

Abstract
During apoptosis induced by various stimuli, cytochrome c is released from mitochondria into the cytosol where it participates in caspase activation. This process has been proposed to be an irreversible consequence of mitochondrial permeability transition pore opening, which leads to mitochondrial swelling and rupture of the outer mitochondrial membrane. Here we present data demonstrating that NGF-deprived sympathetic neurons protected from apoptosis by caspase inhibitors possess mitochondria which, though depleted of cytochrome c and reduced in size, remained structurally intact as viewed by electron microscopy. After re-exposure of neurons to NGF, mitochondria recovered their normal size and their cytochrome c content, by a process requiring de novo protein synthesis. Altogether, these data suggest that depletion of cytochrome c from mitochondria is a controlled process compatible with function recovery. The ability of sympathetic neurons to recover fully from trophic factor deprivation provided irreversible caspase inhibitors have been present during the insult period, has therapeutical implications for a number of acute neuropathologies.
Keywords: apoptosis, Bax, cytochrome c, mitochondria, caspases
Mitochondria have been found to be essential in controlling at least certain apoptosis pathways (Green and Reed, 1998). The mechanisms by which they exert this function include release of caspase activators as cytochrome c (Liu et al., 1996) and apoptosis-inducing factor (Susin et al., 1996), and disruption of electron transport and oxidative phosphorylation (Hockenbery et al., 1993; Kane et al., 1993; Sarafian and Bredesen, 1994; Greenlund et al., 1995; Zamzami et al., 1995; Adachi et al., 1997; Garcia-Ruiz et al., 1997). Cytochrome c has been reported to be released from mitochondria into the cytosol of many cell types undergoing apoptosis (Chauhan et al., 1997; Kluck et al., 1997; Yang et al., 1997). Moreover, mitochondrial cytochrome c release has been shown to be required for apoptosis to occur in sympathetic neurons deprived of NGF (Neame et al., 1998). In the cytosol, cytochrome c participates in caspase activation through binding to Apaf-1 and caspase 9 (Liu et al., 1996; Li et al., 1997). The redistribution of cytochrome c during apoptosis can be prevented by overexpression of the anti-apoptotic protein Bcl-2 (Kluck et al., 1997; Yang et al., 1997). In contrast, just overexpression of the pro-apoptotic protein Bax has been shown to trigger cytochrome c efflux from mitochondria (Eskes et al., 1998; Rossé et al., 1998). Altogether, these results suggest that the release of mitochondrial cytochrome c is tightly regulated by Bcl-2 family members. However, the mechanisms by which cytochrome c is released from mitochondria is still unclear. It has been proposed that opening of the permeability transition pore (PTP)1 (Zoratti and Szabo, 1995; Bernardi and Petronilli, 1996; Beutner et al., 1996; Nicolli et al., 1996; Halestrap et al., 1997) which leads to mitochondrial swelling and possibly to rupture of the mitochondrial outer membrane (Vander Heiden et al., 1997) allows the passive release of caspase-activating proteins from the intermembrane space of mitochondria into the cytosol (Marchetti et al., 1996; Susin et al., 1996; Green and Reed, 1998). However, it has also been reported that, in many cell types, the release of cytochrome c occurs before or in the absence of a change in mitochondrial permeability suggesting that this process involves mechanisms other than (or in addition to) opening of the PTP (Kluck et al., 1997; Yang et al., 1997; Bossy-Wetzel et al., 1998; Green and Reed, 1998). Here we report that in apoptosis of sympathetic neurons induced by NGF deprivation, cytochrome c is released from mitochondria in the absence of mitochondrial swelling. Moreover, we show that addition of NGF back to neurons rescued by Boc-aspartyl(Ome)-fluoromethylketone (BAF) leads to restoration of normal cytochrome c content by mitochondria.
Materials and Methods
Culture of Sympathetic Neurons
Sympathetic neurons from newborn rat cervical superior ganglia were cultured as previously described (Martinou et al., 1995). For electron microscopy studies, neurons were fixed with 1% glutaraldehyde in PBS for 48 h at 4°C and washed in PBS before pre-embedding.
Subcellular Fractionation
Sympathetic neurons (2 × 105 in 3.5-cm-diam Petri dish) were harvested in 100 μl of isotonic buffer (210 mM mannitol, 70 mM sucrose, 1 mM EDTA, and 10 mM Hepes, pH 7.5) supplemented with protease inhibitors cocktail Complete (Boehringer Mannheim) and homogenized with a Dounce homogenizer. Samples were transferred to Eppendorf centrifuge tubes, centrifuged at 900 g for 5 min to remove nuclei, and then followed by centrifugation at 10,000 g for 30 min at 4°C to obtain the heavy membrane pellet (HM) enriched in mitochondria. The HM material was resuspended in 20 μl PBS, 0.2% Triton X-100. The protein concentration was determined by the method of Bradford (1976) in both the HM and soluble fractions. 5 μg (HM fraction) and 8 μg (soluble fraction) were used for Western blotting.
