibril. The apparent disorganization indicated by an arrowhead is an artifact of preparation, found occasionally in wild-type. (H) Detail of the filament array. D41 (I–L) and D45 (M–P) are mutations near the nucleotide entry site in MHC. (I) Sagittal view of a hdp2;D41/+ male. There are persistent structural defects towards the posterior side of muscles e and f. (J) Detail of the fibrils. Note the incomplete definition of Z and M bands towards the edge of the fibril (double arrows). (K) Cross section of fibrils showing the disorganized array at the periphery (star). (L) Detail of the filament array. Occasionally, thick filaments are absent or a thin filament substitutes for thick (squares). (M) Sagittal section of an hdp2;D45/+ male. As with all other suppressors, gross structural defects persist toward the posterior site of e and f muscle, although, in this case, d is also visibly affected. (N) Sarcomeres with incomplete restoration of Z and M lines. (O) Cross view of fibrils. (P) Detail of filament array showing a double thick filament (square). Bar: (A, E, I, and M) 330 μm; (B, F, J, and N) 775 nm; (C, G, K, and O) 430 nm; (D, H, L, and P) 144 nm.
Wild-type sarcomere structure and length in hdp2 individuals is recovered to different degrees as a result of each D mutation. The M line reappears in all four cases but the sarcomere length is best restored by D41. Organization of Z bands is better in D1 and D62 than with the other two alleles (Fig. 4, B, F, J, and N). The number of thick filaments per fibril averages 950 in D45, 830 in D41, 750 in D62, and 650 in D1. These are 5–35% below normal. In spite of nearly normal numbers of thick filaments, D41 fibrils appear particularly unstable at the periphery, where the lattice collapses (Fig. 4 K). These features, and those reported for second site suppressor D3 (Prado et al., 1995), point toward differential sensitivity of the center versus the periphery of the fibril. The arrangement of thick and thin filaments found in the suppressed condition include various types of abnormalities, e.g., absence of a thick filament, excess thin filaments, substitution of thick by thin filaments, or doublets of thick filaments (Fig. 4, D, H, L, and P). These perturbations do not induce major defects in the surrounding structure.
We also studied the effects of the D-series suppressors upon flight muscle function in the absence of hdp2 mutation. The suppressors show dominant effects upon flight muscle function (Table II). D1 is least disruptive, with 85% of adults flying upward or horizontally, compared with 90% in wild-type. D62 is most disruptive, with only 16% flying upward or horizontally (Table II). We determined whether the wild-type Mhc gene could rescue defects in flight ability by crossing each suppressor strain to a stock containing an Mhc transgene (Cripps et al., 1994). No rescue was observed (Table II), consistent with our observation that suppressor alleles produce stable MHC proteins which interfere with myofibril function.
Table II.
Flight Ability Is Impaired by the D-series Suppressors and this Phenotype Is Not Rescued by an Additional Copy of the Mhc Gene
| Mhc genotype | Number tested | Direction of flight | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Up | Horizontal | Down | Not at all | |||||||
| +/+ | 100 | 87 | 3 | 7 | 3 | |||||
| +/+/+ | 119 | 86 | 8 | 4 | 2 | |||||
| D1/+ | 60 | 70 | 15 | 10 | 5 | |||||
| D1/+/+ | 111 | 82 | 8 | 5 | 5 | |||||
| D41/+ | 57 | 32 | 53 | 5 | 10 | |||||
| D41/+/+ | 120 | 37 | 34 | 8 | 21 | |||||
| D45/+ | 90 | 67 | 20 | 3 | 10 | |||||
| D45/+/+ | 93 | 53 | 22 | 15 | 10 | |||||
| D62/+ | 72 | 4 | 12 | 42 | 42 | |||||
| D62/+/+ | 177 | 4 | 20 | 34 | 42 | |||||
| Mhc5/+ | 142 | 14 | 44 | 18 | 24 | |||||
| Mhc5/+/+ | 167 | 17 | 37 | 23 | 23 | |||||
Allelic Interactions and Specificity of Suppressed Phenotypes
To investigate the unique nature of each suppressor's action, we tested all pairwise combinations of D mutants in a hdp2 male background. We expect an additive or synergistic effect when two Mhc mutations are suppressed by different mechanisms. If the same mechanism of suppression is employed by two different suppressors, we expect a phenotype similar to that of flies with a single suppressor. Only combinations over D1 resulted in viable adults, and the structure of the resulting a or b fiber from their DLMs is illustrated in Fig. 5. In the three cases of transheterozygotes, muscle structure is closer to normal than in each of the four independent D mutants. In addition, hdp2;D1/ D41 flies are able to jump while the D/+ mutants are not. Interestingly, the D1/D62 combination exhibits a high number of double thick filaments. This abnormal feature is rarely seen in D1/+ or D62/+ muscles. The synergistic effects of D1 suppression observed in combination with each of the other alleles suggests that D1 employs a unique suppression mechanism compared with the other D-series Mhc alleles.
