

Abstract
The ER/Golgi soluble NSF attachment protein receptor (SNARE) membrin, rsec22b, and rbet1 are enriched in ∼1-μm cytoplasmic structures that lie very close to the ER. These appear to be ER exit sites since secretory cargo concentrates in and exits from these structures. rsec22b and rbet1 fused to fluorescent proteins are enriched at ∼1-μm ER exit sites that remained more or less stationary, but periodically emitted streaks of fluorescence that traveled generally in the direction of the Golgi complex. These exit sites were reused and subsequent tubules or streams of vesicles followed similar trajectories. Fluorescent membrin- enriched ∼1-μm peripheral structures were more mobile and appeared to translocate through the cytoplasm back and forth, between the periphery and the Golgi area. These mobile structures could serve to collect secretory cargo by fusing with ER-derived vesicles and ferrying the cargo to the Golgi. The post-Golgi SNAREs, syntaxin 6 and syntaxin 13, when fused to fluorescent proteins each displayed characteristic patterns of movement. However, syntaxin 13 was the only SNARE whose life cycle appeared to involve interactions with the plasma membrane. These studies reveal the in vivo spatiotemporal dynamics of SNARE proteins and provide new insight into their roles in membrane trafficking.
Keywords: vesicle trafficking, fluorescent proteins, SNAREs, membrane proteins, endoplasmic reticulum
Distinct membrane compartments of the secretory pathway are maintained despite the continuous anterograde and retrograde flow of proteins and lipids throughout the cell. This is accomplished by an intricate series of membrane trafficking decisions including the concentration and packaging of cargo and the organized fusion of specific membranes. Understanding the molecular mechanisms underlying membrane trafficking decisions will greatly further our appreciation of cellular regulatory mechanisms.
One particularly interesting aspect of membrane trafficking is the specificity and mechanism of the membrane fusion process. Studies in yeast (Alto et al., 1993; Brennwald et al., 1994) and the mammalian nerve terminal have defined a set of proteins critical for membrane fusion (Bennett and Scheller, 1993). Vesicle-associated membrane protein (VAMP),1 syntaxin, and synaptosome-associated protein of 25 kD (SNAP-25), collectively referred to as soluble NSF attachment protein receptors (SNAREs) (Söllner et al., 1993a), are the prototypes of a class of integral membrane (VAMP and syntaxin) or lipid modified (SNAP-25) proteins of the vesicle and plasma membrane. The binding of vesicle- and target-SNAREs (v- and t-SNAREs, respectively) from opposite membranes (Nichols et al., 1997) is critical in the fusion process because formation of this complex brings the bilayer in close proximity and possibly even drives the fusion process itself (Hanson et al., 1997; Lin and Scheller, 1997).
The v- and t-SNARE complex referred to as the core complex dissociates in a two-step process. First, two soluble factors, αSNAP and N-ethylmaleimide-sensitive factor, bind the core complex, resulting in a 20S particle. Upon ATP hydrolysis by N-ethylmaleimide-sensitive factor, the v-SNARE dissociates from the complex (Söllner et al., 1993b), allowing the SNAREs to participate in another round of membrane fusion (Mayer et al., 1996). Recent studies have uncovered a large number of VAMP and syntaxin homologues and these proteins are specifically localized to many different compartments within the cell (Wang et al., 1997; Advani et al., 1998). The possibility that the specific pairing of v- and t-SNAREs may be a critical determinant of the specificity of membrane fusion has been widely discussed (Rothman and Warren, 1994) but remains a largely untested hypothesis.
Although many other proteins, thought to function upstream of core complex formation, are critical in the vesicle trafficking process their specific roles are less well understood. For example, members of the Rab family of GTPases are specifically localized on many different membranes and have been demonstrated to be critical in the trafficking process (Pfeffer, 1996; Novick and Zerial, 1997). In addition, the sec1 family of syntaxin binding proteins is required for membrane fusion. While the sec1 family appears to be smaller than the syntaxin family, clear specificity in the associations between members of these protein families has been demonstrated (Pevsner et al., 1994). Also synaptotagmin and CAPS have been proposed to represent sensors for Ca2+ that regulates the final step of exocytosis (Südhof, 1995; Loyet et al., 1998). Further studies of these molecules promises to illuminate the biochemical mechanisms of membrane trafficking and exocytosis.
While genetic, biochemical, and immunohistochemical techniques provide important mechanistic insight into membrane trafficking, they provide little information on the dynamics of organelles in living cells. Several studies have investigated the dynamics of membrane trafficking in living cells using fluorescent dyes. Some of these dyes specifically stain particular domains of the secretory pathway. For example, NBD-ceramide, a vital stain of the TGN, has been used to reveal dynamic tubulovesicular processes that emerge from the TGN extending along microtubules where they contact adjacent Golgi elements (Cooper et al., 1990). More recently, green fluorescent protein (GFP) has been fused to proteins that travel through the secretory pathway in order to observe their trafficking pathways. A particularly useful marker for following trafficking pathways has been the temperature-sensitive mutant viral glycoprotein VSVGtsO45 (Bergmann, 1989) fused to GFP (ts-G-GFP). At the nonpermissive temperature this construct is misfolded and retained in the ER, whereas at the permissive temperature the ts-G-GFP is correctly routed through the Golgi complex to the plasma membrane (Presley et al., 1998). Imaging of ts-G-GFP uncovered an extensive network of tubulovesicular membranes interposed between the ER and the Golgi complex. These studies also revealed large ∼1-μm structures called vesicular tubular clusters (VTCs) that move from the periphery to the Golgi region (Balch et al., 1994; Presley et al., 1997; Sciaky et al., 1997; Cole et al., 1998). Similar techniques have also been used to demonstrate sequential roles of COPII and COPI coats in ER to Golgi trafficking (Scales et al., 1997).