Isolation of Mouse Liver Mitochondria and Incubation with Bax
Mitochondria were isolated by sucrose density gradient centrifugation as previously described (Eskes et al., 1998). Mitochondria were incubated with 5 μM BaxΔTm for 30 min at 30°C in a buffer containing 125 mM KCl, 4 mM MgCl2, 5 mM Na2HPO4, 5 mM succinate, 5 μM rotenone, 0.5 mM EGTA, 15 mM Hepes-KOH, pH 7.4. For electron microscopy, pellets of mitochondria were fixed in 1.5% glutaraldehyde in Sorensen phosphate buffer for 1 h at 4°C and processed as indicated.
Electron Microscopy Studies
Pellets of glutaraldehyde-fixed neurons and isolated mitochondria were pre-embedded into low viscosity agarose, washed with Sorensen phosphate buffer, and then postfixed in 2% OsO4 in phosphate buffer for 1 h at room temperature. Then the samples were washed again in phosphate buffer, dehydrated in alcohol and propylene oxide, and then embedded in Epon. Ultrathin sections of comparable thickness were prepared with a Leica Ultracut ultramicrotome and placed on formvar carbon-coated copper grids. The grids were stained with uranyl acetate and lead citrate and observed with a Philips CM10 transmission electron microscope at 80 kV using a 30–40-μm objective aperture.
Immunocytochemistry
Neurons were fixed with 4% paraformaldehyde in PBS, permeabilized for 10 min with PBS containing 0.2% Triton X-100, incubated for 2 h with an anti–cytochrome c monoclonal antibody (dilution 1:15 in PBS with 5% normal goat serum; PharMingen), and then revealed with a fluorescein-labeled goat anti–mouse antibody.
Results
Cytochrome C Release from NGF-deprived Sympathetic Neurons
We have studied the distribution of cytochrome c in cultured sympathetic neurons from rat superior cervical ganglia (SCG) undergoing apoptosis induced by NGF deprivation. Cytochrome c was analyzed by Western blotting in both soluble cytosolic and the mitochondria enriched heavy membrane (HM) fractions obtained from SCG neurons at 8, 15, and 24 h after NGF. Fig. 1, a and b, highlights a significant decrease of cytochrome c in the HM fraction of neurons deprived of NGF for 8, 15, and 24 h (Fig. 1 b), accompanied by an increase of cytochrome c in the cytosolic fraction (Fig. 1 a). This result was confirmed by immunostaining studies (Fig. 2, a and b). Although all neurons cultured in the presence of NGF displayed a punctate staining (Fig. 2 a), ∼50–75% of those deprived of NGF for 15 h displayed a diffuse cytosolic pattern (Fig. 2 b) and >95% of the latter had a condensed apoptotic nucleus (data not shown). Immunocytochemistry studies revealed that mitochondria present in neurites preserved their cytochrome c content longer than mitochondria present in soma (data not shown).
Figure 1.

Release of cytochrome c from mitochondria of sympathetic neurons. (a and b) Sympathetic neurons from newborn rat SCG were cultured for 5 d in the presence of NGF: on day 5, NGF was withdrawn from the culture medium and the cultures were harvested 8, 15, or 24 h thereafter. Neurons were homogenized in isotonic buffer and the cell extract was separated into soluble (a) and heavy membrane fractions (b). Both fractions were then analyzed for cytochrome c by Western blotting. Lactate dehydrogenase (LDH) and cytochrome oxidase subunit IV (COX IV) were used as gel loading control. (c) Sympathetic neurons were cultured for 5 d in the presence of NGF. On day 5, some cultures were maintained for an additional 5 d either in the continued presence of NGF (lane 1) or in the absence of NGF and presence of 100 μM BAF (lane 2). Some cultures deprived of NGF but supplemented with BAF for 3 d were re-exposed for 2 d to NGF alone (lane 4) or to NGF + 2 μM cycloheximide (lane 3). (d) Sympathetic neurons cultured for 5 d with NGF were deprived of NGF and treated with BAF for 2 d before NGF was added back for 2 and 4 d.
Figure 2.

