

Abstract
In this article, we show that, in transfected COS-1 cells, protein tyrosine phosphatase (PTP)-PEST translocates to the membrane periphery following stimulation by the extracellular matrix protein fibronectin. When plated on fibronectin, PTP-PEST (−/−) fibroblasts display a strong defect in motility. 3 h after plating on fibronectin, the number and size of vinculin containing focal adhesions were greatly increased in the homozygous PTP-PEST mutant cells as compared with heterozygous cells. This phenomenon appears to be due in part to a constitutive increase in tyrosine phosphorylation of p130CAS, a known PTP-PEST substrate, paxillin, which associates with PTP-PEST in vitro, and focal adhesion kinase (FAK). Another effect of this constitutive hyperphosphorylation, consistent with the focal adhesion regulation defect, is that (−/−) cells spread faster than the control cell line when plated on fibronectin. In the PTP-PEST (−/−) cells, an increase in affinity for the SH2 domains of Src and Crk towards p130CAS was also observed. In (−/−) cells, we found a significant increase in the level of tyrosine phosphorylation of PSTPIP, a cleavage furrow–associated protein that interacts physically with all PEST family members. An effect of PSTPIP hyperphosphorylation appears to be that some cells remain attached at the site of the cleavage furrow for an extended period of time. In conclusion, our data suggest PTP-PEST plays a dual role in cell cytoskeleton organization, by promoting the turnover of focal adhesions required for cell migration, and by directly or indirectly regulating the proline, serine, threonine phosphatase interacting protein (PSTPIP) tyrosine phosphorylation level which may be involved in regulating cleavage furrow formation or disassembly during normal cell division.
Keywords: migration, cytokinesis, PTP-PEST, focal adhesion, PSTPIP
The process of cell migration is crucial for the correct development of a mammalian embryo. In an adult organism, cell migration plays an important role in events such as the invasion of a wounded space by fibroblasts and endothelial cells, as well as translocation of lymphocytes and neutrophils to an inflammatory site. In cancer, tumor cells migrate to reach the circulatory system and metastasize throughout the organism (for review see Lauffenburger and Horwitz, 1996). One of the driving forces for cell mobility is actin polymerization/depolymerization, which is partly under the control of the Rho family of small GTPase proteins (Nobes and Hall, 1995; Tapon and Hall, 1997).
Focal adhesions are the sites of contact between the extracellular matrix and the cytoskeleton through the integrin family of transmembrane proteins (Burridge and Chrzanowska-Wodnicka, 1996). They contain many structural proteins such as talin, tensin, and α-actinin, as well as tyrosine kinases such as focal adhesion kinase (FAK),1 Src, and Csk, that are activated upon extracellular matrix binding. Multimeric protein complexes are then formed which contain many different adapter proteins, including p130CAS, Shc, Grb2, Crk, and Nck. These interactions confer an important role to focal adhesions in signal transduction pathways (Hanks and Polte, 1997; Schlaepfer et al., 1997). They also represent a major site of tyrosine phosphorylation in a cell, to a point where, following plating of cells on extracellular components, focal adhesions can be clearly visualized using antiphosphotyrosine antibodies. If the identification of proteins involved in the formation and disassembly of focal adhesion proceeds at a rapid rate (Gilmore and Burridge, 1996), the roles that each play have only begun to be clarified. Recent results show a direct correlation between FAK tyrosine kinase activity and migration rate in vitro, and this effect was correlated with the hyperphosphorylation of p130CAS by the Src kinase (Ilìc et al., 1995; Cary et al., 1996, 1998). The viral form of Src, v-Src, also increases focal adhesion turnover in the transformed cell, and its catalytic activity was shown to be required for this effect (Fincham and Frame, 1998).
Much less is known about the roles of protein tyrosine phosphatases (PTP) in cell migration. Recently, SHP-2 (Yu et al., 1998) was shown to regulate focal adhesions, cell spreading, and migration. Overexpression of a dominant negative mutant of PTP1B resulted in defect in integrin-mediated adhesion and signaling in fibroblasts (Arregui et al., 1998). LAR, a transmembrane PTP, was found to localize to focal adhesions and postulated to regulate their disassembly (Serra-Pages et al., 1995). Another receptor-like PTP, PTP-α, was shown to regulate the activity of Src and thus regulate cell–substratum adhesion (Harder et al., 1998). Another example is the bacterial PTP, Yersinia YopH, that, once translocated into a cell, dephosphorylates p130CAS and FAK, and destabilizes focal adhesions (Black and Bliska, 1997; Persson et al., 1997). Another PTP that dephosphorylates p130CAS is PTP-PEST (Garton et al., 1996). PTP-PEST contains the typical tyrosine phosphatase catalytic domain flanked by proline-rich regions that were shown to interact with several signaling proteins including Grb2 (Charest et al., 1997), paxillin (Shen et al., 1998; Côté, J.-F., C.E. Turner, and M.L. Tremblay, manuscript submitted for publication), as well as p130CAS (Garton et al., 1997) and its two related family members, Sin and Hef1 (Côté et al., 1998). Furthermore, we found that an NPLH motif on the COOH-terminal tail of PTP-PEST was responsible for a constitutive association with the PTB domain of the adaptor protein Shc (Charest et al., 1996). Dephosphorylation of p130CAS can counteract the migration effects of the FAK and Src kinases, and PTP-PEST is therefore a potential regulator of cell motility.
A cleavage furrow–associated protein that is also a substrate for another PEST-like phosphatase, PTP-HSCF, was recently cloned (Spencer et al., 1997). This protein, called PSTPIP, associates via its putative coiled-coil domain with a proline-rich COOH-terminal domain on PTP-HSCF that is conserved in all PEST-like PTPs. Alanine-scanning mutants of PSTPIP demonstrated that Trp in the putative coiled-coil region was essential for in vivo binding to PTP-HSCF, and that this mutant was still capable of association with the cortical cytoskeleton (Dowbenko et al., 1998). More recently, the Src homology-3 (SH3) domain of PSTPIP was shown to associate with the Wiskott-Aldrich syndrome protein (WASP), another protein involved with the regulation of the actin-rich cortical cytoskeleton. This interaction can be disrupted by tyrosine phosphorylation within the SH3 domain of PSTPIP, one of the two tyrosines modified by phosphorylation in response to v-Src cotransfection or pervanadate treatment (Wu et al., 1998b). PSTPIP is homologous to Schizosaccharomyces pombe CDC15p, a phosphorylated protein involved in mediating the cytoskeletal rearrangements of F-actin required for cytokinesis (Fankhauser et al., 1995). When overexpressed in exponentially growing S. pombe, PSTPIP colocalizes with actin in the cleavage furrow and acts as a dominant negative inhibitor of cytokinesis (Spencer et al., 1997).
The present report shows that when COS-1 cells were transfected with PTP-PEST, we could follow a translocation of this PTP to the membrane periphery following plating on the extracellular matrix fibronectin. We then analyzed, using a fibroblast cell line where PTP-PEST was removed by gene targeting (Côté et al., 1998), migration capabilities, and the number of focal adhesions in cell lines homozygous (−/−) and heterozygous (+/−) for the PTP-PEST mutation. Since the (−/−) cells showed a marked decrease in migration and increase in focal adhesions, we then verified the phosphorylation state of important tyrosine phosphorylated proteins found in focal adhesions. In addition to p130CAS, a known substrate of PTP-PEST, other focal adhesion proteins were found to be hyperphosphorylated, FAK and paxillin. Paxillin was recently shown to be associated with PTP-PEST (Shen et al., 1998) but does not seem to be a direct physiological substrate for this PTP (Côté, J.-F., C.E. Turner, and M.L. Tremblay, manuscript submitted for publication). The constitutive hyperphosphorylation of these focal adhesion proteins also seems to increase the spreading rate of (−/−) cells on fibronectin. Interestingly, we found an abnormal incidence of cytokinesing cells which were linked at their cleavage furrow site in the PTP-PEST (−/−) fibroblasts. Coincidentally, PSTPIP was found to be hyperphosphorylated in the same cell line. From these results, we conclude that PTP-PEST plays an important role in cell migration, possibly by helping the breakdown of focal adhesions via the dephosphorylation of paxillin and/or p130CAS, as well as in cytoskeletal rearrangement during cytokinesis, potentially by regulating the phosphorylation level of PSTPIP.
Materials and Methods
Antibodies
Mouse polyclonal anti–PSTPIP-2 antibody is described in Wu et al. (1998a). Rabbit polyclonal anti–PTP-PEST antibody is described in Charest et al. (1995). Mouse monoclonal anticortactin is a gift from Dr. J. Thomas Parsons, University of Virginia, Charlottesville, VA. The following antibodies were purchased as indicated: rabbit anti–FAK (A17) from Santa Cruz Biotechnology, Inc.; mouse antipaxillin from Transduction Laboratories; mouse antivinculin (hVIN-1) from Sigma Chemical Co.; HRP-conjugated anti–mouse and –rabbit IgG antibodies and FITC-conjugated anti–mouse antibody from Jackson ImmunoResearch Laboratories, Inc. The antiphosphotyrosine antibodies used were mouse HRP-conjugated antiphosphotyrosine antibody (PY20) from Transduction Laboratories and the mouse monoclonal 4G10.