In this study we use a variety of antibodies and GFP fusion proteins to study the dynamics of SNARE proteins and the trafficking pathways they mediate within the secretory pathway (see Fig. 1). We had generated previously a useful set of mAbs and pAbs to the cytoplasmic domains of several SNARE proteins (Hay et al., 1998). As additional markers to identify the ER, we used antibodies that recognized calnexin and Bip. We tracked the flow of cargo using ts-G-GFP. The dynamics of individual SNARE proteins was investigated in vivo using either blue, cyan, green, or yellow fluorescent proteins (BFP, CFP, GFP, YFP, respectively) fused to the lumenal or cytoplasmic domains of the SNAREs. We reveal rsec22b- and rbet1-enriched ER exit sites from which vesicles and/ or tubules emanate. Movement of these vesicles/tubules is generally directed towards the Golgi complex. Other pre-Golgi SNAREs displayed different behaviors. Membrin is concentrated in dynamic 1-μm structures, whereas GOS-28 appears restricted to the Golgi and remains relatively static. Post-Golgi SNAREs show distinct and extremely dynamic trafficking pathways. By using cyan and yellow fluorescent fusion proteins transfected into the same cell the intersection of the distinct trafficking pathways is observed. The unique dynamics of each SNARE-FP–labeled organelle likely requires specific coupling to motors and cytoskeletal tracks. Consequently, this process plays an important role in determining the specificity of protein trafficking and membrane fusion.
Figure 1.

Schematic of reagents used in this study. mAbs and pAbs against the cytoplasmic domains of several SNARE proteins were characterized previously. Antibodies against Bip and calnexin were used to identify the ER. The temperature-sensitive form of VSV-G coupled to fluorescent proteins serves as a secretory pathway cargo visible in living cells. B, C, G, and YFP were fused to a variety of SNARE proteins. Coupling to the NH2-terminal of the SNARE results in a fluorescent protein exposed to the cytoplasm, whereas fusion to the COOH-terminal end places the fluorescent protein within the lumen.
Materials and Methods
Immunofluorescence Microscopy of Fixed Normal Rat Kidney (NRK) Cells
NRK cells were maintained in DME containing 5 g glucose per liter, 10% FCS, and antibiotics in a humidified 5% CO2 incubator. For transfection with ts-G-GFP (slight modification of Scales et al., 1997) all of the following occurred: one barely confluent 10-cm dish of NRK cells, ∼107 cells, was removed from the plate with trypsin-EDTA, washed once with PBS, resuspended in 0.6 ml of PBS, and incubated for 10 min on ice with 10 μg of plasmid DNA in a 0.4-cm gap electroporation cell (Bio-Rad Laboratories). The cells were electroporated using 960 μF and 0.25 kV in an electroporator (Bio-Rad Laboratories) with capacitance extender, and plated on 2.2-cm2 glass coverslips. After 5 h at 37°C, sodium butyrate was added to 5 mM and the cells were shifted to 40°C for 12 h. For microscopy, coverslips (from control nontransfected cells at 37°C or transfected cells at 40°C) were either dropped directly into a well containing 4% paraformaldehyde, 0.1 M sodium phosphate, pH 7.0, or dropped into wells of medium preequilibrated at 32°C, and incubated for various lengths of time before fixation. After 30 min of fixation, coverslips were moved to wells containing 0.1 M glycine in PBS for >10 min, and equilibrated in permeabilization buffer (PBS containing 0.4% saponin, 1% BSA, and 2% normal goat serum). Staining was carried out at room temperature for 1 h in a permeabilization solution containing combinations of a calnexin polyclonal antiserum (Hammond and Helenius, 1994), a commercial rabbit polyclonal antibody to Bip (Stressgen), the affinity-purified rabbit anti–rat membrin 2-125 (Hay et al., 1998) or rbet1 mAb 16G6 (Hay et al., 1998). Coverslips were washed three times for 15 min each in permeabilization buffer and incubated for 30 min with anti–rabbit or anti–mouse secondary antibodies conjugated to FITC or Texas red. For staining with one of these antisera relative to GFP-G protein, Texas red secondary was used for staining so that it could be detected relative to the inherent green fluorescence of the GFP-G protein. After washing with permeabilization solution as above, coverslips were mounted in mounting medium (Vectashield; Vector Laboratories) and viewed using a fluorescence microscope (Olympus IX70; Olympus Optical Co.) with a 100× oil immersion lens, an image acquisition system (DeltaVision; Applied Precision), and a computer (model O2; Silicon Graphics). Images were captured to disk using filter sets appropriate for FITC (for FITC or GFP fluorescence) or Texas red (for Texas red fluorescence). Images were cropped, adjusted, and arranged using Adobe Photoshop and printed on a photodigital printer (Fujix Pictography; Fuji Photo Film Co.). Colocalization was quantitated blindly by placing puncta into three categories: SNARE alone, ts-G-VSV alone, or SNARE and ts-G-VSV. From each time point at least 50 puncta were categorized.
Immunogold Labeling of Ultrathin Cryosections
HepG2 cells were fixed in a mixture of 2% formaldehyde and 0.2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, and prepared for cryosectioning and double-immunogold labeling according to the protein A gold method as described (Slot et al., 1991; Liou et al., 1996; Hay et al., 1998).
Construction of Fluorescent Protein Fusions
Coding regions of SNAREs were amplified using PCR and cloned in frame into pEGFP-N3 or pEGFP-C1 (CLONTECH Laboratories, Inc.). EYFP, ECFP, and EBFP (CLONTECH Laboratories, Inc.) coding regions were amplified using PCR and cloned into pEGFP-N3 and pEGFP-C1 lacking GFP to produce pEYFP-N3, pEYFP-C1, pECFP-N3, pECFP-C1, pEBFP-N3, and pEBFP-C1 mammalian expression vectors (collectively called pEFP). Coding regions from each SNARE were cut from pEGFP-N3-SNARE and pEGFP-C1-SNARE and ligated into various pEFP vectors. All PCR-amplified inserts were sequenced to ensure no mutations were introduced. VSV-G-ts045 coding region was cut from VSV-G-ts-045-GFP (Scales et al., 1997) and ligated into various pEFP-N3 constructs.
Time-Lapse Imaging
NRK cells were electroporated with 7 μg plasmid DNA for single-transfected or 5 μg of each plasmid for double-transfected cells and plated onto glass coverslips one day before imaging as previously described. Coverslips with transfected cells were transferred to an imaging chamber (Warner Instruments) containing DME without phenol red supplemented with 25 mM Hepes, pH 7.4, 10% FCS, 1× penicillin/streptomycin. Cells were visualized with an inverted microscope (Olympus IX70; Olympus America Inc.) and a 60× or 100× oil immersion objective. The entire microscope was enclosed in a plastic chamber and prewarmed to either 32°C for VSV-FP movies, or 37°C for movies not involving VSV. Images were acquired using a CCD camera and DeltaVision software every 1.5–4 s with 0.3–1.0-s exposure times for up to 100 exposures for single- and 200 exposures for double-transfected cells. An FITC filter set was used for GFP movies and a custom made CFP/YFP excitation/emission filter set and polychroic was used for two-color CFP and YFP movies (Chroma Technologies Corp.). Resulting movies were analyzed on a computer (octane; Silicon Graphics) and DeltaVision image analysis software.