Figure 5.

Double suppressor combinations. Longitudinal (A, C, and E) and transverse (B, D, and F) views of D1/ D45 (A and B), D1/D41 (C and D) and D1/D62 (E and F) dorsolongitudinal muscles in males carrying the hdp2 mutation. Note the improved restoration of muscle structure in cases of D41 and D45 combinations with D1 suggesting independent and additive mechanisms of suppression. In the case of the D1/ D62 combination, although there is an additional improvement in the restoration of sarcomere and filament array, there is a new structural feature: the frequent assembly of thick filaments in pairs. Bar: (A, C, and E) 775 nm; (B, D, and F) 144 nm.
Next, we tested whether other Mhc alleles are capable of suppression of hdp2 phenotypes, either alone or in combination with D-series suppressors (Table III). Three point mutations and the H20 deficiency chromosome were chosen to observe effects of specific amino acid changes or reduction in MHC levels upon the hdp2 phenotypes. Homyk and Emerson (1988) had previously described a negative interaction between two of these alleles (Mhc5 and Mhc8) and hdp2. Our data corroborated that Mhc5 is lethal in combination with hdp2/Y, but showed a reduced viability, rather than complete lethality, between Mhc8 and hdp2/Y (Table III). The heldup phenotype was maintained in viable organisms in the latter case. This was also seen for the Mhc6 point mutation and the deficiency chromosome. These results indicate that underexpression of MHC or non-D point mutations known to cause a dominant flightless phenotype do not suppress the heldup wing phenotype associated with specific troponin I allele hdp2.
Table III.
Interactions Between D Suppressors and other Mhc Mutations in hdp2 Males*
| Mhc5 | Mhc6 | Mhc8 | + | Df(2)H20 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| D1 | Lethal | + | hdp | + | Viable | |||||
| D41 | hdp | + | Lethal‡ | + | Lethal | |||||
| D45 | hdp | 50% hdp | Poorly viable | + | Lethal | |||||
| D62 | hdp | + | Lethal‡ | + | Lethal | |||||
| + | Lethal | hdp | Poorly viable hdp | hdp | hdp | |||||
| Df(2)H20 | Viable/ | Viable/ | Lethal | Viable/ | Lethal | |||||
| flightless | flightless | flightless |
Minimum of 100 offspring per cross were screened.
Lethal also in females hdp2/+.