To raise polyclonal antibodies against PSTPIP and p130CAS, rabbits were injected intramuscularly with either 100 μg of purified GST PSTPIP (aa 247–415) or GST p130CAS (aa 21–521) in PBS emulsified in complete Freund's adjuvant. Rabbits were boosted with the immunogens emulsified in incomplete Freund's adjuvant every 3 wk. Between each injection, samples of serums from rabbits were collected and analyzed for their abilities to detect either PSTPIP or p130CAS in Western blotting experiments. When the titer of the antibodies in the serum was judged satisfactory, the antibodies were further purified by protein A–Agarose columns (Bio-Rad) for use in Western blotting or used directly for immunoprecipitation experiments (5 μl). The anti-PSTPIP antibody was conjugated using a peroxidase labeling kit (Boehringer Mannheim Corp.).
Cell Culture
The PTP-PEST (+/−) and (−/−) cell lines are described in Côté et al. (1998). PTP-PEST (−/−), PTP-PEST (+/−), and COS-1 cells were maintained in DME supplemented with 10% FBS and penicillin/streptomycin.
Stable Overexpression of PTP-PEST in PTP-PEST (−/−) Cells
Full-length, wild-type PTP-PEST was inserted in a vector containing a selectable Zeocin resistance marker, pcDNA3.1/Zeo (+) (Invitrogen Corp.). Subconfluent PTP-PEST (−/−) cells were transfected with this recombinant vector using Lipofectamine Plus (GIBCO BRL). Cells were first cultured in DME plus 10% serum for 48 h before selection with 25 μg/ml of Zeocin (Invitrogen Corp.). Zeocin-resistant clones were isolated and screened for expression of PTP-PEST by immunoblot analysis as described below.
PTP-PEST Translocation in COS-1 Cells
Exponentially growing COS-1 cells from a 15-cm tissue culture dish were transfected with 40 μg of pACTAG PTP-PEST by electroporation as described in Charest et al. (1995). The cells were then grown for 48 h on Falcon chamber slides (Becton Dickinson and Co.) in DME supplemented with 10% FBS and antibiotics. For EGF treatment, cells were serum starved by incubation in DME 0.1% FBS for 16 h followed by 10 min of treatment in 0.1% medium containing 100 ng/ml EGF (Upstate Biotechnology Inc.). For integrin activation, the cells were trypsinized and washed twice with DME containing 10% FBS and plated on fibronectin-coated slides (described above) for 45 min before fixing. Immunofluorescence was performed as described below, using 12CA5 (1:1,000) as a primary antibody.
Migration Assays
The capacity of each cell line to migrate on fibronectin was monitored by two different assays. In the wound healing assay, Falcon chamber slides were coated overnight at 4°C with a solution of fibronectin (10 μg/ml) in PBS, 10 mM sodium phosphate, 140 mM NaCl, pH 7.4. Cells were plated at 60% confluence in normal (10% serum) medium. After attachment, the monolayers were wounded by scoring with a sterile plastic 200 μl micropipette tip. Each well was then washed and fed daily with normal medium. After 24 h, cells were fixed with 4% paraformaldehyde (PFA) in PBS for 5 min at room temperature and photographed using a low-magnification phase-contrast microscope. The extent of migration into the wound area was evaluated qualitatively.
Transwell® chambers (Corning-Costar) migration assays were performed as described in Klemke et al. (1998). In brief, the under surface of the polycarbonate membrane (pore size, 8 μm) was precoated with fibronectin (10 μg/ml in PBS) overnight at 4°C. The membrane was washed to remove excess ligand and the lower chamber filled with DME containing 10% serum. PTP-PEST (+/−), PTP-PEST (−/−), and PTP-PEST (−/−) stably overexpressing PTP-PEST (105 in 100 μl of DME containing 10% serum) were added to the upper chamber, and left for 5 h at 37°C before fixing with 95% methanol in PBS. Unmigrated cells on the upper side of the membrane were removed with a cotton tip applicator, and the migrated cells were stained with methylene blue and counted using an inverted microscope (×40). Each determination represents the average of four individual wells and error bars represent SD. As a control, the lower side of the membrane was coated with 0.5% BSA. The background level of cell migration represented <0.1% of the individual experiments.
Immunofluorescence
Cells were plated on fibronectin-coated slides (described above) for 20 min or 3 h. They were then fixed 20 min with 4% (wt/vol) PFA in PBS, and permeabilized with 0.1% (vol/vol) Triton X-100 in 4% PFA for another 20 min. The slides were blocked with 1% (wt/vol) BSA in PBS for 20 min. The cells were then incubated with the antivinculin antibody (1: 400 dilution in PBS) for 1 h at room temperature, washed three times with PBS, and stained with a mixture of FITC-conjugated anti–mouse antibody (1:200) and 4 μl/well of TRITC-conjugated phalloidin (Molecular Probes Inc.). After being washed three times with PBS, the coverslips were mounted in a 1:1 mixture of glycerol and 2.5% 1,4-diazabicyclo [2.2.2]octane (DABCO; Sigma Chemical Co.) in PBS. The cells were visualized with a Nikon fluorescence microscope. The number of focal adhesions in each cell was evaluated from photographs of the vinculin staining.
For cleavage furrow staining, the cells were plated at low confluence on uncoated glass slides and left overnight in 10% serum containing medium. The cells were then fixed, permeabilized, blocked, and stained as described above. The primary antibody used was PY20.
Protein Immunoprecipitation and Immunoblotting
Dishes of cells were washed with PBS supplemented with 1 mM of sodium vanadate, harvested, lysed, and evaluated for protein content using the Bio-Rad protein assay. For immunoprecipitations of paxillin and cortactin, cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40). For FAK, PSTPIP, and PSTPIP2 immunoprecipitations, cells were lysed by sonication in HNMETG (50 mM Hepes, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 10% glycerol). For vinculin and p130CAS, the lysis buffer used was TNE buffer (10 mM Tris HCl, pH 7.5, 1 mM EDTA, 150 mM NaCl, 1% Nonidet P-40). Lysis buffers were supplemented with 1 mM of sodium vanadate and Complete™ protease inhibitor mixture (Boehringer Mannheim GmbH).
For each immunoprecipitation, 500 μg of total cell lysate was incubated with 1 μg of antibody and 30 μl of protein G–Agarose (GIBCO BRL) to a total volume of 1 ml in their respective lysis buffer (supplemented with vanadate and protease inhibitors) at 4°C for 90 min. The beads were then washed three times (the last wash for 15 min at 4°C) in their respective buffers, except FAK, PSTPIP, and PSTPIP2 which were washed in HNTG buffer (10 mM Hepes-KOH, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 10% glycerol), and resuspended in 30 μl of SDS-PAGE loading dye.
Western blots were performed following common procedures. Proteins were resolved on SDS-PAGE, transferred onto a PVDF membrane, blocked, and probed with the corresponding antibody in the same blocking buffer (1% BSA, 1% [vol/vol] goat serum, 0.1% Tween, in PBS). The following dilutions were used for the primary antibodies: 1:3,000 for 4G10 and anti–PTP-PEST; 1:10,000 for antipaxillin; 1:1,000 for antivinculin, anti-p130CAS, anticortactin, and the HRP-conjugated pY20; 1:100 for anti-FAK. Secondary antibodies were used at 1:10,000. The Renaissance™ chemiluminescence kit (New England Nuclear Life Science Products) was used for detection.
Spreading Assay
3.5-cm tissue culture dishes were coated with 1 μg/cm2 of fibronectin overnight at 4°C. Plates were washed with PBS, and 1 ml of normal medium was prewarmed in a humidified 37°C incubator. Cells were trypsinized, counted, and 5 × 105 cells were added with medium to the plates for a final volume of 2 ml. Random fields were photographed after 10, 15, and 30 min using a low magnification phase-contrast microscope. Photographs were evaluated for the percentage of spread cells. Unspread cells were described as phase-bright and punctual, whereas spread cells were not phase-bright, with extensive visible membrane protrusions. The two kinds of cells were distinguishable enough so that two independent counts of the same field gave the same result ±2%. Four fields were counted for each cell line and, in each field, >300 cells were counted. The experiment was repeated four times.
In Vitro Protein Binding Assays
In vitro binding studies using glutathione S-transferase (GST) fusion proteins were performed as described elsewhere (Charest et al., 1996). In brief, 0.5 mg of each cell lysate was precleared with GST bound to glutathione–Sepharose beads (Pharmacia Biotech, Inc.) for 1 h at 4°C. The supernatants were then incubated with the GST-fusion protein prebound to glutathione–Sepharose beads in a total of 1 ml of TNE buffer supplemented with 0.1 mM vanadate for 90 min at 4°C, washed three times with TNE buffer, and resuspended in an equal volume of 2× SDS-PAGE loading buffer. Protein complexes were separated by SDS-PAGE and detected by immunoblotting as described above. The GST-Grb2 FL construct is described in Charest et al. (1997). The GST-SH2 domains of Crk and Src are a generous gift from Dr. Park (McGill University).