Results
Punctuate Structures Containing rbet1 and Membrin Are ER Exit Sites
Previous immunofluorescence studies (Hay et al., 1998) localized SNAREs (syntaxin 5, membrin, rsec22b, and rbet1) to the ER, Golgi, and numerous punctate structures throughout the cytoplasm. Immuno-EM established the identity of the punctate structures as VTCs of the peripheral and Golgi-adjacent ER–Golgi intermediate compartment (IC). To further characterize these elements and investigate their role as intermediates in secretory cargo transport, we employed double-label immunofluorescence microscopy experiments using ts-G-GFP and antibodies that specifically recognize SNAREs (Fig. 1).
First, we examined the relative distributions of endogenous rbet1 and membrin in fixed NRK cells. As shown in Fig. 2 (top), both proteins heavily labeled the juxtanuclear Golgi area as well as small punctate structures throughout the cytoplasm. These steady-state, 37°C structures are similar to those that were observed to contain rbet1, rsec22b, and membrin after incubations at 15°C. The difference is that at 15°C, these peripheral structures become markedly more intense relative to juxtanuclear staining and their size increases (not shown). As documented previously (Hay et al., 1998), rbet1 and rsec22b are present at higher levels in the ER than membrin. Although many bright cytoplasmic structures containing rbet1 also contained membrin (Fig. 2, top, arrows), VTCs were also visible that contained one but not the other of the two, suggesting some differentiation in their functions (see arrowheads).
Figure 2.
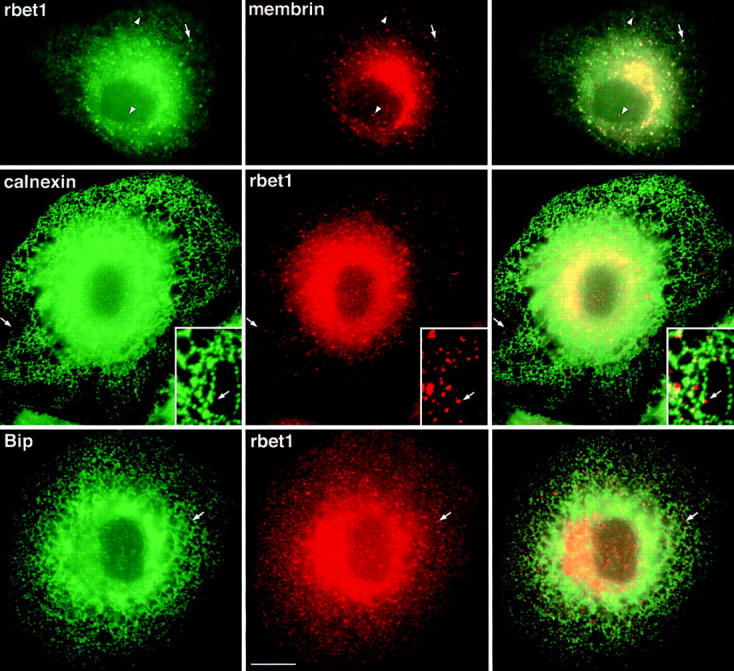
Peripheral structures containing both rbet1 and membrin are closely associated with, yet distinct from, ER tubules in fixed NRK cells. (Top row) Wild-type NRK cells grown at 37°C were fixed and stained for rbet1 (left) and membrin (middle) as described in Materials and Methods. Arrows represent peripheral structures positive for both proteins. Arrowheads represent peripheral structures positive for one, but not the other protein. (Middle row) Wild-type NRK cells grown at 37°C were fixed and stained for calnexin (left) and rbet1 (middle). (Bottom row) Wild-type NRK cells grown at 37°C were fixed and stained for Bip (left) and rbet1 (middle). Arrows in the middle and bottom rows point out peripheral rbet1- or membrin-positive structures very near, but distinct from, ER tubules. For Figs. 2, 4, and 5, the third column represents the merging of the first and second images. Bars, 10 μm (relative to objects in the first and second columns) and 4.3 μm (relative to insets). Insets show a higher magnification image from a similar cell.
Next we investigated whether the VTCs containing rbet1 and membrin were closely associated with ER elements by costaining with antibodies against resident ER proteins. We focused on peripheral VTCs because the density of membranes in the central Golgi area of the cell is too high to observe distinct VTCs. As shown in Fig. 2 (middle) many rbet1-containing elements were closely associated with ER tubules marked with calnexin, an integral membrane ER chaperone. Some of the rbet1 foci actually exclude or were de-enriched for calnexin (see arrows). Despite their apparent distinctness, the exclusive rbet1 elements and ER tubules appeared conspicuously associated, since the rbet1 spots were usually arranged in very close proximity to ER tubules. This is consistent with at least a portion of the rbet1-containing spots representing structures situated downstream of the sorting of secretory cargo away from ER resident proteins. These VTCs may originate adjacent to the ER by local fusion of ER-derived vesicles. We occasionally observed rbet1 spots that were not immediately adjacent to ER tubules (see Fig. 4, middle), perhaps representing more mature VTCs en route to the Golgi area. Another ER marker, Bip, displayed a similar overall staining pattern as calnexin; it also shared only limited overlap with rbet1 in the peripheral VTCs (Fig. 2, bottom, arrows).
Figure 4.