Cytochrome c immunostaining. Cytochrome c immunostaining of neurons cultured in the following conditions: (a and b) Neurons cultured in the presence of NGF for 5 d followed by 15 h in the continued presence (a) or absence (b) of NGF; (c) neurons cultured for 5 d in the presence of NGF followed by 2 d without NGF but in the presence of 100 μM BAF; (d–f) neurons as in (c) were re-exposed for 1 d to NGF alone (d), to NGF + 5 μM colchicine (e) or to NGF + 2 μM cycloheximide (f) before immunostaining for cytochrome c. In b and c, we have deliberately chosen a field containing a neuron that still shows a normal cytochrome c pattern in order to illustrate more clearly the appearance of neurons with cytochrome c depleted mitochondria. Bar, 30 μm.
We also analyzed the cytochrome c distribution in sympathetic neurons rescued from NGF-deprivation with the caspase peptide inhibitor BAF. In the presence of 100 μM BAF, >80% neurons were able to survive after NGF deprivation for 5 d, although they had markedly smaller somas and atrophic neurites (Fig. 3 c). In agreement with previous data (Deshmukh et al., 1996), we found that BAF was able to inhibit caspase activity as assessed by immunoblot analysis of poly-(ADP ribose) polymerase (data not shown). A major decrease in cytochrome c levels was detected in the HM fraction of those neurons (Fig. 1 c), a finding confirmed by cytochrome c immunostaining (Fig. 2 c). These data indicate that, during apoptosis, the release of cytochrome c from mitochondria of sympathetic neurons does not require caspase activity.
Figure 3.
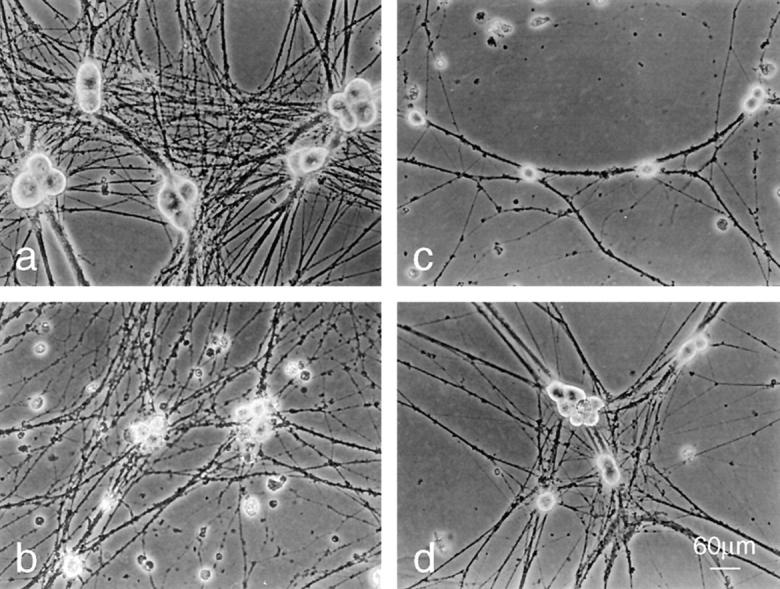
Morphology of BAF-protected neurons before and after re-exposure to NGF. Phase–contrast images of neurons were cultured in the following conditions: (a) neurons cultured for 6 d in the presence of NGF. (b and c) Neurons cultured 5 d in the presence of NGF followed by 24 h in the absence of NGF (b) or 48 h in the absence of NGF and presence of 100 μM BAF (c). (d) Neurons were cultured as in c and re-exposed to NGF for 48 h. Bar, 60 μm.
Cytochrome C Recovery by Mitochondria of NGF-rescued Neurons Re-exposed to NGF
The major objective of this article was to test whether mitochondria from BAF-rescued neurons could recover their cytochrome c content following re-exposure of neurons to NGF. Cytochrome c analysis in HM fraction of neurons deprived of NGF in the presence of BAF for 2 d and then re-exposed to NGF for 3 d, revealed a significant increase in the level of mitochondrial cytochrome c compared with neurons cultured in the absence of NGF and the presence of BAF alone for 5 d (Fig. 1 c). A kinetic analysis revealed a progressive recovery of cytochrome c by mitochondria which was almost complete after a 4-d NGF treatment (Fig. 1 d). Consistent with this finding, immunocytochemistry studies showed reappearance of a punctate mitochondrial cytochrome c staining in almost all neurons (>80%, n = 3) re-exposed to NGF for 24 h (Fig. 2 d). The effect of re-exposure to NGF was blocked by cycloheximide (Fig. 1 c and Fig. 2 f), suggesting the requirement of de novo protein synthesis for its action. It is possible that synthesis of cytochrome c precursor apocytochrome c was a prerequisite for cytochrome c recovery by mitochondria as only apocytochrome c has been shown to be imported into mitochondria (Mayer et al., 1995). In contrast, the recovery of normal cytochrome c levels by mitochondria was not prevented by the microtubule disrupting agent colchicine (Fig. 2 e), therefore excluding the possibility that reappearance of a normal cytochrome c staining pattern in the cell body was due to microtubule associated migration of still intact mitochondria from neurites to the soma.