Results
Following Fibronectin-induced Integrin Activation, PTP-PEST Relocalizes from the Cytoplasm to the Membrane Periphery in COS-1 Cells
Because p130CAS is known to translocate to focal adhesions when phosphorylated by the integrin pathway (Harte et al., 1996), and since PTP-PEST and p130CAS associate via an SH3 proline-rich domain interaction (Garton et al., 1997), we first verified if PTP-PEST could translocate within a cell following integrin stimulation. COS-1 cells were transfected with a PTP-PEST construct tagged with the hemagglutinin antigen (HA) epitope, allowing visualization by indirect immunofluorescence (Fig. 1). Untransfected cells showed no immunofluorescence staining (Fig. 1 b), demonstrating the absence of nonspecific staining with the 12CA5 antibody. In transfected cells (Fig. 1 d), PTP-PEST localization was diffuse in the cytoplasm, as previously described (Charest et al., 1995). When the cells were stimulated with 0.1 μg/ml of EGF, no significant change in PTP-PEST staining was observed (Fig. 1 f), although PTP-PEST was shown previously to associate with the EGF receptor via Grb2 (Charest et al., 1997). However, for a significant percentage of attached transfected cells plated on fibronectin-coated slides for 45 min, staining for the HA epitope showed a clear relocalization to the membrane periphery even with lower levels of protein expression (Fig. 1 g). These results prove that, in COS-1 cells, PTP-PEST can relocalize to the membrane periphery following integrin, but not EGF receptor, stimulation.
Figure 1.








PTP-PEST translocates to the membrane periphery following fibronectin stimulation of COS-1 cells. a, c, e, and g are phase-contrast images while b, d, f, and h are HA epitope indirect immunofluorescence. (a and b) Untransfected cells. (c and d) Transfected, unstimulated cells. (e and f) Transfected cells starved for 16 h in 0.1% serum, then stimulated for 10 min with 100 ng/ml of EGF. (g and h) Transfected cells plated on fibronectin slides for 45 min before fixing and staining (see Materials and Methods). The arrow in h shows an example of membrane periphery translocation of the immunofluorescence staining. Bar, 20 μm.
PTP-PEST (−/−) Cells Have a Decreased Motility on Fibronectin
Two recent reports suggested a correlation between p130CAS phosphorylation level and cell migration rate. In both studies, a kinase was transfected in the cells. One used v-Src, for which p130CAS is a direct substrate (Fincham and Frame, 1998), while the other overexpressed FAK, that, once activated, binds both c-Src and its substrate p130CAS (Cary et al., 1998). In both studies, hyperphosphorylation of p130CAS was associated with an increase in rate of migration. Since p130CAS is a substrate for PTP-PEST (Garton et al., 1996), removal of this PTP results in hyperphosphorylation of p130CAS (Côté et al., 1998). We decided to investigate whether the absence of this PTP would result in a change in the motility of these cells. The two cell lines used were heterozygous and homozygous for the PTP-PEST deletion. To minimize any dominant negative effects from the targeted allele, comparisons were made between the homozygous and the heterozygous cell lines.
One of the simplest assays to qualitatively compare cell migration is the wound healing migration assay. Fibroblast monolayers plated on fibronectin-coated slides were wounded at 37°C and fixed after 24 h. Fig. 2 shows a typical region of the wound for each cell line obtained after five independent experiments. The PTP-PEST (+/−) cells were able to migrate into the wound at a rate greater than the front of cells pushed in by proliferation (Fig. 2 b), whereas the (−/−) cells showed a complete absence of chemokinetism (Fig. 2 d) and were not able to actively invade the wounded area.
Figure 2.


Gene targeting of the PTP-PEST suppresses fibroblast motility on the extracellular matrix fibronectin. Monolayers of each cell line were wounded (a and b) and maintained at 37°C for 24 h before fixing (c and d). The ability to migrate into the wound was monitored by phase-contrast microscopy of unstained cells which were photographed (×100). The aspect of each wound represents the typical result obtained after five independent experiments. In a chamber-type assay (e), the bottom side of the polycarbonate membrane was coated with fibronectin, and 105 cells were added to the top chamber. After 5 h, the cells that translocated to the bottom side of the membrane were counted and the result is shown as an average of eight fields from four independent experiments (error bars: standard deviation). PTP-PEST (−/−) cells stably overexpressing wild-type PTP-PEST were also tested by the same method. (Inset) Western blotting against PTP-PEST in the two cell lines. The band that appears represents overexpression since the antibody is known not to detect endogenous levels of PTP-PEST. Bar, 200 μm.
A more quantitative assay for migration is the chamber mobility assay, where the ability of cells to translocate to the fibronectin-coated side of a perforated membrane is measured (see Materials and Methods). Once again, we compared the migration of PTP-PEST (+/−) and PTP-PEST (−/−) cells. As in the wound-healing assay, a significant decrease in motility was observed in the knockout cells (Fig. 2 e). To ensure that this effect is indeed due to the absence of PTP-PEST, PTP-PEST (−/−) cells were transfected with PTP-PEST and a stably overexpressing clone was obtained. When tested in the same assay, these cells showed an increase in migration that was significant but still not comparable with wild-type levels. One possibility is that overexpression of PTP-PEST could also have deleterious effects on cell migration, as is the case with PTP1B (Liu et al., 1998). To test this hypothesis, we are currently trying to obtain clones expressing more physiological levels of PTP-PEST, as well as different constructs and catalytically inactive mutants of the phosphatase, and verify the effects on this phenotype and the others described below.
The two migration assays are complementary, since each has different limitations. For instance, the wound healing assay is affected by the rates of cell proliferation and by cell–cell adhesive interactions which restrict the ability of the cells to move into the wounded area. In the chamber assay, differences in cell shape and size may alter the ability of the cells to pass through the pores. Taken together, the data suggest that the loss of PTP-PEST results in motility defects in embryonic fibroblast cell lines that can be reincreased by overexpression of PTP-PEST in the (−/−) cells.
PTP-PEST (−/−) Cells Have an Increase in Number of Focal Adhesions
The cytoskeleton of the cell plays an important role in cell motility. We investigated whether this observed migration impairment of homozygous PTP-PEST mutant cells could be caused by an abnormal organization of the actin filaments, or by a visible difference in focal adhesions. Cells from each cell line were plated on fibronectin. The actin filaments were stained with a rhodamine-conjugated phalloidin (Fig. 3, a, c, e, and g), and the focal adhesions were highlighted by indirect immunofluorescence using mouse mAb against vinculin (Fig. 3, b, d, f, and h). 25 min after plating, both cell lines showed membrane ruffles and filopodia (Fig. 3, a and c), suggesting that the defect in migration was not caused by an incapacity to polymerize actin or to organize these structures. Large, immature focal adhesions were also found in each cell line (Fig. 3, b and d), showing that the initial pathways forming these contacts were intact in PTP-PEST (−/−) cells.
Figure 3.



Actin and focal adhesions staining in PTP-PEST (+/−) (a, b, e, and f) and (−/−) (c, d, g, and h) cells plated on fibronectin. After 20 min (a–d), there are no qualitative differences on the actin filaments, stained using a rhodamine-phalloidin conjugate (a and c) or in focal adhesions, stained with an antivinculin antibody and highlighted using a FITC-conjugated second antibody (b and d). However, when the cells were left for 3 h before fixing, the (+/−) cells became rounded (e) and only formed punctual focal adhesions at their periphery (f), whereas the (−/−) cells continued to spread, forming numerous stress fibers (g), and numerous focal adhesion plaques scattered throughout their ventral surface (h). The number of focal adhesions after 3 h in each cell was counted from photographs and the results are shown in i. Bar, 20 μm.
However, when cells were left for 3 h on fibronectin before fixing and staining, the homozygous cells remained well spread, with numerous focal adhesions scattered throughout the ventral surface of the cell (Fig. 3 h). Surrounding the cells were small, hair-like actin fibers that characterize cells in the early stages of apolar migration (Fig. 3 g). The PTP-PEST (+/−) cells, in contrast, were rounded, with well-defined edges and fewer focal contacts concentrated at the cell periphery (Fig. 3, e and f). The (−/−) cells also contained a higher number of stress fibers, a state that is incompatible with cell migration. A quantitative evaluation of the number of focal adhesions after 3 h, as counted on vinculin-staining photographs, is shown in Fig. 3 i. PTP-PEST (−/−) cells have, on average, >85 focal contacts per cell, compared with ∼25 in a (+/−) cell. Thus, the migration defect observed in the wound healing assay above may be in part due to disregulation of focal adhesion assembly and/or disassembly in the PTP-PEST cell lines.