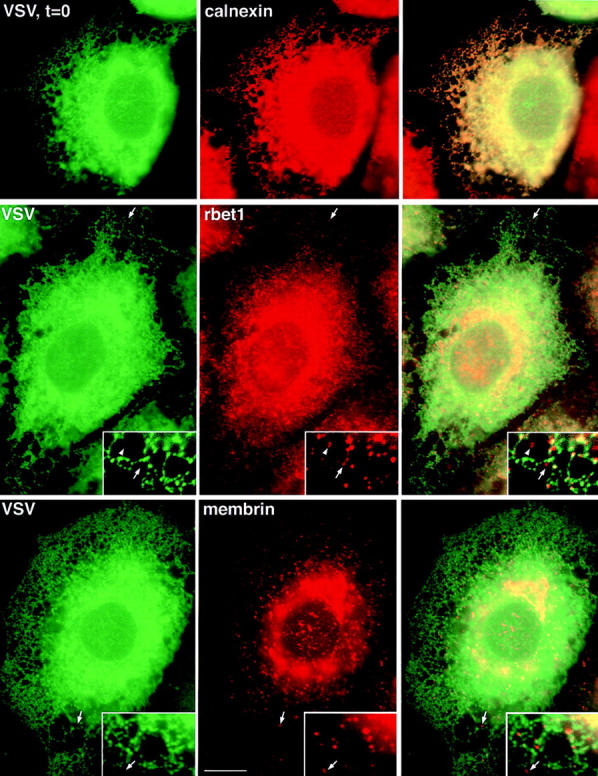
Secretory cargo retained in the ER has access to a subset of peripheral structures containing rbet1 and membrin. (Top row) ts-G-GFP– transfected NRK cells grown at 40°C were fixed and the subcellular distribution of ts-G-GFP protein (left) was compared to the calnexin staining pattern (middle). (Middle row) ts-G-GFP–transfected NRK cells grown at 40°C were fixed and the subcellular distribution of ts-G-GFP (left) was compared to the rbet1 staining pattern (middle). (Bottom row) ts-G-GFP–transfected NRK cells grown at 40°C were fixed and the subcellular distribution of ts-G-GFP (left) was compared to the membrin staining pattern (middle). Right column represents an overlay of the previous two images in the row. Arrows point out peripheral rbet1- or membrin-containing structures that did not contain a significant amount of ts-G-GFP protein, although they were adjacent to ER tubules. The arrowheads demonstrate that some of the exclusive structures were not immediately adjacent to ER tubules, perhaps representing mobile VTCs en route to the Golgi complex. Bars: (frames) 10 μm; (inset) 4.3 μm.
The organization of these SNARE-enriched foci was investigated at higher resolution using immuno-EM and examples are shown of the labeling observed with rsec22b and COPII antibodies (Fig. 3). COPII coat proteins are present on anterograde-directed transport vesicles that bud from the ER. Several conclusions are drawn from these micrographs. First, no difference is observed in the organization of these sites when they are labeled with antibodies that recognize rbet1, rsec22b, or membrin, consistent with the colocalization of the spotty immunoreactivity at the light level. Second, sites close to the nucleus and those located close to the plasma membrane have the same general organization. Third, the structures are comprised of an intricate network of vesicles and/or tubules, and many are immunoreactive for both the SNAREs and COPII. It is not possible to discern whether long tubules emanate from the ER traveling in and out of the plane of section, or, alternatively, if the observed elements represent a myriad of individual vesicles. Finally, as is noted at the light level, these structures are adjacent to clearly recognizable ER cisternae.
Figure 3.
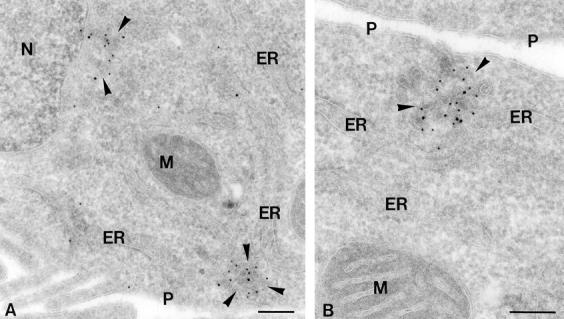
COPII and rsec22b immunogold labeling of peripheral ER exit sites. Ultrathin cryosections of HepG2 cells double-immunogold–labeled for COPII (10 nm gold) and rsec22b (15 nm gold). (A) ER exit sites (arrowheads) with similar morphology are present near the nucleus, N, and close to the plasma membrane, P. (B) A higher magnification of a peripheral ER exit site showing the characteristic tubulovesicular morphology. M, mitochondrion. Bars, 200 nm.
To help clarify the relationship of the SNARE-containing structures to secretory cargo, we used a GFP-tagged version of VSV-G-ts045 (ts-G-GFP) as a model ER-to-Golgi cargo protein (Fig. 1). The temperature-sensitive nature of ts045 allows abrupt release of the cargo G protein from the ER, enabling morphological characterization of sequential transport intermediates (Plutner et al., 1992). NRK cells were transfected with a ts-G-GFP protein construct (Scales et al., 1997) and held at 40°C to accumulate the cargo in the ER. At 40°C, ts-G-GFP was found to largely colocalize with calnexin in the ER (Fig. 4, top). It often appeared that calnexin was enriched in different sections of ER tubules, whereas the ts-G-GFP was more homogeneously distributed. However, both of these proteins clearly resided in contiguous regions of the same ER tubules. As expected, rbet1 peripheral structures only partially overlapped with the ER-retained cargo. Fig. 4 (middle) demonstrates that although many rbet1 spots fall along ER tubules and contain the green cargo, many also lie just adjacent to (see arrows) or even some distance (arrowheads) from the ER structures and lack ts-G-GFP. Hence, it appears that cargo in the ER does not have access to a portion of the rbet1-containing structures, indicating that sorting and/or vesicle transport is required to move from the ER into these structures. A similar relationship is observed between VSV-G and membrin under these conditions (Fig. 4, bottom, arrows). Before releasing ts-G-VSV from the ER 18% of the rbet1 puncta and 4% of the membrin puncta colocalize with ts-G-VSV.
When released from the ER by shifting to 32°C, the ts-G-GFP cargo quickly began entering peripheral structures distinct from the ER. As seen in Fig. 5 (top), after 5 min at the permissive temperature, the cargo was still very apparent in the ER, but was also present in calnexin-negative puncta (see arrows). This is similar to those that contained the ER/Golgi SNAREs. 5 min after release of cargo from the ER, 57% of rbet1 and 27% of membrin puncta colocalized with ts-G-GFP. The cargo became more concentrated in these puncta as transport continued. By 10 min after release from the ER, the ER staining was weak and the peripheral puncta and Golgi area staining was intense. At this time 42% of rbet1 and 52% of membrin puncta colocalized with ts-G-GFP (Fig. 5, rbet1, middle; membrin, bottom), The data indicate that these SNARE-containing structures represent bona fide intermediates in cargo transport and are suggestive of a sequential localization, first in rbet1 and later in membrin puncta. Concentrated cargo colocalized with SNAREs both in foci in close proximity to and some distance away from ER tubules.