We studied the non-suppressor Mhc point mutants in more detail in an attempt to clarify their ability or inability to interact with the hdp2 mutation. Each mutant accumulates substantial levels of MHC in adult thoraces: Mhc 5 homozygotes at 88% of wild-type levels, Mhc6 homozygotes at nearly 100% (Kronert et al., 1995), and Mhc8/+ (which is recessive lethal) at 79% (Mogami et al., 1986). Mhc6 is a point mutation (Arg to His) in the rod of the myosin molecule (Kronert et al., 1995). We determined molecular defects in the other two mutants by sequencing clones containing PCR-amplified copies of their Mhc genes. As suspected, these mutations result from single amino acid changes. In the case of Mhc5, amino acid 200 is mutated from a Gly to Asp (resulting from an A to G transition in exon 4). On the three-dimensional crystal structure, this residue is located near the base of loop 1 of the molecule, at the beginning of a long helix that appears to interact with the bound nucleotide (Fig. 2). Interestingly, the mutated amino acid in Mhc5 is quite close to residue 261, which is mutated in suppressor strain D45. The Mhc8 mutation is located in the region that binds regulatory light chain, at residue 832 (Fig. 2). The C to T mutation in exon 12 results in a change from Tyr to His. This portion of the molecule is part of the lever arm that is proposed to move during the myosin power stroke, due to pivoting about a point near the active site (Holmes, 1997; Dominguez et al., 1998).
These three Mhc point mutations exhibit very different effects when tested in combination with the D mutations in a hdp2 background (Table III). D1 is lethal when over Mhc5, but viable over the other two Mhc alleles and the deficiency chromosome (Df(2)H20). In contrast, Mhc8 is lethal or poorly viable over D41, D45, or D62, but not over D1. The Mhc6 mutation has no effect on viability in combination with suppressor mutations or on their ability to suppress heldup wing phenotype, except for a reduction in suppression with the D45 allele.
Finally, we tested the troponin I allele specificity of heldup wing suppression by D-series mutations. We used hdp3 or hdp2/hdp3 as alternative backgrounds. The hdp3 point mutation causes abnormal RNA splicing, resulting in failure of a specific subset of troponin I isoforms to accumulate in the indirect flight muscles (Barbas et al., 1993). hdp3 mutants display a paucity of thin filaments and severely disrupted myofibrils (Beall and Fyrberg, 1991). We detected no suppression in hdp3 or hdp2/hdp3 backgrounds, indicating that D-series alleles suppress a specific molecular defect in hdp2 mutation.
Taken together, our genetic studies demonstrate that suppression of the heldup wing phenotype in the hdp2 point mutant can only result from specific modifications of MHC structure, as opposed to other perturbations in MHC structure or reductions in myosin concentration. Conversely, structural defects in DLMs caused by depletion of certain troponin I isoforms cannot be suppressed by these single amino acid changes in MHC.
Discussion
In this paper, we identified an unexpected interrelationship between myosin and troponin I through the use of a mutational screen for increased muscle function and integrity. We demonstrated that specific mutations in Mhc revert the heldup wings phenotype and muscle degeneration displayed by flies carrying the hdp2 allele of troponin I. This reversion is allele specific, both for troponin I mutations and mutations in myosin, indicating that our approach identifies a novel type of functional interaction between the muscle proteins. Our data demonstrate that suppressive effects of D-series mutations do not arise simply from a reduction in myosin. This is based on accumulation of MHC in the mutant lines, as well as the failure of Mhc null mutations to suppress hdp2.
The role of the amino acid mutated in hdp2 may be inferred from recent structural and functional studies on this region of the protein in vertebrate troponin I. The hdp2 mutation affects the NH2-terminal α-helical portion of the protein shown to interact with troponin C (Farah et al., 1994; Tripet et al., 1997; Leszyk et al., 1998; Vassylyev et al., 1998). Rabbit skeletal muscle troponin I/troponin C cocrystal structure shows hydrophobic interactions between residue 25, which corresponds to the site of hdp2 mutation, and troponin C (Vassylyev et al., 1998). Although interaction between troponin I and troponin C appeared stable (Farah et al., 1994), the NH2-terminal fragment is now proposed to be released upon Ca2+ binding to troponin C (Tripet et al., 1997; Vassylyev et al., 1998). This release permits binding of an inhibitory domain of troponin I to troponin C, allowing the tropomyosin strand to move from its position blocking actin–myosin interaction. A reasonable model for hdp2 defect is that the mutation hastens release of the α helix at lower Ca2+ concentrations, resulting in more ready binding of troponin I's inhibitory domain to troponin C. Unregulated actin–myosin interaction would result. The hypercontracted sarcomeres and muscle degeneration observed are consistent with this model (Fig. 1), as is the requirement for thick filaments for the degenerative phenotype (Beall and Fyrberg, 1991).