Constitutive Hyperphosphorylation of Paxillin and FAK in PTP-PEST (−/−) Cells
Attempting to understand the differences observed in the size and number of focal adhesions between the two cell lines which differ only by the presence or absence of PTP-PEST, we analyzed the tyrosine phosphorylation state of specific focal adhesion proteins (Fig. 4 a). The adapter protein p130CAS was previously shown to be hyperphosphorylated (Côté et al., 1998), since it is a physiological substrate for PTP-PEST (Garton et al., 1996). The other proteins immunoprecipitated and analyzed for their phosphotyrosine status with an antiphosphotyrosine antibody included paxillin and cortactin. Paxillin was shown to be hyperphosphorylated in FAK null cell lines, a mutation that was also associated with a decrease in cell mobility and increase in numbers of focal adhesions (Ilìc et al., 1995). Paxillin phosphorylation was also shown recently to be required for cell spreading and focal adhesion formation (Richardson et al., 1997). In the present study, paxillin was found in a hyperphosphorylated state (Fig. 4 a). Interestingly, paxillin was shown to physically associate with PTP-PEST (Shen et al., 1998), but experiments using trapping mutants suggest that paxillin is not a direct substrate for PTP-PEST (Côté, J.-F., C.E. Turner, and M.L. Tremblay, manuscript submitted for publication). Cortactin, like p130CAS, is a pp60Src substrate (Wu et al., 1991), although it is not associated with focal adhesions. It interacts with the actin cytoskeleton (Wu and Parsons, 1993) and is tyrosine phosphorylated following integrin-mediated cell adhesion to extracellular matrix (Vuori and Ruoslahti, 1995). However, Fig. 4 a shows that cortactin is not constitutively tyrosine hyperphosphorylated in the PTP-PEST (−/−) cells, suggesting that it is not a direct or indirect substrate for this phosphatase.
Figure 4.

(a) Constitutive hyperphosphorylation of FAK and paxillin in PTP-PEST (−/−) cells. Left column shows extent of tyrosine phosphorylation of immunoprecipitates as detected by the 4G10 antiphosphotyrosine mAb; right column shows evaluation of the amount of protein loaded using the same antibody as the immunoprecipitation. (b) Rescue of p130CAS hyperphosphorylation in PTP-PEST (−/−) overexpressing wild-type PTP-PEST. p130CAS was already shown to be a substrate for PTP-PEST (Garton et al., 1996) and to be hyperphosphorylated in the PEST (−/−) cells (Côté et al., 1998). Left panel shows phosphorylation levels of p130CAS in each cell line; right panel shows loading control.
Vinculin is a structural protein that links talin and actin when focal adhesions form. It can be tyrosine phosphorylated, but this phosphorylation does not seem to be implicated in focal adhesion formation, the latter due to a conformational change in vinculin following phosphatidyl inositol biphosphate binding (Burridge and Chrzanowska-Wodnicka, 1996). There was no detectable basal level of tyrosine phosphorylation of vinculin in both cell lines, potentially due to the fact that the cells were not stimulated.
Another focal adhesion component to be investigated was FAK. The role of p130CAS in migration was shown to reside in the pathways triggered by FAK (Cary et al., 1998), which is associated with the integrins and becomes active by transphosphorylation when they cluster. Phosphorylation of Y397 on FAK, which is the binding site of the Src kinase, was shown to be crucial for its role in cell migration (Cary et al., 1998). Fig. 4 a shows that FAK is slightly hyperphosphorylated in the PTP-PEST null cells, suggesting that this PTP may regulate its phosphorylation level. Since FAK and PTP-PEST were never shown to interact directly, it is possible that this hyperphosphorylation is a consequence of p130CAS or paxillin hyperphosphorylation.
Also, to follow our results showing that p130CAS is constitutively hyperphosphorylated in PTP-PEST (−/−) cells (Côté et al., 1998), we verified if this hyperphosphorylation was decreased in the clone overexpressing wild-type PTP-PEST used in Fig. 2 e. p130CAS was immunoprecipitated from lysates of PTP-PEST (+/−), (−/−), and (−/−) overpressing PTP-PEST. The phosphorylation levels of p130CAS in each cell line were then evaluated as for the focal adhesion proteins described above. Fig. 4 b shows that the phosphorylation of p130CAS is decreased to normal levels in the PTP-PEST (−/−) cells overexpressing PTP-PEST. Taken together, these data suggest that PTP-PEST mediates the dephosphorylation of a specific subset of focal adhesion–associated proteins.
PTP-PEST (−/−) Cells Spread Faster on Fibronectin
This hyperphosphorylation of p130CAS, paxillin, and FAK is constitutive, meaning the cells were not stimulated with growth factors or extracellular ligands. Also, cell spreading was associated with paxillin tyrosine phosphorylation (Richardson et al., 1997). Finally, overexpression of PTP1B, a PTP for which p130CAS is a substrate (Liu et al., 1996), caused inhibition of cell spreading (Liu et al., 1998). These facts led us to investigate whether the PTP-PEST (−/−) cells could be, on the contrary, more prone to attach on fibronectin since p130CAS, paxillin, and FAK are already hyperphosphorylated. Normally growing cells were trypsinized and plated on fibronectin-coated tissue culture plates. After 10, 15, and 30 min, random fields were photographed and the cells were counted for the extent of cell spreading. The results of two independent experiments with different populations of the two same PTP-PEST (−/−) and (+/−) clones that were tested in the preceding experiments are shown in Fig. 5. After 10 min, more PTP-PEST (−/−) are opaque, which is a characteristic of spread cells under phase-contrast microscopy (Fig. 5, a and b). Photographs of fields after 30 min (Fig. 5, c and d) as well as a quantitative curve of the spreading time course (Fig. 5 e) are also shown.
Figure 5.
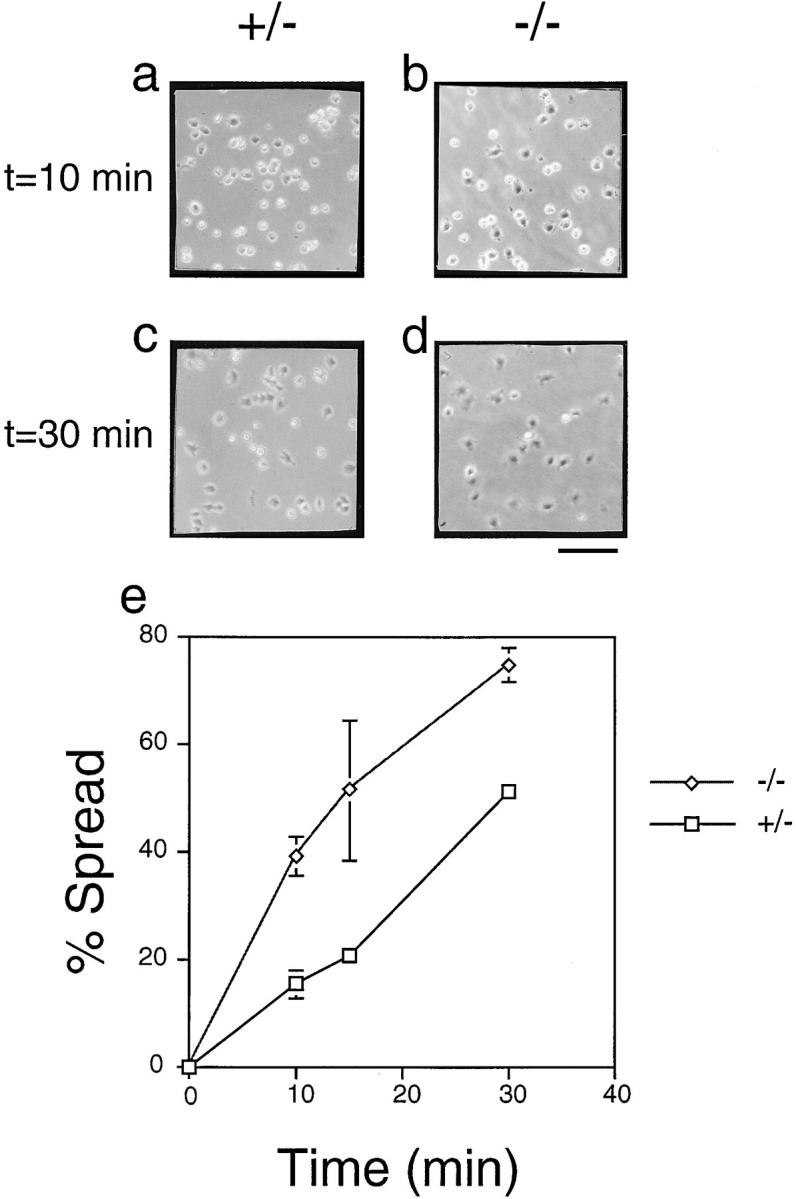
Analysis of cell spreading following PTP-PEST targeting in fibroblasts. Both PTP-PEST (+/−) and (−/−) were allowed to spread on fibronectin-coated tissue culture dishes, and the number of spread cells was assayed after 10, 15, and 30 min. Phase-contrast of random fields after 10 (a and b) and 30 (c and d) min are shown for PTP-PEST (+/−; a and c) and PTP-PEST (−/−; b and d) cells. PTP-PEST (−/−) cells showed significantly higher cell spreading. A quantitative evaluation was expressed as the number of cells spread as a percentage of the total number of cells in the field (e). Average number of cells per field: 300; n = 8. Error bars represent the standard deviation of the mean. Bar, 200 μm.