Figure 5.
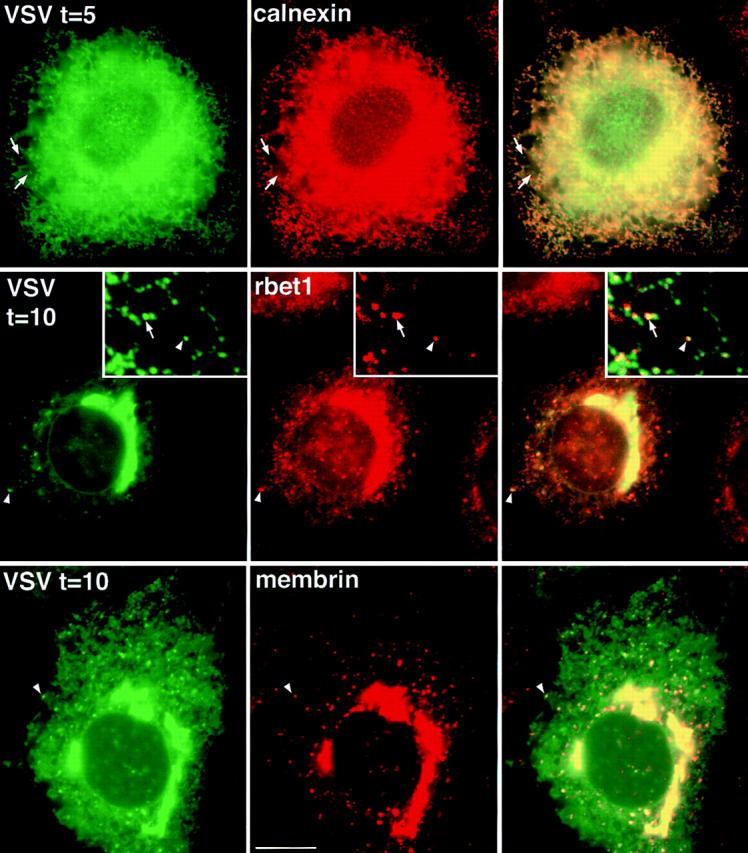
GFP-labeled secretory cargo rapidly enters and accumulates in punctate rbet1- and membrin-containing structures when released from the ER. (Top row) ts-G-GFP–transfected NRK cells grown at 40°C and incubated for 5 min at 32°C were fixed and the subcellular distribution of ts-G-GFP (left) was compared to the calnexin staining pattern (middle). (Middle row) ts-G-GFP–transfected NRK cells grown at 40°C and incubated for 10 min at 32°C were fixed, and the subcellular distribution of ts-G-GFP (left) was compared to the rbet1 staining pattern (middle). (Bottom row) ts-G-GFP–transfected NRK cells grown at 40°C and incubated for 10 min at 32°C were fixed and the subcellular distribution of ts-G-GFP (left) was compared to the membrin staining pattern (middle). Right column respresents an overlay of the previous two images in the row. Arrows and arrowheads point out peripheral rbet1-containing structures that do not appear to overlap ER tubules, yet clearly contain the ts-G-GFP cargo. The arrowheads in the inset point to a structure enriched for both SNAREs and cargo that is clearly distinct from the ER, perhaps representing a mobile VTC en route to the Golgi complex. Bars: (frames) 10 μm; (insets) 4.3 μm.
By 60 min after release from the ER, cargo was visible predominantly in the Golgi and plasma membrane, with little ER or peripheral IC staining (not shown). Overall the data suggest that the cargo moves from broadly dispersed locations within the ER into the SNARE-containing peripheral sites where it exits these domains and travels to the Golgi apparatus.
SNARE-containing Organelles Revealed by Fluorescent Protein Fusions
To better understand the dynamics of transport intermediates containing SNARE proteins we transfected cells with various fluorescent proteins (FPs) fused to either the NH2- or COOH-terminal ends of the SNAREs (Fig. 1). Either stable or transient expression in NRK cells was used to observe the localization of the SNARE-FP fusion proteins. Consistent with our previous experience, the transfected SNARE-FPs localized in very similar, if not identical, patterns to those of the endogenous proteins (Fig. 6).
Figure 6.