The four suppressor alleles within the Mhc gene may identify specific molecular interactions between troponin I and myosin. Direct interaction between the troponin complex and the myosin head in insect flight muscle is structurally feasible, since antibody labeling of troponin complexes show they occur at some sites of rigor crossbridge attachment (Reedy et al., 1994). Myosin interaction may occur directly with the wild-type troponin I residue identified by the hdp2 mutation, perhaps aiding release of the surrounding α-helical region during Ca2+ binding by troponin C. This would facilitate actomyosin interactions, allowing the thin filament to progress to a fully active state. When poor regulation occurs in the hdp2 mutant, the suppressor mutation could prevent or alter myosin interaction with the troponin I molecule. This would decrease the mutant troponin I's ability to release from troponin C, allowing the blocking action of troponin I on actomyosin interaction to continue at low Ca2+ concentrations. More normal muscle structure and function would result. Thus, while the troponin I mutation could alter the equilibrium among the three states of the thin filament proposed by McKillop and Geeves (1993) and Vibert et al. (1997), this equilibrium could be reestablished through a compensating mutation in the myosin head. The observation by Lin et al. (1996), that troponin mutations can alter cycling of crossbridges, supports this possibility.
Direct interaction between mutated residues in troponin I and the myosin head is feasible for the residues identified by the D62 Mhc mutation. Biochemical (Mornet et al., 1981; Sutoh, 1982), structural (Rayment et al., 1993a,b), and chimeric molecule studies (Uyeda et al., 1994; Rovner et al., 1995) indicate that residues deleted from the actin binding loop of MHC in mutation D62 normally interact with the thin filament during the crossbridge cycle. For suppressor mutation D1, changes in orientation of the actin-binding loop could result from amino acid alteration at the loop's base. Instead of revealing a direct interaction between troponin I and MHC, D1 or D62 could affect crossbridge cycling and indirectly compensate for the troponin I mutation. The mechanism of action of these two suppressors may be similar. However, the synergistic effect of D1 when combined with the other D suppressors, and the peculiar effect of D1 in combination with other Mhc alleles (Table III), suggests that this suppressor elicits a different, albeit unknown, functional change.
Direct interaction between the MHC regions identified by the other two suppressor mutations (D41 and D45) and troponin I is not as obvious a possibility. However, it is important to realize that crystal structures of the myosin head represent static pictures of particular stages of the mechanochemical cycle. Thus, other contacts between thick and thin filaments are possible. A more likely explanation involves nucleotide exchange. Since both mutations are located near the nucleotide entry site of the molecule, it is reasonable to postulate that they would affect the ATPase cycle by regulating nucleotide entry or exit from the binding pocket (Murphy and Spudich, 1998; Sweeney et al., 1998). ADP release is the rate-limiting step in unloaded shortening of some muscles (Siemankowski et al., 1985). If suppressor mutations reduce the rate of ADP release, myosin's dissociation from actin, which occurs upon subsequent binding of ATP, would be inhibited. This could dampen the unregulated actomyosin interactions that appear to occur in the hdp2 mutant, since the ability of the myosin molecule to bind ATP and go through another step of the mechanochemical cycle would be reduced.
Another consideration for the mechanism of suppression is that myosin could act through a third protein to regulate troponin I. In this situation, troponin I would interact indirectly with myosin, through another protein or protein complex (such as tropomyosin or other components of the troponin complex). When troponin I has an abnormal interaction with this partner in the hdp2 mutant, the partner is unable to productively interact with myosin, unless a specific interacting site (the location of the suppressor mutation) is altered. Actin is an obvious possibility for such an intermediary protein, since it interacts with the troponin/tropomyosin complex, as well as with myosin.