In agreement with previous data (Deshmukh et al., 1996), we observed that addition of NGF back to BAF-protected SCG neurons caused an increase in soma diameter and neurite extension (Fig. 3 d). Altogether, these results indicate that the NGF receptors TRKA (tyrosine kinase receptor) and their signaling components remained functional in neurons deprived of NGF for at least 5 d. Interestingly, addition of NGF alone (without BAF) back to BAF-rescued neurons was sufficient to promote mitochondria recovery, regrowth of neurons, and long-term survival, suggesting that the caspases which had been activated during apoptosis had been irreversibly inhibited by BAF.
Ultrastructure of Mitochondria
The reappearance of cytochrome c in mitochondria of BAF-rescued neurons re-exposed to NGF suggested that the ultrastructure of mitochondria had been preserved. This hypothesis was tested by electron microscopic studies. Fig. 4 a shows that mitochondria from neurons cultured continuously in the presence of NGF appeared elongated or oval-shaped, with sparse cristae (mean cross-sectional area ± SEM was 0.110 ± 0.007 μm2, n = 115, Fig. 5 a). 24 h after NGF deprivation (Fig. 4 b) most mitochondria were round in shape, smaller than normal (mean cross-sectional area: 0.082 ± 0.004 μm2, mean ± SEM, n = 124, Fig. 5 a) with a hyperdense matrix. No obvious rupture of the outer mitochondrial membrane has been observed. Mitochondria from BAF-rescued neurons (Fig. 4 c) were even smaller (mean cross-sectional area: 0.071 ± 0.005 μm2, mean ± SEM, n = 80, Fig. 5 a), often forming aggregates surrounded by lysosomes (data not shown). After addition of NGF back to BAF-rescued neurons (Fig. 4 d), mitochondria recovered the shape and size typical of neurons continuously cultured in the presence of NGF (0.134 ± 0.08 μm2, mean ± SEM, n = 121, Fig. 5 a). Lysosomes containing myelin figures, lipid droplets, as well as images of autophagy, have also been observed as previously described by Martin et al. (1988).
Figure 4.
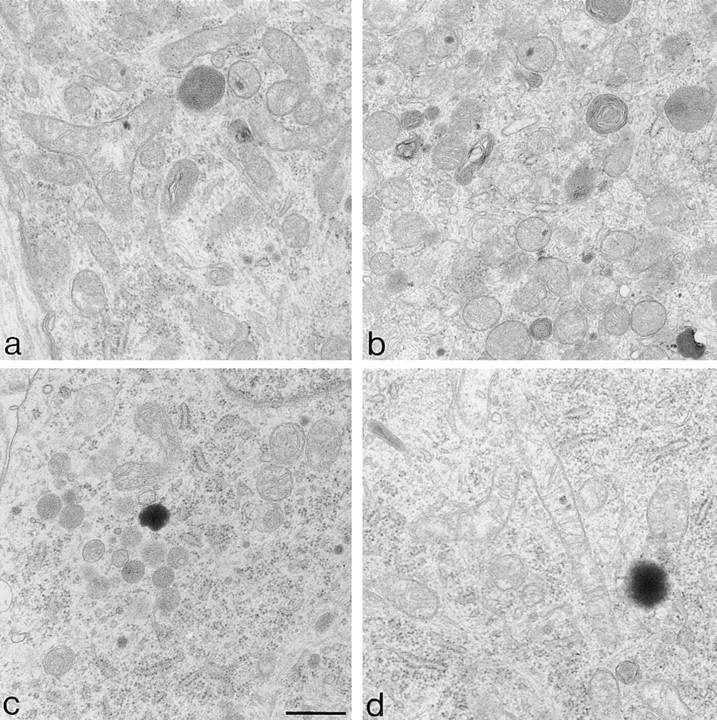
Ultrastructural analysis of mitochondria by electron microscopy. Sympathetic neurons were cultured for 5 d in the presence of NGF. At day 5, some cultures were maintained in the presence of NGF for an additional 2 d (a), or deprived of NGF for 1 d (b) or deprived of NGF but in the presence of BAF (c). After 2 d of treatment with BAF, cultures of neurons protected from apoptosis were re-exposed to NGF for an additional 2 d (d). Bar, 0.5 μm.