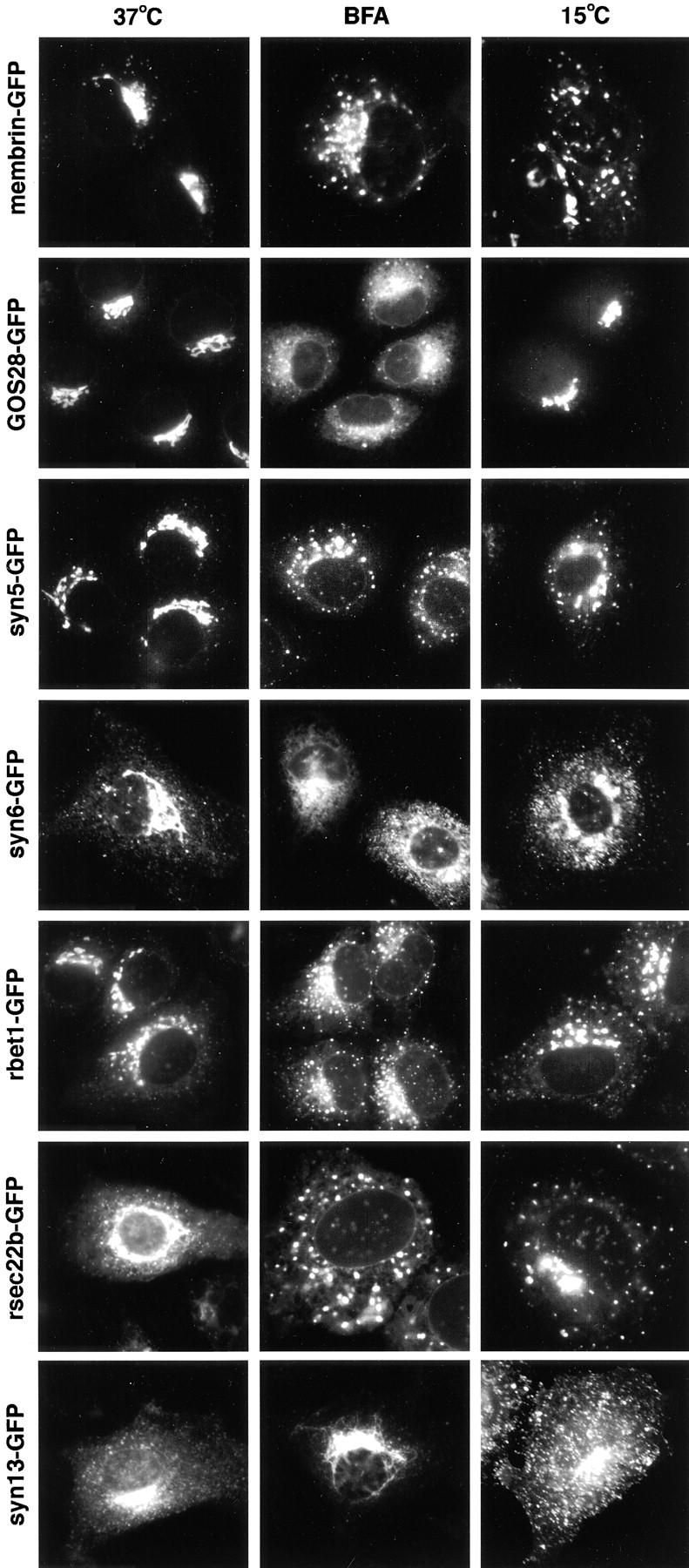
GFP-tagged SNAREs display a localization, sensitivity to BFA, and 15°C behavior similar to that of the endogenous proteins. Seven SNAREs were fused to GFP and images were captured at 37°C, in the presence of BFA for 1 h, or after 1 h at 15°C. The width of each panel is 56 μm.
In transient expression experiments we examined the SNARE-FP localization in cells expressing widely varying levels of the transfected product based on the fluorescence intensity. At all intermediate levels of expression the pattern matched closely with that of the endogenous protein. To further investigate the dynamics of the GFP-tagged SNAREs compared to the native protein we treated transfected cells with Brefeldin A (BFA) or lowered the temperature to 15°C. Again the GFP-tagged SNARE proteins behaved as the wild-type proteins (Fig. 6). For example, GOS-28 is little effected at 15°C, while membrin, rbet1, and Sec22 become dispersed within the cytoplasm into a punctate distribution. Treatment of syntaxin 13–transfected cells with BFA resulted in redistribution into a compact spot whereas the Golgi region staining of sec22b and the other IC SNAREs dispersed into a punctate fluorescence pattern. These data support the notion that the trafficking pathways of the transfected GFP-tagged SNAREs are very similar if not identical to the endogenous proteins.
To see if SNARE-FP incorporates into complexes we used antibodies against syntaxin 5 to immunoprecipitate solubilized membranes from cells transfected with ER/ Golgi SNARE-FPs. SNARE-FPs were found to be present in complexes with endogenous proteins. For example, immunoprecipitation of endogenous syntaxin 5 resulted in coimmunoprecipitation of membrin-GFP (not shown). Also, GFP mAbs (CLONTECH Laboratories) were able to precipitate endogenous syntaxin 5 in membrin-GFP– transfected cells but not GFP-transfected cells. Finally, the presence of GFP-tagged proteins did alter the localization of other endogenous SNAREs (data not shown). Taken together, these data support the hypothesis that the transfected SNARE-FPs have many, if not all, of the properties of the endogenous molecules.
To directly test the hypothesis that cargo moves into rsec22b/rbet1 peripheral sites and exits these sites, we performed time-lapse imaging of cells transfected with spectrally distinct fluorescent proteins (Fig. 1). We used various fluorescent protein combinations and found that although GFP and BFP are spectrally distinct, photodamage of BFP led to rapid diminution of the signal making this combination impossible for imaging studies. YFP and CFP are spectrally distinct and also photostable, allowing imaging of both proteins in a single cell. We did not observe any bleedthrough into the CFP channel in YFP-transfected cells and vice versa. Thus, the CFP-YFP combination of fluorescent proteins is ideal for double-labeled imaging studies. After release of ts-G-YFP from the temperature block we observed accumulation of the cargo protein in a preexisting rsec22b-CFP–labeled site (Fig. 7, A and B). Subsequently, the colabeled sites lose the ts-G-YFP, leaving behind the rsec22b-CFP (Fig. 7, C and D). The ts-G-YFP dynamics observed are similar to those previously described (Presley et al., 1997; Scales et al., 1997). These data confirm our hypothesis suggesting that the peripheral rsec22b/rbet1-enriched sites are indeed ER exit sites. We interpret our data as the direct observation of the filling and exit of cargo from ER exit sites which remain behind at a fixed position within the cell.
Figure 7.
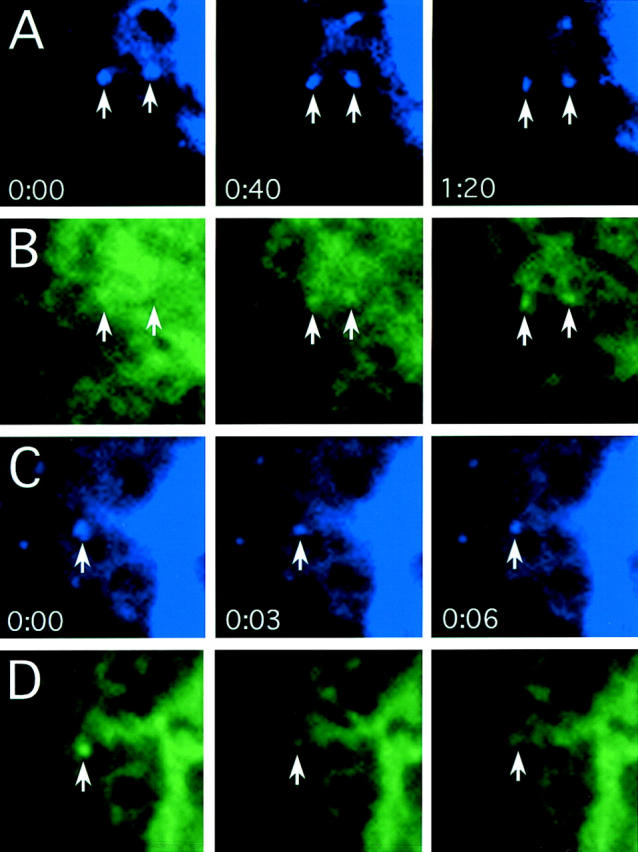
rsec22b sites concentrate cargo that travel to the Golgi complex. (A) rsec22b-CFP labels peripheral sites. Time in minutes:seconds for A and B is indicated. (B) ts-G-YFP is localized in the ER and, upon release from the temperature block, becomes concentrated at preexisting rsec22b-CFP sites. (C) rsec22b-CFP labeling of peripheral sites. Time in minutes:seconds for C and D is indicated. (D) 5 min after release from the temperature block ts-G-GFP is depleted from rsec22b sites. Panel width is 6.2 μm.