A key result of our study is that specific residues on MHC are required for suppression, suggesting they are critical to thick–thin filament interactions. None of the other alleles of Mhc, including point mutations, suppress the heldup wing phenotype (Table III). This includes a mutation in the motor domain (Mhc5), a mutation in the lever arm (Mhc8), and a mutation in the rod (Mhc6). Interestingly, the genotype hdp2;Mhc5/+ results in a lethal interaction (Table III, and Homyk and Emerson, 1988). The location of this mutation close to the site of nucleotide entry/exit, and near D41 and D45 suppressors suggests that Mhc5 might affect the ATPase cycle in the reverse direction of suppressors, thereby exacerbating rather than ameliorating the hdp2 phenotypes. Support for this hypothesis is provided by the observation that lethality, but not heldup phenotype, of the hdp2;Mhc5/+ genotype is eliminated when either the D41, D45, or D62 suppressors replace the wild-type Mhc allele (Table III). D1 is an exception in rescuing lethality of the hdp2;Mhc5 combination. In contrast, MHC of the D1 type is compatible with Mhc8 for viability, but this is not so with D41, D45, or D62 (Table III). The opposite effects of D1 and other suppressor alleles strengthens our conclusion from suppressor heterozygote studies that D1 MHC acts to suppress the hdp2 phenotype by a different mechanism than other suppressors.
Our studies have implications for understanding disease processes in humans. In familial hypertrophic cardiomyopathy, single amino acid changes in a number of contractile proteins affect crossbridge cycling, resulting in myofibrillar disarray and hypertrophy (Towbin, 1998; Watkins et al., 1995). Mutations implicated in this disease include numerous defects in the myosin S1 domain (Rayment et al., 1995) and in troponin I (Kimura et al., 1997). Thus, mutations in both thick and thin filament components can have similar consequences upon human cardiac muscle structure and function. A confounding factor in understanding the basis of disease process, and predicting its severity, is that genetic background influences disease penetrance. Our observations in Drosophila indicate that mutations in other components of the contractile apparatus can either exacerbate or ameliorate muscle dysfunction, and could serve as a model for understanding influences of genetic background upon disease penetrance. Further, our findings suggest suppression of human diseases by a mutated version of a contractile protein might prove useful in developing therapeutic strategies.
Abbreviations used in this paper: D
Su(hdp2)D
- DLM
dorsolongitudinal muscle
- MHC
myosin heavy chain
- Mhc
myosin heavy chain gene
Footnotes
We appreciate the help of Dr. Ronald Milligan (The Scripps Research Institute) in preparation of Fig. 2. We thank Drs. Richard Cripps (University of New Mexico), Larry Tobacman (University of Iowa), and Douglas Swank (San Diego State University) for helpful comments on the manuscript.
This research was supported by grants from the Muscular Dystrophy Association and the National Institutes of Health (GM32443) to S.I. Bernstein, and from the Direccion General de Investigacíon Cientifica y Técnica (Spanish Ministry of Culture; PM96-0006) to A. Ferrús.