These results suggest that the PTP-PEST (−/−) cells are primed for attachment and spreading via integrin-stimulated pathways, and that PTP-PEST has a physiological role in regulating this event. The negative role of PTP-PEST in cell spreading is consistent with its role described above in cell migration via the breakdown of cell–substratum links.
Increased Affinity for the SH2 Domains of Src and Crk toward p130CAS, but Not FAK or Paxillin, in the PTP-PEST (−/−) Cells
To understand where the tyrosine residues hyperphosphorylated on p130CAS, paxillin, and FAK in the PTP-PEST (−/−) cells are, and to verify if these sites could be specific SH2-binding motifs, we examined the physical association of each of these proteins with a panel of SH2 domains in vitro. The three constructs tested were the GST fusion proteins of the Src and Crk SH2 domains, as well as the full-length Grb2 (Charest et al., 1997). These three SH2 domains were shown to bind phosphotyrosines on FAK (Cantley and Songyang, 1994; Schaller et al., 1994; Schlaepfer et al., 1994; Schlaepfer and Hunter, 1996), Src and Crk to p130CAS (Sakai et al., 1994), and Crk to paxillin (Turner and Miller, 1994).
Each cell lysate was first precleared with GST alone to eliminate nonspecific binding (see Materials and Methods). 0.5 mg of the remaining proteins was then incubated with each GST-fusion protein and the bound proteins were resolved by SDS-PAGE (Fig. 6). Between the PTP-PEST (+/−) and (−/−) cell lines, the greatest difference in affinity is observed between the SH2 domain of Src and p130CAS and, to a lesser extent, between the SH2 domain of Crk and p130CAS, strongly suggesting that the tyrosines within these SH2-binding domains on p130CAS are hyperphosphorylated. The other lanes show a small and fairly constant (∼50%) increase in affinity, that may or may not be indirect and could reflect a general constitutive increase in focal complexes in the PTP-PEST (−/−) cells. The exact tyrosines that are hyperphosphorylated on paxillin and FAK are still under investigation. They may not be found in putative SH2-binding domains, but can play other roles in focal adhesion turnover.
Figure 6.

Constitutive interactions of FAK, paxillin, and p130CAS with the SH2-domains of Crk, Grb2, and Src in binding assays. TNE cell lysates were prepared from exponentially growing PTP-PEST (+/−) and PTP-PEST (−/−) cells. Lysates were incubated with the indicated GST fusion proteins coupled to glutathione-agarose as described in Materials and Methods. The precipitates were analyzed by sequential stripping and immunoblotting of the same membrane with the indicated antibodies.
Hyperphosphorylation of PSTPIP in the PTP-PEST (−/−) Cells and Effects on Cleavage Furrow Formation
While we investigated the effects of PTP-PEST targeting in cell migration, another protein involved with the actin cytoskeleton, PSTPIP, was cloned and shown to be a substrate for a PTP of the PEST family, PTP-HSCF (Spencer et al., 1997). A putative coiled-coil region of PSTPIP interacts with the COOH-terminal, proline-rich region of PTP-HSCF (Dowbenko et al., 1998), a region that is conserved in all members of the PEST family, including PTP-PEST.
Using a polyclonal antibody raised against PSTPIP, we analyzed its tyrosine phosphorylation level in the PTP-PEST (−/−) and (+/−) cells by immunoprecipitation, followed by antiphosphotyrosine immunoblotting. Data from Spencer et al. (1997) suggested that PSTPIP tyrosine phosphorylation was controlled by the members of the PEST family of PTPs, and Fig. 7 shows that PSTPIP was hyperphosphorylated in the PTP-PEST knockout cell line. Taken together with the fact that PSTPIP is a direct substrate for PTP-HSCF (Spencer et al., 1997), and that this substrate effect required the conserved COOH-terminal proline-rich binding domain, these results suggest that PSTPIP could be a substrate for PTP-PEST in fibroblasts. The same experiment was performed on PSTPIP2, a protein with high homology to PSTPIP which binds to the PEST type PTPs but does not contain the SH3 domain, and is not a substrate for the PEST family of PTPs (Wu et al., 1998a). The phosphorylation level of PSTPIP2 was similar in the (+/−) and the (−/−) cell lines (data not shown), suggesting the presence of another level of specificity, such as substrate recognition/activity in PTP-PEST.
Figure 7.
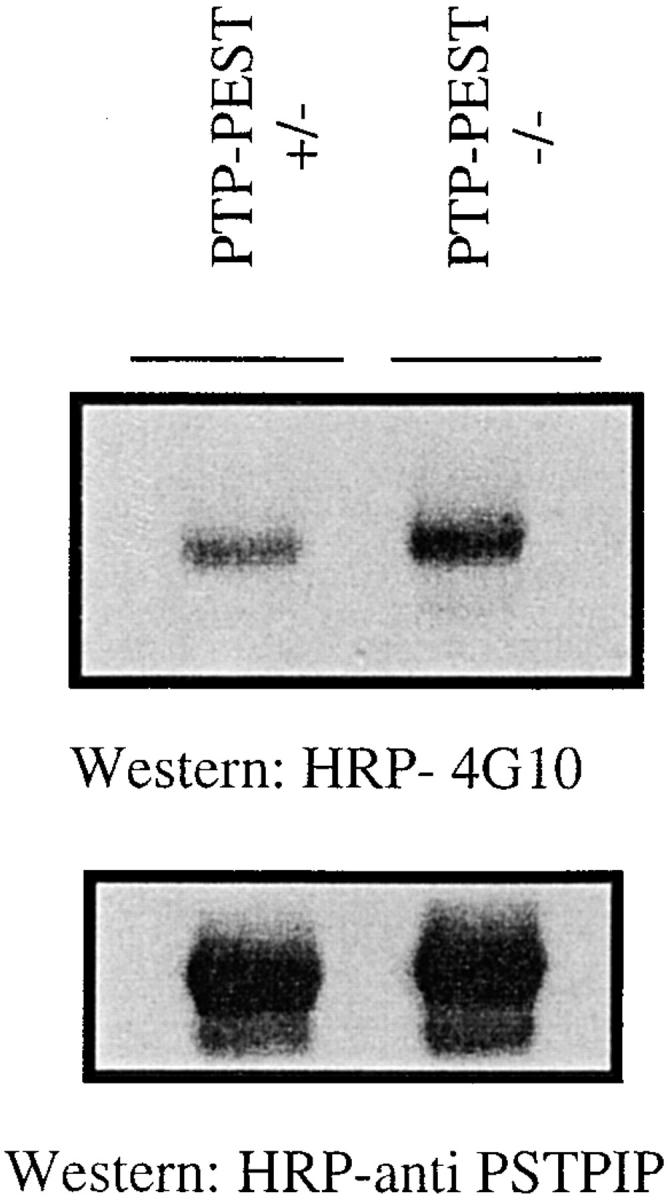
Constitutive hyperphosphorylation of PSTPIP in unsynchronized PTP-PEST (−/−) cells. PSTPIP was immunoprecipitated from PTP-PEST (+/−) and PTP-PEST (−/−) cell lysates and probed with a HRP-conjugated antiphosphotyrosine antibody (top). PSTPIP was hyperphosphorylated in the (−/−) cells. (Bottom) Anti-PSTPIP blot of the same membrane to ensure equal loading of the protein.
PSTPIP is homologous to S. pombe CDC15, an actin-associated protein that is critically involved in the formation of the cleavage furrow during cell division (Fankhauser et al., 1995). During our manipulations of the PTP-PEST (−/−) cells, we observed a high occurrence of cells that seemed blocked in a late stage of cytokinesis, with the two daughter cells almost independent, but still attached to each other by an actin-rich junction which appears to be derived from the cleavage furrow. Fig. 8 a shows the actin cleavage furrow rings of several of these cells as stained by rhodamine-phalloidin. Fig. 8 b represents the same cells showing the indirect antiphosphotyrosine immunostaining and revealing a clear band of tyrosine phosphorylated material at the cleavage furrow. To eliminate the possibility of contamination of the rhodamine fluorescence through the FITC filter, the same antiphosphotyrosine staining was performed in the absence of rhodamine-phalloidin, with the same result. These cells were found in all PTP-PEST (−/−) cultures that we observed, and were virtually absent from any other wild-type cell lines. The rare PTP-PEST (+/−) cells found in this phase were also probed for antiphosphotyrosine and the cleavage furrow did reveal some staining. Fig. 8 b does not represent a phenotype but shows the implication of tyrosine phosphorylation in cytokinesis, which can explain why a PTP could play a role in this event. This phenomenon was postulated to be the result of a longer M phase due to an impairment of the actin rearrangement during division, a process where tyrosine phosphorylation is involved.
Figure 8.




Cytokinesis defect in PTP-PEST (−/−) cells. Unsynchronized PTP-PEST (−/−) cells were plated on uncoated tissue culture glass slides and stained using rhodamine-conjugated phalloidin to highlight the actin-rich cleavage furrows (a). An antiphosphotyrosine immunostaining of cells in the same population is shown in b. Bar, 30 μm.