The Dynamics of SNARE-FP–labeled Organelles in Living Cells
Many types of organelle movements were observed in the video images of living cells transfected with FP-tagged SNAREs. A summary of movements observed at 3-s intervals with a 1-s exposure time is shown in Fig. 8. Each type of event is depicted by a particular combination of symbols so that summaries of organelle movements can be presented in later figures. The first type of movement involves a fluorescent organelle moving in the field without significant morphological changes (Fig. 8 A). Between 0 and 4 s the organelle has moved 3.9 μm and between 4 and 8 s the organelle has moved in the same general direction another 2.4 μm. This movement is summarized by a closed circle with an arrow pointing in the direction that the organelle is moving. Since the organelle is observed in the second frame it is again depicted by a circle followed by another arrow that ends at the position of the organelle in the third frame. We interpret this movement to be a membrane element or cluster of elements maintaining its structure and simply moving from one position to the next. The rate of this type of movement is ∼0.75–1.0 μm/s.
Figure 8.
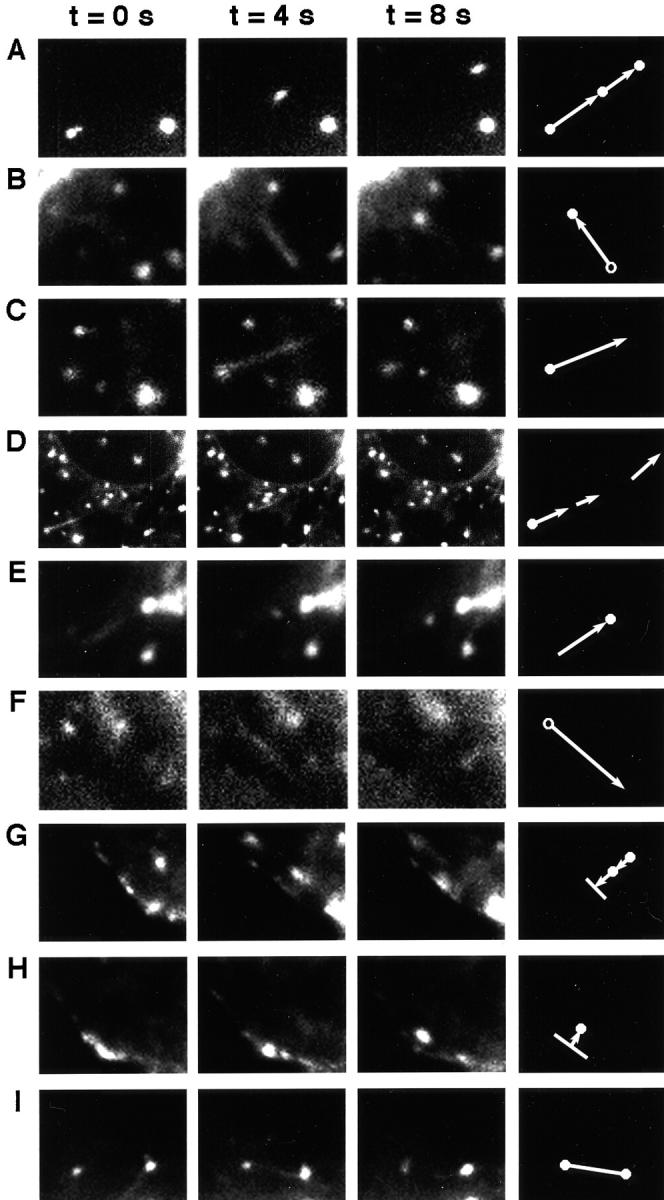
SNARE-GFP fusions undergo a variety of distinct movements. 1-s exposures are taken with 3 s between exposures. Symbols at the far right are used to represent the class of event. (A) Syntaxin 6-GFP; (B) rsec22b-GFP; (C) rsec22b-GFP; (D) lower magnification of C; (E) rsec22b-GFP; (F) rsec22b-GFP; (G) syntaxin 13-GFP; (H) syntaxin 13-GFP; and (I) rbet1-GFP. The width of a panel is 7.9 microns, except in panel D where the length is 22.2 μm.
A similar movement is observed in Fig. 8 B; however, this time a streak of fluorescence is observed emanating from an organelle in frame one and ending at an organelle that appears in frame three. Since the two types of movements (Fig. 8, A and B) may be mechanistically different we illustrate them slightly differently; in the latter case an open circle is connected to a closed circle by an arrow pointing in the direction of the movement. A streak of fluorescence may originate from either a structure moving while the shutter is open, in effect blurring across the CCD array, or from an elongated, tubular organelle. Here we favor the former case since the structure in the original position is not present in the final image and the streak is the same width as the original organelle.
We also observe streaks emanating from round, ∼0.75-μm organelles. However, in this case the streaks either loose their fluorescence or are lost from our field of view in some other fashion (Fig. 8 C). These events are illustrated by a closed circle at the base of an arrow. In many cases we are able to follow the fluorescent streaks emanating from a sphere through several frames (Fig. 8 D; left frame is a lower magnification image of the region shown in Fig. 8 C, middle frame). We depict these events by a closed circle with an arrow emanating in the direction of the streak, as in Fig. 8 C. The subsequent fluorescent streaks are illustrated as arrows (without a circle at the base or the end) pointing in the direction of the movement and proportional to the length of the streak (Fig. 8 D). Rates of movements seen with this type of event are 1.3 ± 0.7 μm/s. These dynamics we interpret as extending tubules or vesicle streams because the streaks are thinner than the organelle from which they originate and at least a fraction of the original organelle remains behind at the initial position. In addition, in images collected with 0.5-s exposure times with no dark time between exposures we see partial overlap in different exposures along the length of the tubule (not shown). In other words, the end of a tubule in one exposure may have moved to the midpoint of the tubule in the next image.