References
- al-Khayat HA, Yagi N, Squire JM. Structural changes in actin-tropomyosin during muscle regulation: computer modelling of low-angle X-ray diffraction data. J Mol Biol. 1995;252:611–632. doi: 10.1006/jmbi.1995.0524. [DOI] [PubMed] [Google Scholar]
- Barbas JA, Galceran J, Torroja L, Prado A, Ferrús A. Abnormal muscle development in the heldup3 mutant of Drosophila melanogasteris caused by a splicing defect affecting selected troponin I isoforms. Mol Cell Biol. 1993;13:1433–1439. doi: 10.1128/mcb.13.3.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall CJ, Fyrberg E. Muscle abnormalities in Drosophila melanogasterheldup mutants are caused by missing or aberrant troponin-I isoforms. J Cell Biol. 1991;114:941–951. doi: 10.1083/jcb.114.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein SI, Milligan RA. Fine tuning a molecular motor: the location of alternative domains in the Drosophilamyosin head. J Mol Biol. 1997;271:1–6. doi: 10.1006/jmbi.1997.1160. [DOI] [PubMed] [Google Scholar]
- Collier VL, Kronert WA, O'Donnell PT, Edwards KA, Bernstein SI. Alternative myosin hinge regions are utilized in a tissue-specific fashion that correlates with muscle contraction speed. Genes Dev. 1990;4:885–895. doi: 10.1101/gad.4.6.885. [DOI] [PubMed] [Google Scholar]
- Cripps RM, Becker KD, Mardahl M, Kronert WA, Hodges D, Bernstein SI. Transformation of Drosophila melanogasterwith the wild-type myosin heavy-chain gene: rescue of mutant phenotypes and analysis of defects caused by overexpression. J Cell Biol. 1994;126:689–699. doi: 10.1083/jcb.126.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez R, Freyzon Y, Trybus KM, Cohen C. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell. 1998;94:559–571. doi: 10.1016/s0092-8674(00)81598-6. [DOI] [PubMed] [Google Scholar]
- Drummond DR, Hennessey ES, Sparrow JC. Characterisation of missense mutations in the Act88F gene of Drosophila melanogaster. . Mol Gen Genet. 1991;226:70–80. doi: 10.1007/BF00273589. [DOI] [PubMed] [Google Scholar]
- Farah CS, Reinach FC. The troponin complex and regulation of muscle contraction. FASEB J. 1995;9:755–767. doi: 10.1096/fasebj.9.9.7601340. [DOI] [PubMed] [Google Scholar]
- Farah CS, Miyamoto CA, Ramos CH, da Silva AC, Quaggio RB, Fujimori K, Smillie LB, Reinach FC. Structural and regulatory functions of the NH2- and COOH-terminal regions of skeletal muscle troponin I. J Biol Chem. 1994;269:5230–5240. [PubMed] [Google Scholar]
- Gengyo-Ando K, Kagawa H. Single charge change on the helical surface of the paramyosin rod dramatically disrupts thick filament assembly in Caenorhabditis elegans. . J Mol Biol. 1991;219:429–441. doi: 10.1016/0022-2836(91)90184-8. [DOI] [PubMed] [Google Scholar]
- Greenwald IS, Horvitz HR. Dominant suppressors of a muscle mutant define an essential gene of Caenorhabditis elegans. . Genetics. 1982;101:211–225. doi: 10.1093/genetics/101.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings GA, Emerson CP., Jr Myosin functional domains encoded by alternative exons are expressed in specific thoracic muscles of Drosophila. . J Cell Biol. 1991;114:263–276. doi: 10.1083/jcb.114.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess NK, Bernstein SI. Developmentally regulated alternative splicing of Drosophilamyosin heavy chain transcripts: in vivo analysis of an unusual 3′ splice site. Dev Biol. 1991;146:339–344. doi: 10.1016/0012-1606(91)90235-u. [DOI] [PubMed] [Google Scholar]
- Holmes KC. The swinging lever-arm hypothesis of muscle contraction. Curr Biol. 1997;7:R112–R118. doi: 10.1016/s0960-9822(06)00051-0. [DOI] [PubMed] [Google Scholar]
- Homyk T, Jr, Emerson CP., Jr Functional interactions between unlinked muscle genes within haploinsufficient regions of the Drosophilagenome. Genetics. 1988;119:105–121. doi: 10.1093/genetics/119.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowett, T. 1986. Preparation of nucleic acids. In Drosophila, a Practical Approach. D.B. Roberts, editor. IRL Press, Oxford. 275–286.
- Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 1997;16:379–382. doi: 10.1038/ng0897-379. [DOI] [PubMed] [Google Scholar]
- Kronert WA, O'Donnell PT, Fieck A, Lawn A, Vigoreaux JO, Sparrow JC, Bernstein SI. Defects in the Drosophilamyosin rod permit sarcomere assembly but cause flight muscle degeneration. J Mol Biol. 1995;249:111–125. doi: 10.1006/jmbi.1995.0283. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leszyk J, Tao T, Nuwaysir LM, Gergely J. Identification of the photocrosslinking sites in troponin-I with 4-maleimidobenzophenone labelled mutant troponin-Cs having single cysteines at positions 158 and 21. J Muscle Res Cell Motil. 1998;19:479–490. doi: 10.1023/a:1005352324741. [DOI] [PubMed] [Google Scholar]
- Lin D, Bobkova A, Homsher E, Tobacman LS. Altered cardiac troponin T in vitro function in the presence of a mutation implicated in familial hypertrophic cardiomyopathy. J Clin Invest. 1996;97:2842–2848. doi: 10.1172/JCI118740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley, D.L., and G. Zimm. 1992. The Genome of Drosophila melanogaster. Academic Press, San Diego. 1133 pp.
- Mardahl M, Cripps RM, Rinehart RR, Bernstein SI, Harris GL. Introduction of y+ onto a CyOchromosome. Drosophila Inform Serv. 1993;72:141–142. [Google Scholar]
- McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger JM. Myosin binding-induced cooperative activation of the thin filament in cardiac myocytes and skeletal muscle fibers. Biophys J. 1995;68:1430–1442. doi: 10.1016/S0006-3495(95)80316-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerman DG, Plurad S, Waterston RH, Baillie DL. Mutations in the unc-54 myosin heavy chain gene of Caenorhabditis elegansthat alter contractility but not muscle structure. Cell. 1982;29:773–781. doi: 10.1016/0092-8674(82)90439-1. [DOI] [PubMed] [Google Scholar]
- Mogami K, O'Donnell PT, Bernstein SI, Wright TR, Emerson CP., Jr Mutations of the Drosophilamyosin heavy-chain gene: effects on transcription, myosin accumulation, and muscle function. Proc Natl Acad Sci USA. 1986;83:1393–1397. doi: 10.1073/pnas.83.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornet D, Bertrand R, Pantel P, Audemard E, Kassab R. Structure of the actin-myosin interface. Nature. 1981;292:301–306. doi: 10.1038/292301a0. [DOI] [PubMed] [Google Scholar]
- Murphy CT, Spudich JA. Dictyostelium myosin 25–50K loop substitutions specifically affect ADP release rates. Biochemistry. 1998;37:6738–6744. doi: 10.1021/bi972903j. [DOI] [PubMed] [Google Scholar]
- O'Donnell PT, Bernstein SI. Molecular and ultrastructural defects in a Drosophilamyosin heavy chain mutant: differential effects on muscle function produced by similar thick filament abnormalities. J Cell Biol. 1988;107:2601–2612. doi: 10.1083/jcb.107.6.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell PT, Collier VL, Mogami K, Bernstein SI. Ultrastructural and molecular analyses of homozygous-viable Drosophila melanogastermuscle mutants indicate there is a complex pattern of myosin heavy-chain isoform distribution. Genes Dev. 1989;3:1233–1246. doi: 10.1101/gad.3.8.1233. [DOI] [PubMed] [Google Scholar]
- Park EC, Horvitz HR. C. elegans unc-105mutations affect muscle and are suppressed by other mutations that affect muscle. Genetics. 1986;113:853–867. doi: 10.1093/genetics/113.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckham M, Molloy JE, Sparrow JC, White DC. Physiological properties of the dorsal longitudinal flight muscle and the tergal depressor of the trochanter muscle of Drosophila melanogaster. . J Muscle Res Cell Motil. 1990;11:203–215. doi: 10.1007/BF01843574. [DOI] [PubMed] [Google Scholar]