Discussion
FAK activation and focal adhesions formation are closely related events. Until recently, the exact order in which they occur following integrin stimulation was greatly debated. Recent experiments (Wilson et al., 1995; Chrzanowska-Wodnicka and Burridge, 1996) showed that FAK activation is a result of focal adhesion formation. Due to the physical tension caused by the stress fiber formation in a cell, under the control of the Rho GTPase, the associated integrins cluster and associate with other structural proteins like α-actinin and tensin (Burridge and Chrzanowska-Wodnicka, 1996). FAK proteins are associated with integrins and can transphosphorylate in a manner similar to receptor tyrosine kinases following extracellular ligand binding. This phosphorylation activates FAK and provides docking sites for other signal transduction proteins that propagate integrin-triggered pathways.
However, the relationship between focal adhesion formation and FAK activation is apparently not strictly linear. Among the signals sent by FAK, some were shown to feed back and play a role in the turnover rate of focal adhesions. The existence of such signals was concluded when the FAK null cell lines showed an increase in focal adhesion size and number (Ilìc et al., 1995). However, these results did not take into account the effects of other members of the FAK family that were later identified, such as Pyk2 (Sasaki et al., 1995). Recently, FAK was shown to play a role in migration, an event that requires focal adhesion formation and breakdown. This increase in migration occurs via the tyrosine phosphorylation of an adapter protein, p130CAS (Cary et al., 1998). Also, gene disruption of a cytoplasmic tyrosine phosphatase with two SH2 domains, SHP-2, was shown to impair the ability of fibroblasts to spread and migrate on fibronectin (Yu et al., 1998). This phenotype was associated with a decrease in FAK dephosphorylation following cell detachment and suspension. In this paper, we examined another cell line with a gene inactivated by homologous recombination, cytosolic PTP-PEST. A major substrate for this enzyme in both mice and humans was shown to be p130CAS (Garton et al., 1996; Côté et al., 1998), and we show in Fig. 1 that, in COS-1 cells, PTP-PEST can translocate to the membrane periphery following integrin activation, possibly in order to reach the p130CAS substrate which also translocates to focal adhesions when FAK is activated (Nakamoto et al., 1997).
The effects of PTP-PEST removal on cell migration and focal adhesion number were similar to the phenotype of the FAK mutant cells (Ilìc et al., 1995). Both showed a decrease in fibroblast migration using fibronectin as an extracellular matrix, and the presence of numerous immature focal adhesions scattered on the ventral face of the cell (Figs. 2 and 3). Even though FAK and PTP-PEST have opposite catalytic activities on a common substrate, p130CAS, PTP-PEST does not seem to antagonize the effects of FAK in cell migration, but rather to potentiate these effects. This suggests a mechanism where both a formation and a breakdown pathway are triggered at the same time to increase the turnover rate of the focal adhesion structures. Without the pathway required for focal adhesion breakdown, the adhesive contacts between the extracellular matrix and the cell are too strong, and the cell adheres and does not migrate (Schwarzbauer, 1997). In FAK (−/−) cells, the actin stress fibers were dense around the cell periphery rather than in the center, where they can exert the transcellular tension necessary for cell migration (Lauffenburger and Horwitz, 1996). In contrast, in the PTP-PEST (−/−) cells, central stress fibers are omnipresent, suggesting that the cells cannot turn them over. This state is also incompatible with cell migration. The absence of differences in the early stages of cell attachment to fibronectin (Fig. 3, a–d) suggests that PTP-PEST is not implicated in the initial formation of stress fibers or focal adhesions. However, at equilibrium, clear differences appear (Fig. 3, e–h). The focal adhesions in the PTP-PEST (−/−) cells are numerous and scattered throughout the ventral (i.e., substrate-attached) membrane of the cell, which can in part explain the absence of motility of the cells when tested in the wound healing assay. In contrast, the heterozygous cells became rounded, their focal adhesions were punctate and found only at the tips of their membranes, which is consistent with a migrating cell. This led us to believe that PTP-PEST is implicated in focal adhesion and stress fiber breakdown, a role that is the counterpart of FAK or Src but is required for the successful achievement of the same event, cell migration.
In the PTP-PEST mutant cell line, p130CAS, paxillin, and FAK were found in a hyperphosphorylated state. The phosphorylation of paxillin was shown to be associated with cell spreading on an extracellular matrix (Richardson et al., 1997), which is consistent with data reported here. Paxillin can associate with PTP-PEST (Shen et al., 1998), but it does not constitute a direct physiological substrate for this PTP (Côté, J.-F., C.E. Turner, and M.L. Tremblay, manuscript submitted for publication) and the exact site that is hyperphosphorylated is still not clear.
SH2-domain affinity assays shown in Fig. 4 strongly suggest the sites that are hyperphosphorylated on p130CAS are within the SH2-binding domains of Src and Crk. One hypothesis is that the only tyrosine that is substrate for PTP-PEST is the 762YDYV765 Src SH2-binding region on p130CAS, which binds Src in a tyrosine-dependant manner (Sakai et al., 1994). In PTP-PEST (−/−) cells, this can cause Src to constitutively bind p130CAS and hyperphosphorylate the Crk-binding motifs on p130CAS. This would explain why p130CAS is so hyperphosphorylated in the PTP-PEST (−/−) cells, and why the difference in affinity is greater with the Src SH2 domain compared with the Crk SH2 domain, even though there are 15 putative binding domains for Crk compared with only one for Src on p130CAS. In the same line of thought, this could also be the cause for the small FAK hyperphosphorylation in the PTP-PEST (−/−) cells, since the SH3 domain of p130CAS can bind a proline-rich region on FAK (Polte and Hanks, 1995), and FAK is also a substrate for Src (Calalb et al., 1995, 1996; Schlaepfer and Hunter, 1996). This increased affinity between Crk and p130CAS in the PTP-PEST (−/−) cells can be related to the phenotypes observed in Figs. 2 and 3. The CAS/Crk coupling has been shown to play a role in the induction of cell migration (Klemke et al., 1998). Thus, incorrect regulation of this molecular switch in the absence of PTP-PEST could lead to aberrant cell migration. These results provide a concrete, tyrosine phosphorylation–dependent way for PTP-PEST to regulate cell migration, and strongly suggest the phenotypes observed here are the result of the gene targeting. Still, reintroduction at physiological levels of different PTP-PEST constructs in the knockout cells to differentially rescue the phenotypes described in this paper is currently under way in our laboratory.
Studies using the PTP inhibitor phenylarsine oxide showed that treatment of cells with this compound was sufficient to induce the formation of stress fibers even after starving them for 16 h (Retta et al., 1996). Phenylarsine oxide reacts with two thiol groups of closely spaced cysteine residues in the active site of the phosphatase. The PTP-PEST catalytic domain contains the sequence 231CSAGC235 (Charest et al., 1995), the cysteine at position 231 being crucial for its catalytic activity. These studies also showed that focal adhesion disassembly results in stimulation of phosphatase activity, which could be assayed using FAK and paxillin as substrates. That, and the fact that paxillin is hyperphosphorylated in the PTP-PEST knockout cell line, suggest that PTP-PEST is a candidate PTP involved in the focal adhesion breakdown in the conditions studied in these experiments.
The role of a PTP in focal adhesion breakdown would suggest that overexpression of the PTP would also inhibit cell migration, by impairing the formation of the focal adhesions at the leading edge of the cell. Experiments involving another PTP that can dephosphorylate p130CAS, PTP1B (Liu et al., 1996), showed that overexpression of this PTP in rat fibroblasts decreased cell migration while increasing the time required for the cell to spread on fibronectin (Liu et al., 1998). This was linked to a disordered formation of focal adhesions. In this article, we demonstrated that removal of a PTP that can dephosphorylate p130CAS, PTP-PEST, increased the spreading speed of targeted cells on the same extracellular matrix protein (Fig. 5). Interestingly, the PTP1B-overexpressing cells eventually formed numerous, large focal complexes scattered over their ventral surface, like ones found in the PTP-PEST (−/−) cells. These experiments and the ones presented in this paper suggest that an intermediate level of PTP activity towards p130CAS is required for the formation of normal focal adhesions and for cell migration, which is consistent with a role in focal adhesion turnover.
PTP-PEST may also play a role in the regulation of the cell cytoskeleton, this time via the cleavage furrow–associated protein PSTPIP. PSTPIP was originally identified as a binding partner and a substrate for the phosphatase PTP-HSCF (Spencer et al., 1997), a PEST tyrosine phosphatase. PTP-HSCF dephosphorylates tyrosine residues in PSTPIP that are modified either by coexpression of the v-Src tyrosine kinase or in the presence of the unspecific PTP inhibitor pervanadate. One of these sites, within the SH3 domain of PSTPIP, was shown to regulate binding with the proline-rich region found on WASP (Wu et al., 1998), and to control aspects of the actin cytoskeleton.