Figure 5.
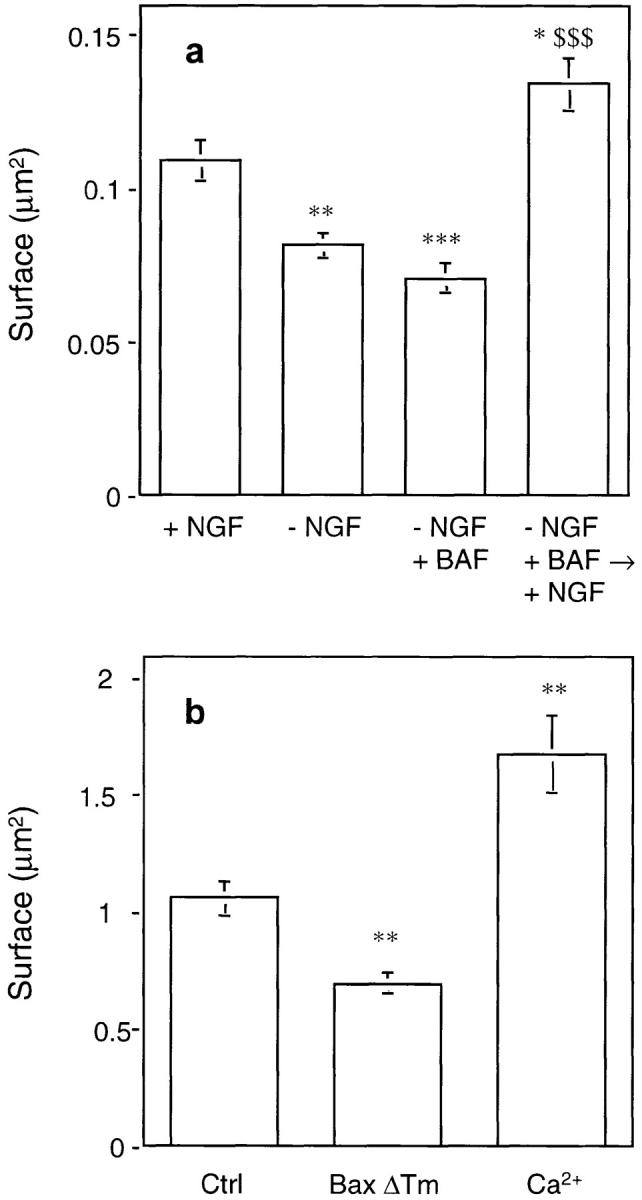
Quantification of mitochondria size. (a) The size of mitochondria from neurons cultured in the conditions described in Fig. 4 was measured using micrographs printed at the same enlargement from negatives taken at original magnification of 11,500. The cross-sectional area of 80–120 mitochondria from five to 10 cells was measured, using NIH Image program (v. 1.58), and the mean area as well as other parameters were calculated using Kaleidagraph program (v. 3.08d). Data represent mean ± SEM for 80–120 mitochondria analyzed. *, P < 0.05; **, P < 0.01; ***, P < 0.001; significantly different from values obtained with + NGF. $$$, P < 0.001; significantly different from values obtained with –NGF + BAF (analysis of variance [ANOVA] followed by Bonferroni's test). (b) Mitochondria from mouse liver were incubated for 30 min at 30°C in the presence of 5 μM BaxΔTm or 100 μM free calcium before ultrastructural analysis (see also Fig. 6). The mitochondrial cross-sectional area was measured as described in a. Results represent mean ± SEM for 60–110 mitochondria analyzed. **, P < 0.01; significantly different from control values (ANOVA followed by Bonferroni's test).
Bax Induces Mitochondrial Shrinking and Triggers Cytochrome C Efflux from Mitochondria
Bax is known to play an essential role in neuronal apoptosis. Sympathetic neurons from Bax-deficient mice can survive more than 20 d in the absence of NGF (Deckwerth et al., 1996). Moreover, Bax is known to trigger cytochrome c release when added directly to isolated mitochondria (Eskes et al., 1998; Jürgensmeier et al., 1998). We therefore wondered whether Bax could also be responsible for the mitochondrial morphological change observed in neurons after NGF deprivation. This hypothesis was tested by electron microscopy using isolated mitochondria that had been incubated for 15 min in the presence of 5 μM BaxΔTm or, for comparison, with 100 μM calcium which stimulates PTP opening (Fig. 5 b and Fig. 6). Although, as previously reported (Petronilli et al., 1993), opening of the PTP with calcium led to mitochondrial swelling, Bax, in contrast, triggered mitochondrial shrinkage resulting in a morphology similar to that observed in apoptotic neurons (Fig. 5 b and Fig. 6).