- Prado A, Canal I, Barbas JA, Molloy J, Ferrús A. Functional recovery of troponin I in a Drosophilaheldup mutant after a second site mutation. Mol Biol Cell. 1995;6:1433–1441. doi: 10.1091/mbc.6.11.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993a;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993b;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- Rayment I, Holden HM, Sellers JR, Fananapazir L, Epstein ND. Structural interpretation of the mutations in the beta-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 1995;92:3864–3868. doi: 10.1073/pnas.92.9.3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedy MC, Reedy MK, Leonard KR, Bullard B. Gold/Fab immuno electron microscopy localization of troponin H and troponin T in Lethocerusflight muscle. J Mol Biol. 1994;239:52–67. doi: 10.1006/jmbi.1994.1350. [DOI] [PubMed] [Google Scholar]
- Rovner AS, Freyzon Y, Trybus KM. Chimeric substitutions of the actin-binding loop activate dephosphorylated but not phosphorylated smooth muscle heavy meromyosin. J Biol Chem. 1995;270:30260–30263. doi: 10.1074/jbc.270.51.30260. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: a Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Siemankowski RF, Wiseman MO, White HD. ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle. Proc Natl Acad Sci USA. 1985;82:658–662. doi: 10.1073/pnas.82.3.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire JM. Architecture and function in the muscle sarcomere. Curr Opin Struct Biol. 1997;7:247–257. doi: 10.1016/s0959-440x(97)80033-4. [DOI] [PubMed] [Google Scholar]
- Sutoh K. An actin-binding site on the 20K fragment of myosin subfragment 1. Biochemistry. 1982;21:4800–4804. doi: 10.1021/bi00262a043. [DOI] [PubMed] [Google Scholar]
- Sweeney HL, Rosenfeld SS, Brown F, Faust L, Smith J, Xing J, Stein LA, Sellers JR. Kinetic tuning of myosin via a flexible loop adjacent to the nucleotide binding pocket. J Biol Chem. 1998;273:6262–6270. doi: 10.1074/jbc.273.11.6262. [DOI] [PubMed] [Google Scholar]
- Towbin JA. The role of cytoskeletal proteins in cardiomyopathies. Curr Opin Cell Biol. 1998;10:131–139. doi: 10.1016/s0955-0674(98)80096-3. [DOI] [PubMed] [Google Scholar]
- Tripet B, Van Eyk JE, Hodges RS. Mapping of a second actin-tropomyosin and a second troponin C binding site within the C terminus of troponin I, and their importance in the Ca2+-dependent regulation of muscle contraction. J Mol Biol. 1997;271:728–750. doi: 10.1006/jmbi.1997.1200. [DOI] [PubMed] [Google Scholar]
- Uyeda TQ, Ruppel KM, Spudich JA. Enzymatic activities correlate with chimaeric substitutions at the actin-binding face of myosin. Nature. 1994;368:567–569. doi: 10.1038/368567a0. [DOI] [PubMed] [Google Scholar]
- Van Eyk JE, Thomas LT, Tripet B, Wiesner RJ, Pearlstone JR, Farah CS, Reinach FC, Hodges RS. Distinct regions of troponin I regulate Ca2+-dependent activation and Ca2+sensitivity of the acto-S1-TM ATPase activity of the thin filament. J Biol Chem. 1997;272:10529–10537. doi: 10.1074/jbc.272.16.10529. [DOI] [PubMed] [Google Scholar]
- Vassylyev DG, Takeda S, Wakatsuki S, Maeda K, Maeda Y. Crystal structure of troponin C in complex with troponin I fragment at 2.3-Å resolution. Proc Natl Acad Sci USA. 1998;95:4847–4852. doi: 10.1073/pnas.95.9.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J Mol Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- Watkins H, Seidman JG, Seidman CE. Familial hypertrophic cardiomyopathy: a genetic model of cardiac hypertrophy. Hum Mol Genet. 1995;4:1721–1727. doi: 10.1093/hmg/4.suppl_1.1721. [DOI] [PubMed] [Google Scholar]