The possibility that tyrosine phosphorylation is involved in furrow development and the signaling events coordinating nuclear division was reported by Cool et al. (1992). Their data show that BHK cells overexpressing a truncated PTP, namely T cell PTP, become highly multinucleate, apparently through a failure in cytokinesis. This deletion mutant lacked the COOH-terminal extension responsible for its proper localization, and the redistribution of the enzyme to the soluble fraction caused both a furrowing defect and an asynchronous entry into S phase of two nuclei within the same syncytial cell.
It has not been shown whether PTP-PEST binds directly to PSTPIP, but peptides derived from the conserved proline-rich COOH terminus of PTP-PEST can compete the binding of PTP-HSCF to PSTPIP (Spencer et al., 1997), consistent with the suggestion that PTP-PEST binds to PSTPIP in a manner similar to PTP-HSCF binding. In addition, while no previous data had shown that PTP-PEST can dephosphorylate PSTPIP, the fact that PSTPIP is hyperphosphorylated in the PTP-PEST (−/−) cells (Fig. 7) strongly suggests that PTP-PEST plays a direct role in modulating the tyrosine phosphorylation level of PSTPIP. PTP-HSCF and another member of the PEST family, PTP-PEP, are generally not expressed in fibroblasts, and it is possible that PTP-PEST regulates PSTPIP in fibroblasts the way PTP-HSCF does in hematopoietic cells. The exact site that is hyperphosphorylated on PSTPIP in PTP-PEST (−/−) cells is not known, and the effects of this hyperphosphorylation on binding, for example, to WASP (Wu et al., 1998b) are still under investigation. One of the observable effects appears to be an increase in the relative amount of cells found in the last stages of cytokinesis in a field of unsynchronized cells (Fig. 8), possibly due to the role of PSTPIP in cleavage furrow assembly or disassembly. It is also possible that the increase of PSTPIP tyrosine phosphorylation is the result, and not the cause, of the increase of the number of cells in this phase.
The fact that PTP-PEST (−/−) cells still divide and grow at rates comparable to other cells suggests the presence of other mechanisms involved in cell division. Isolation and characterization of cytokinesis-deficient mutants in Dictyostelium discoideum, a highly motile organism that undergoes cell cleavage much like higher eukaryotes, provided examples of cell division occurring with impaired cytokinesis (Vithalani et al., 1996). In particular, a mutation in the myosin gene that prevents the protein assembly in thick filaments resulted in organisms clearly defective in the contractile events involved in cytokinesis (Fukui et al., 1990). Another mutant, called 10BH2, also had a complete defect in cytokinesis (Vithalani et al., 1996). However, both mutants, which showed cell division defects when grown in suspension, were able to divide when plated on solid substratum by pinching off a portion of their cytoplasm in a process known as traction-mediated cytofission (Fukui et al., 1990). It is possible that the PTP-PEST (−/−) cells in part rely on this event to normally grow, even if the defect in chemokinesis observed in the wound-healing assay could impair the required migration. Interestingly, another D. discoideum mutant, in the single gene encoding calmodulin, formed and constricted a contractile cleavage furrow ring, but the midbody linking the daughter cells failed to completely close (Liu et al., 1992). The resultant population contained cells resembling cells observed in the PTP-PEST (−/−) population. This cytoplasmic bridge could be broken by shear forces when the cells were grown in suspension cultures and the cells could multiply normally. This suggests that cells in this state require little force to successfully complete division, and could explain the fact that the PTP-PEST (−/−) cells can still divide.
In this article, we reported two roles that a specific PTP, PTP-PEST, could play in regulating the cytoskeleton of fibroblasts. The first, possibly via its capacity to dephosphorylate p130CAS, is to break down focal adhesions, an event which is required for cell migration on an extracellular matrix like fibronectin. PTP-PEST was shown to localize at the membrane periphery when COS-1 cells were plated on fibronectin and this confers to PTP-PEST a physiological role in cell migration that is not a secondary effect of overexpression. The fact that PTP-PEST is mostly found in a cytoplasmic pool (Charest et al., 1995) and can be recruited to the plasma membrane after fibronectin-mediated attachment provides the cell a mechanism to increase its focal adhesions turnover rate proportional to the stimulus, even when FAK, p130CAS, or v-Src is overexpressed (Akasaka et al., 1995; Cary et al., 1998; Fincham and Frame, 1998). PTP-PEST also plays a role in modulating the phosphorylation level of PSTPIP, a protein that associates with the cytoskeleton (Spencer et al., 1997). The role of this PTP-PEST activity is not known, but it may involve the binding of WASP to PSTPIP (Wu et al., 1998b). Since WASP has been shown to regulate actin fiber assembly and cytokinesis in both yeast (Li, 1997) and mammalian (Symons et al., 1996) cells. These data suggest that this interaction may be somehow modulated in the PTP-PEST (−/−) cells.
Acknowledgments
The authors would like to thank T. Parsons for the generous gift of anticortactin mAb, M. Park for the two SH2-GST fusion proteins, as well as J. Wagner and A. Cheng for critical review of the manuscript.
This work was supported by a grant from the Medical Research Council of Canada (MRC) to M.L. Tremblay. A. Angers-Loustau is a recipient of a fonds pour la Formation des Chercheurs et l'Aide à la Recherche (FCAR) studentship. J.-F. Côté is a recipient of a Cancer Research Society studentship (CRS). A. Charest is a recipient of a NCI post-doctoral fellowship. M.L. Tremblay is a Chercheur boursier from Les Fonds De La Recherche En Santé Du Québec (FRSQ).
Abbreviations used in this paper
- FAK
focal adhesion kinase
- GST
glutathione S-transferase
- HA
hemagglutinin antigen
- PSTPIP
proline, serine, threonine phosphatase interacting protein
- PTP
protein tyrosine phosphatase
- SH
Src homology
- WASP
Wiskott-Aldrich syndrome protein
References
- Akasaka T, van Leeuwen RL, Yoshinaga IG, Mihm MC, Jr, Byers HR. Focal adhesion kinase (p125FAK) expression correlates with motility of human melanoma cell lines. J Investig Dermatol. 1995;105:104–108. doi: 10.1111/1523-1747.ep12313396. [DOI] [PubMed] [Google Scholar]
- Arregui CO, Balsamo J, Lilien J. Impaired integrin-mediated adhesion and signaling in fibroblasts expressing a dominant-negative mutant PTP1B. J Cell Biol. 1998;143:861–873. doi: 10.1083/jcb.143.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DS, Bliska JB. Identification of p130CAS as a substrate of YersiniaYopH (Yop51), a bacterial protein tyrosine phosphatase that translocates into mammalian cells and targets focal adhesions. EMBO (Eur Mol Biol Organ) J. 1997;16:2730–2744. doi: 10.1093/emboj/16.10.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calalb MB, Zhang X, Polte TR, Hanks SK. Focal adhesion kinase tyrosine-861 is a major site of phosphorylation by Src. Biochem Biophys Res Commun. 1996;228:662–668. doi: 10.1006/bbrc.1996.1714. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Songyang Z. Specificity in recognition of phosphopeptides by src-homology 2 domains. J Cell Sci Suppl. 1994;18:121–126. doi: 10.1242/jcs.1994.supplement_18.18. [DOI] [PubMed] [Google Scholar]
- Cary LA, Chang JF, Guan JL. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J Cell Sci. 1996;109:1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- Cary LA, Han DC, Polte TR, Hanks SK, Guan JL. Identification of p130CASas a mediator of focal adhesion kinase–promoted cell migration. J Cell Biol. 1998;140:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest A, Wagner J, Shen SH, Tremblay ML. Murine protein tyrosine phosphatase-PEST, a stable cytosolic protein tyrosine phosphatase. Biochem J. 1995;308:425–432. doi: 10.1042/bj3080425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest A, Wagner J, Jacob S, McGlade CJ, Tremblay ML. Phosphotyrosine-independent binding of SHC to the NPLH sequence of murine protein-tyrosine phosphatase-PEST. Evidence for extended phosphotyrosine binding/phosphotyrosine interaction domain recognition specificity. J Biol Chem. 1996;271:8424–8429. doi: 10.1074/jbc.271.14.8424. [DOI] [PubMed] [Google Scholar]
- Charest A, Wagner J, Kwan M, Tremblay ML. Coupling of the murine protein tyrosine phosphatase PEST to the epidermal growth factor (EGF) receptor through a Src homology 3 (SH3) domain-mediated association with Grb2. Oncogene. 1997;14:1643–1651. doi: 10.1038/sj.onc.1201008. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool DE, Andreassen PR, Tonks NK, Krebs EG, Fischer EH, Margolis RL. Cytokinetic failure and asynchronous nuclear division in BHK cells overexpressing a truncated protein-tyrosine-phosphatase. Proc Natl Acad Sci USA. 1992;89:5422–5426. doi: 10.1073/pnas.89.12.5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté J-F, Charest A, Wagner J, Tremblay ML. Combination of gene targeting and substrate trapping to identify substrates of protein tyrosine phosphatases using PTP-PEST as a model. Biochemistry. 1998;37:13128–13137. doi: 10.1021/bi981259l. [DOI] [PubMed] [Google Scholar]