Figure 6.
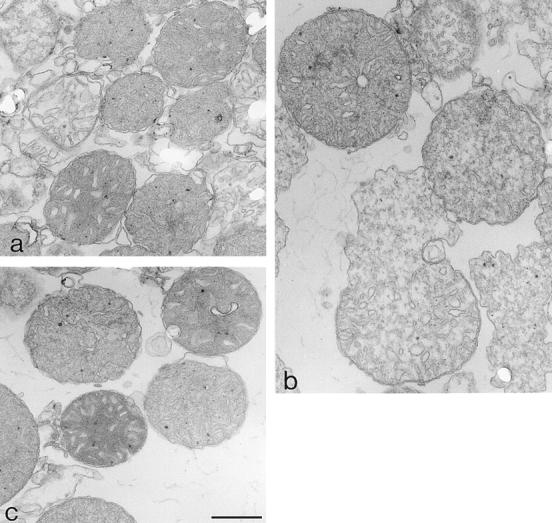
Ultrastructure of isolated mitochondria. Mitochondria treated with Bax (a) (conditions described in Fig. 5 b legend) had, on average, smaller cross-section areas than control mitochondria (c). However, after incubation with calcium (b), mitochondria were generally larger, often showing swelling lesions. Bar, 0.5 μm.
Discussion
We report that in sympathetic neurons undergoing apoptosis triggered by NGF deprivation, mitochondria are reduced in size, display a hyperdense matrix, and are depleted of cytochrome c. A similar morphology of mitochondria lacking cytochrome c was observed in neurons protected from NGF deprivation by the caspase peptide inhibitor BAF. Interestingly, upon re-exposure to NGF, the mitochondria from BAF-rescued neurons recovered both a normal size and cytochrome c content. Altogether, these data do not support the hypothesis according to which cytochrome c release is the result of opening of the PTP (Marchetti et al., 1996; Susin et al., 1996; Green and Reed, 1998) and subsequent mitochondrial swelling and rupture of the outer mitochondrial membrane (Vander Heiden et al., 1997) since neither of these two features were observed in apoptotic neurons. Instead, our data agree with previous morphological studies of various cell types undergoing apoptosis showing preservation of mitochondrial membrane ultrastructure (Wyllie et al., 1980) and a reduction in total mitochondrial volume (Mannweiler and Bernhard, 1957; Rouiller, 1957; Svoboda and Higginson, 1963; Wachstein and Besen, 1964; Hackenbrock, 1968). More recently, ultracondensation of mitochondria has also been observed in apoptotic nodal myocytes (James et al., 1993), in lymphoblastic leukemic cells undergoing tumor necrosis factor-induced apoptosis (Lia et al., 1997), in a colon carcinoma cell line treated with herbimycin A (Mancini et al., 1997) and in etoposide-treated THP.1 cells (Zhuang et al., 1998). In some of these studies, a condensed conformation of mitochondria associated with hyperdensity of the matrix, as described here, has been postulated to be the result of water and ion loss from the matrix and to correspond to low-energy states of mitochondria (Hackenbrock, 1968).
Bax, a pro-apoptotic member of the Bcl-2 family essential for neuronal apoptosis (Deckwerth et al., 1996), can form ion channels in synthetic lipid membranes (Antonsson et al., 1997; Schlesinger et al., 1997) and therefore could be a likely candidate responsible for these mitochondrial changes during apoptosis. In support of this hypothesis, addition of Bax directly to isolated mitochondria triggers the release of cytochrome c (Eskes et al., 1998; Jürgensmeier et al., 1998) by a mechanism that may involve pore formation. We now show that this effect is accompanied by a reduction in mitochondrial volume. However, the mechanisms of action of Bax are still unclear. It has been recently reported that Bax can interact with the adenine nucleotide translocator (ANT), a component of the mitochondrial PTP (Marzo et al., 1998). Moreover, in yeast, the ANT appears to be required for the Bax killing function (Marzo et al., 1998). However, the importance of the ANT in apoptosis of mammalian cells has not yet been demonstrated.
One of the most striking observations reported here is the ability of mitochondria from BAF-rescued neurons to recover a normal size and cytochrome c content after re-exposure of neurons to NGF. The transition from small to large mitochondria induced by NGF deserves particular attention. This may be the result of mitochondrial fusion, an hypothesis that we are currently testing. We cannot exclude the possibility that proliferation of mitochondria may also take part in the complete recovery of neurons associated with the ability of neurons to grow in size, to extend neurites and to survive over long periods after re-exposure to NGF.