- Dowbenko D, Spencer S, Quan C, Lasky LA. Identification of a novel polyproline recognition site in the cytoskeletal associated protein, proline serine threonine phosphatase interacting protein. J Biol Chem. 1998;273:989–996. doi: 10.1074/jbc.273.2.989. [DOI] [PubMed] [Google Scholar]
- Fankhauser C, Reymond A, Cerutti L, Utzig S, Hofmann K, Simanis V. The S. pombecdc15 gene is a key element in the reorganization of F-actin at mitosis. Cell. 1995;82:435–444. doi: 10.1016/0092-8674(95)90432-8. [DOI] [PubMed] [Google Scholar]
- Fincham VJ, Frame MC. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO (Eur Mol Biol Organ) J. 1998;17:81–92. doi: 10.1093/emboj/17.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui Y, De Lozanne A, Spudich JA. Structure and function of the cytoskeleton of a Dictyosteliummyosin-defective mutant. J Cell Biol. 1990;110:367–378. doi: 10.1083/jcb.110.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton AJ, Flint AJ, Tonks NK. Identification of p130(cas) as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol Cell Biol. 1996;16:6408–6418. doi: 10.1128/mcb.16.11.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton AJ, Burnham MR, Bouton AH, Tonks NK. Association of PTP-PEST with the SH3 domain of p130(Cas) a novel mechanism of protein tyrosine phosphatase substrate recognition. Oncogene. 1997;15:877–885. doi: 10.1038/sj.onc.1201279. [DOI] [PubMed] [Google Scholar]
- Gilmore AP, Burridge K. Molecular mechanisms for focal adhesion assembly through regulation of protein–protein interactions. Structure. 1996;4:647–651. doi: 10.1016/s0969-2126(96)00069-x. [DOI] [PubMed] [Google Scholar]
- Hanks SK, Polte TR. Signaling through focal adhesion kinase. Bioessays. 1997;19:137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- Harder KW, Moller NPH, Peacock JW, Jirik FR. Protein-tyrosine phosphatase alpha regulates Src family kinases and alters cell–substratum adhesion. J Biol Chem. 1998;273:31890–31900. doi: 10.1074/jbc.273.48.31890. [DOI] [PubMed] [Google Scholar]
- Harte MT, Hildebrand JD, Burnham MR, Bouton AH, Parsons JT. p130CAS, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J Biol Chem. 1996;271:13649–13655. doi: 10.1074/jbc.271.23.13649. [DOI] [PubMed] [Google Scholar]
- Ilìc D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Klemke RL, Leng J, Molander R, Brooks PC, Vuori K, Cheresh DA. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Li R. Bee1, a yeast protein with homology to Wiscott-Aldrich syndrome protein, is critical for the assembly of cortical actin cytoskeleton. J Cell Biol. 1997;136:649–658. doi: 10.1083/jcb.136.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Hill DE, Chernoff J. Direct binding of the proline-rich region of protein tyrosine phosphatase 1B to the Src homology 3 domain of p130(Cas) J Biol Chem. 1996;271:31290–31295. doi: 10.1074/jbc.271.49.31290. [DOI] [PubMed] [Google Scholar]
- Liu F, Sells MA, Chernoff J. Protein tyrosine phosphatase 1B negatively regulates integrin signaling. Curr Biol. 1998;8:173–176. doi: 10.1016/s0960-9822(98)70066-1. [DOI] [PubMed] [Google Scholar]
- Liu T, Williams JG, Clarke M. Inducible expression of calmodulin antisense RNA in Dictyosteliumcells inhibits the completion of cytokinesis. Mol Biol Cell. 1992;3:1403–1413. doi: 10.1091/mbc.3.12.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto T, Sakai R, Honda H, Ogawa S, Ueno H, Suzuki T, Aizawa S, Yazaki Y, Hirai H. Requirements for localization of p130(cas) to focal adhesions. Mol Cell Biol. 1997;17:3884–3897. doi: 10.1128/mcb.17.7.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans. 1995;23:456–459. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- Persson C, Carballeira N, Wolf-Watz H, Fallman M. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130CASand FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO (Eur Mol Biol Organ) J. 1997;16:2307–2318. doi: 10.1093/emboj/16.9.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polte TR, Hanks SK. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130CAS . Proc Natl Acad Sci USA. 1995;92:10678–10682. doi: 10.1073/pnas.92.23.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retta SF, Barry ST, Critchley DR, Defilippi P, Silengo L, Tarone G. Focal adhesion and stress fiber formation is regulated by tyrosine phosphatase activity. Exp Cell Res. 1996;229:307–317. doi: 10.1006/excr.1996.0376. [DOI] [PubMed] [Google Scholar]
- Richardson A, Malik RK, Hildebrand JD, Parsons JT. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: a role for paxillin tyrosine phosphorylation. Mol Cell Biol. 1997;17:6906–6914. doi: 10.1128/mcb.17.12.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, Yazaki Y, Hirai H. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO (Eur Mol Biol Organ) J. 1994;13:3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Nagura K, Ishino M, Tobioka H, Kotani K, Sasaki T. Cloning and characterization of cell adhesion kinase beta, a novel protein-tyrosine kinase of the focal adhesion kinase subfamily. J Biol Chem. 1995;270:21206–21219. doi: 10.1074/jbc.270.36.21206. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Broome MA, Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-Src complex: involvement of the Grb2, p130CAS, and Nck adaptor proteins. Mol Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbauer JE. Cell migration: may the force be with you. Curr Biol. 1997;7:R292–R294. doi: 10.1016/s0960-9822(06)00140-0. [DOI] [PubMed] [Google Scholar]
- Serra-Pages C, Kadersha N, Fazikas L, Medly Q, Debant A, Struli M. The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR interacting protein co-localize at focal adhesions. EMBO (Eur Mol Biol Organ) J. 1995;14:2827–2838. doi: 10.1002/j.1460-2075.1995.tb07282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Schneider G, Cloutier JF, Veillette A, Schaller MD. Direct association of protein-tyrosine phosphatase PTP-PEST with paxillin. J Biol Chem. 1998;273:6474–6481. doi: 10.1074/jbc.273.11.6474. [DOI] [PubMed] [Google Scholar]
- Spencer S, Dowbenko D, Cheng J, Li WL, Brush J, Utzig S, Simanis V, Lasky LA. PSTPIP a tyrosine phosphorylated cleavage furrow– associated protein that is a substrate for a PEST tyrosine phosphatase. J Cell Biol. 1997;138:845–860. doi: 10.1083/jcb.138.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons M, Derry JM, Karlak B, Jiang S, Lemahieu V, McCormick F, Francke U, Abo A. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996;84:723–734. doi: 10.1016/s0092-8674(00)81050-8. [DOI] [PubMed] [Google Scholar]
- Tapon N, Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- Turner CE, Miller JT. Primary sequence of paxillin contains putative SH2 and SH3 domain binding motifs and multiple LIM domains: identification of a vinculin and pp125Fak-binding region. J Cell Sci. 1994;107:1583–1591. doi: 10.1242/jcs.107.6.1583. [DOI] [PubMed] [Google Scholar]
- Vithalani KK, Shoffner JD, De Lozanne A. Isolation and characterization of a novel cytokinesis-deficient mutant in Dictyostelium discoideum. . J Cell Biochem. 1996;62:290–301. doi: 10.1002/(sici)1097-4644(199608)62:2<290::aid-jcb16>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Vuori K, Ruoslahti E. Tyrosine phosphorylation of p130CASand cortactin accompanies integrin-mediated cell adhesion to extracellular matrix. J Biol Chem. 1995;270:22259–22262. doi: 10.1074/jbc.270.38.22259. [DOI] [PubMed] [Google Scholar]
- Wilson L, Carrier MJ, Kellie S. pp125FAK tyrosine kinase activity is not required for the assembly of F-actin stress fibres and focal adhesions in cultured mouse aortic smooth muscle cells. J Cell Sci. 1995;108:2381–2391. doi: 10.1242/jcs.108.6.2381. [DOI] [PubMed] [Google Scholar]
- Wu H, Parsons JT. Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol. 1993;120:1417–1426. doi: 10.1083/jcb.120.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Reynolds AB, Kanner SB, Vines RR, Parsons JT. Identification and characterization of a novel cytoskeleton-associated pp60src substrate. Mol Cell Biol. 1991;11:5113–5124. doi: 10.1128/mcb.11.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dowbenko D, Lasky LK. PSTPIP 2, a second tyrosine phosphorylated, cytoskeletal-associated protein that binds a PEST-type protein-tyrosine phosphatase. J Biol Chem. 1998a;273:30487–30496. doi: 10.1074/jbc.273.46.30487. [DOI] [PubMed] [Google Scholar]
- Wu Y, Spencer SD, Lasky LA. Tyrosine phosphorylation regulates the SH3-mediated binding of the Wiskott-Aldrich syndrome protein to PSTPIP, a cytoskeletal-associated protein. J Biol Chem. 1998b;273:5765–5770. doi: 10.1074/jbc.273.10.5765. [DOI] [PubMed] [Google Scholar]
- Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein-tyrosine phosphatase SHP-2 regulates cell spreading, migration and focal adhesion. J Biol Chem. 1998;273:21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]