Recovery of function after protection by caspase inhibitors does not apply to all cell types. Indeed, it has previously been reported that inhibition of caspases in diverse types of apoptosis is incompatible with long-term survival, suggesting that in those cells caspases are activated after the cells become committed to apoptosis (McCarthy et al., 1997). Moreover, it has been shown that overexpression of Bax in Jurkat cells leads to mitochondrial dysfunction and caspase-independent apoptosis (Xiang et al., 1996). In the case of sympathetic neurons undergoing apoptosis induced by NGF deprivation, the situation is different as caspase activation appears to represent the point at which cells become committed to die (Deshmukh et al., 1996). Consistent with this, caspase inhibitors can inhibit apoptosis induced by overexpression of Bax (Vekrellis et al., 1997; Martinou et al., 1998) or Bak (Martinou et al., 1998) in these neurons. The difference between sympathetic neurons and other cell types may reside in intrinsic specificities of their apoptotic pathway, in specific properties of their mitochondria or could be related, at least in vitro, to their ability to produce ATP through glycolysis rather than through oxidative phosphorylation. The ability of mitochondria to recover fully their function when homeostatic conditions are restored may be specific for mitochondria from neurons. This could explain why in both caspase 3– and caspase 9–deficient mice, only neurons are protected from apoptosis during development (Kuida et al., 1996, 1998; Kakem et al., 1998).
Abbreviations used in this paper
- ANT
adenine nucleotide translocator
- BAF
Boc-aspartyl(Ome)-fluoromethylketone
- HM
heavy membrane fraction enriched in mitochondria
- PTP
permeability transition pore
- SCG
sympathetic neurons from newborn rat cervical superior ganglia
Footnotes
We are very grateful to S. Catsicas (University of Lausanne, Switzerland) for having made possible the collaboration between S. Fakan (University of Lausanne) and J.-C. Martinou and for his interest in this work; to S. Arkinstall and K. Maundrell (SPRI, Geneva, Switzerland) for critical reading of the manuscript; T. Wells for encouraging support; J. Fakan, V. Mamin, and F. Voinesco (University of Lausanne) for excellent technical assistance; N. Ruchonnet and S. Tapia (University of Lausanne) for help with morphometrical and quantitative evaluation of the results; and C. Hebert (SPRI, Geneva, Switzerland) for artwork.
Work performed at the Center of Electron Microscopy (University of Lausanne, Lausanne, Switzerland) was supported by funds from Ares Serono. Part of this work was also supported by grants from the European Community (Biotech grant BIO4CT96 0774 to J.-C. Martinou).
References
- Adachi S, Cross AR, Babior BM, Gottlieb RA. Bcl-2 and the outer mitochondrial membrane in the inactivation of cytochrome c during Fas-mediated apoptosis. J Biol Chem. 1997;272:21878–21882. doi: 10.1074/jbc.272.35.21878. [DOI] [PubMed] [Google Scholar]
- Antonsson B, Conti F, Ciavatta AM, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod J-J, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou J-C. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel; a critical apraisal. J Bioenerg Biomembr. 1996;28:129–136. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- Beutner G, Rueck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin, and adenylate translocator in rat brain resemble the permeability transition pore. FEBS (Fed Eur Biochem Soc) Lett. 1996;396:189–195. doi: 10.1016/0014-5793(96)01092-7. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur Mol Biol Organ) J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Pandey P, Ogata A, Teoh G, Krett N, Halgren R, Rosen S, Kufe D, Kharbanda S, Anderson K. Cytochrome c-dependent and -independent induction of apoptosis in multiple myeloma cells. J Biol Chem. 1997;272:29995–29997. doi: 10.1074/jbc.272.48.29995. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EMJ, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EMJ. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskes R, Antonsson B, Osen-Sand A, Montessuit S, Richter C, Sadoul R, Mazzei G, Nichols A, Martinou J-C. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ions. J Cell Biol. 1998;143:217–224. doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. J Biol Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Greenlund LJS, Deckwerth TL, Johnson EMJ. Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed cell death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- Hackenbrock CR. Chemical and physical fixation of isolated mitochondria in low-energy and high-energy states. Proc Natl Acad Sci USA. 1968;61:598–605. doi: 10.1073/pnas.61.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Woodfield K-Y, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocator. J Biol Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- Hockenbery DM, Oltvai ZN, Yin X-M, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- James TN, Terasaki F, Pavlovich ER, Vikhert AM. Apoptosis and pleomorphic micromitochondriosis in the sinus nodes surgically excised from five patients with the long QT syndrome. J Lab Clin Med. 1993;122:309–323. [PubMed] [Google Scholar]
- Jürgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Örd T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan CK, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan C-Y, Gu Y, Taya C, Karasuyama H, Su MS-S, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lia J, Dourmashkin RR, Newland AC, Kelsey SM. Mitochondrial ultracondensation, but not swelling, is involved in TNF alpha-induced apoptosis in human T-lymphoblastic leukaemic cells. Leuk Res. 1997;21:973–983. doi: 10.1016/s0145-2126(97)00078-7. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Mancini M, Anderson BO, Caldwell E, Sedghinasab M, Paty PB, Hockenbery DM. Mitochondrial proliferation and paradoxical membrane depolarization during terminal differenciation and apoptosis in a human colon carcinoma cell line. J Cell Biol. 1997;138:449–469. doi: 10.1083/jcb.138.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannweiler K, Bernhard W. Recherches ultrastructurales sur une tumeur rénale experimentale du hamster. J Ultrastruct Res. 1957;1:158–169. doi: 10.1016/s0022-5320(57)80004-5. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Haeffner A, Hirsch F, Geuskens M, Kroemer G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med. 1996;184:1155–1160. doi: 10.1084/jem.184.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EMJ. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou I, Fernandez PA, Missotten M, White E, Allet B, Sadoul R, Martinou J-C. Viral proteins E1B19K and p35 protect sympathetic neurons from cell death induced by NGF deprivation. J Cell Biol. 1995;128:201–208. doi: 10.1083/jcb.128.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou I, Missotten M, Fernandez PA, Sadoul R, Martinou J-C. Bax and Bak proteins require caspase activity to trigger apoptosis in sympathetic neurons. Neuroreport. 1998;9:15–19. doi: 10.1097/00001756-199801050-00004. [DOI] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HLA, Prévost M-C, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Mayer A, Neupert W, Lill R. Translocation of apocytochrome c across the outer membrane of mitochondria. J Biol Chem. 1995;270:12390–12397. doi: 10.1074/jbc.270.21.12390. [DOI] [PubMed] [Google Scholar]
- McCarthy NJ, Whyte MKB, Gilbert CS, Evan GI. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A-sensitive channel. J Biol Chem. 1996;271:2185–2192. doi: 10.1074/jbc.271.4.2185. [DOI] [PubMed] [Google Scholar]
- Petronilli V, Cola C, Massari S, Colonna R, Bernardi P. Physiological effectors modify voltage sensing by the cyclosporin A-sensitive permeability transition pore of mitochondria. J Biol Chem. 1993;268:21939–21945. [PubMed] [Google Scholar]
- Rossé T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Rouiller C. Contribution de la microscopie electronique a l'étude du foie normal et pathologique. Ann Anat Pathol. 1957;2:548–562. [PubMed] [Google Scholar]
- Sarafian TA, Bredesen DE. Is apoptosis mediated by reactive oxygen species? . Free Rad Res. 1994;21:1–8. doi: 10.3109/10715769409056549. [DOI] [PubMed] [Google Scholar]
- Schlesinger PH, Gross A, Yin X-M, Yamamoto K, Saito M, Waksman G, Korsmeyer SJ. Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2. Proc Natl Acad Sci USA. 1997;94:11357–11362. doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda DJ, Higginson J. Ultrastructural hepatic changes in rats on a necrogenic diet. Am J Pathol. 1963;43:477–495. [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden, M.G., N.S. Chandel, E.K. Williamson, P.T. Schumacker, and C.B. Thompson. Bcl-xLregulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Vekrellis K, McCarthy MJ, Watson A, Whifield J, Rubin LL, Ham J. Bax promotes neuronal cell death and is downregulated during the development of the nervous system. Development (Camb) 1997;124:1239–1249. doi: 10.1242/dev.124.6.1239. [DOI] [PubMed] [Google Scholar]
- Wachstein M, Besen M. Electron microscopy of renal coagulative necrosis due to dl-serine, with special reference to mitochondrial pyknosis. Am J Pathol. 1964;44:383–400. [PMC free article] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Xiang J, Chao DT, Korsmeyer ST. Bax-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Dinsdale D, Cohen GM. Apoptosis, in human monocytic THP.1 cells, results in the release of cytochrome c from mitochondria prior to their ultracondensation, formation of outer membrane discontinuities and reduction in inner membrane potential. Cell Death Differ. 1998;5:953–962. doi: 10.1038/sj.cdd.4400440. [DOI] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]