Another type of movement observed is a streak that ends in a spherical organelle, depicted by an arrow with a closed circle only at the arrow head (Fig. 8 E). Fluorescent spheres are also observed to emit a streak that appears to consume the fluorescence of the sphere. This is depicted by an arrow pointing in the direction of the streak with an open circle at the origin (Fig. 8 F). These movements could either be tubules emanating from the sphere or the sphere itself moving while losing fluorescence. Lengths of the arrows are proportional to the distance of the migration during the time interval or the length of the fluorescent streak.
Post-Golgi SNARE proteins exhibited additional events as illustrated in Fig. 8, G and H. Organelles are observed approaching the plasma membrane followed by an increase in the fluorescence along the membrane, suggesting an exocytotic membrane fusion event. Conversely, we observe bright fluorescent regions of the membrane followed by the appearance of an intracellular organelle, which suggests an endocytic event. These events are depicted by a line along the plasma membrane followed by an arrow pointing either toward (exocytosis), or away (endocytosis) from the plasma membrane. At this level of resolution we cannot rule out the possibility that the organelles are only in close proximity to the membrane and that we are not observing actual exo- or endocytic events. Finally, using several labeled SNAREs we have observed two organelles that appear to become connected by a tubule, suggesting the transfer of contents between the structures. To illustrate these events we connected two circles with a line (Fig. 8 I).
ER and Golgi SNARE Dynamics
To follow the dynamics of individual organelles and/or transport intermediates labeled with SNARE-FP we imaged cells and plotted the movements according to the key outlined in Fig. 8. Movements of SNARE-GFP organelles are shown in Fig. 9, where each color represents the pathway of a separate element plotted at up to nine positions.
Figure 9.
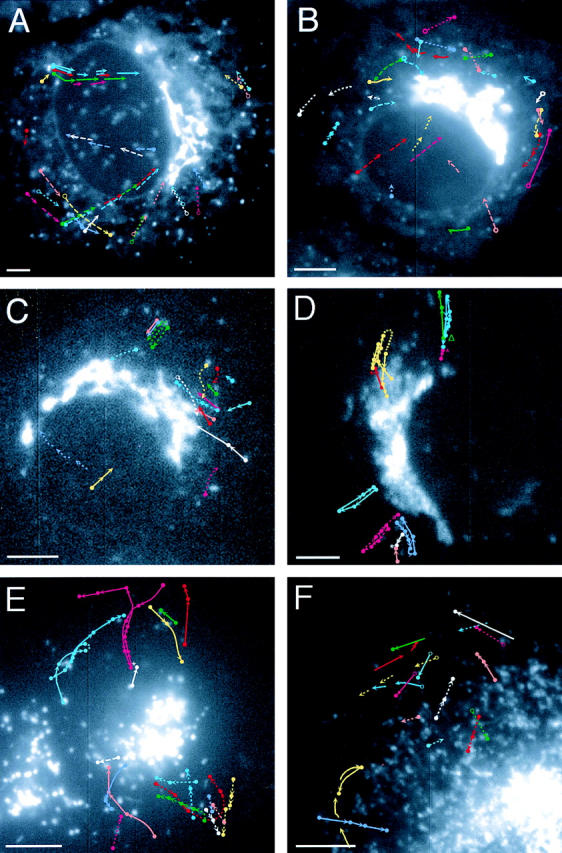
SNARE dynamics. (A) Sec22b-GFP–transfected cell with the pathways of individual organelles illustrated by different colors according to the key in Fig. 8. 1-s exposures were taken every 3 s. (B) rbet1-GFP. (C) syn5-GFP. (D) Membrin-GFP. An asterisk illustrates two structures that appear to associate with each other, whereas an open triangle marks two structures that appear to dissociate from each other. (E) Syntaxin 6-GFP is shown with 0.5-s exposures followed by 2.5 s of dark time. (F) Syntaxin 13-GFP. 0.5-s exposures followed by 1.5 s of dark time. Bars, 5 μm.
rsec22b-GFP and rbet1-GFP are observed at peripheral sites, consistent with our previous observations of a significant contingent of these SNAREs in the ER (Fig. 9, A and B). Peripheral fluorescent sites are most easily observed with rsec22b-GFP and are largely stationary with the exception of some small movements that might be due to Brownian motion. Far fewer back and forth movements are observed with rsec22b than with membrin (see below). Importantly, these relatively stationary structures are the points of origin of streaks of fluorescence that often travel across the cytoplasm toward the Golgi region (note the solid line with red, green, and blue traces emanating from a single structure in Fig. 9 A). 29% of the movements observed were of this type (C in Fig. 8) and 40% of the movements were migrating fluorescent streaks (D in Fig. 8). We hypothesize that these streaks represent tubules or closely aligned rows of vesicles carrying cargo from ER exit sites to the Golgi area. Since distinct streaks emanating from a particular site at different times follow a similar pathway, the organelles are likely traveling on the same or parallel microtubule tracks. Other types of rsec22b-GFP movements were as follows: 16% (Fig. 8 A), 10% (Fig. 8 F), and 4% (Fig. 8 I). Relatively little motion is seen in the retrograde direction, suggesting that the recycling of rsec22b occurs via a process that is rare or difficult for us to observe. In the example of a retrograde movement in Fig. 9 A (dashed white and purple arrows pointing away from the Golgi region), the site is consumed. While quite similar motions are recorded for rsec22b-GFP and rbet1-GFP, it was more difficult to follow the rbet1-GFP particles for long distances, perhaps because the fluorescence intensity is fainter. We observed a few rbet1-GFP sites near the Golgi region that appear to connect to each other via tubules (Fig. 9 B).
Syntaxin 5-GFP displays less frequent movement events in our recordings. The events observed are similar in character to the other ER and Golgi SNAREs although fewer long distance movements from peripheral sites were recorded (Fig. 9 C). The cell in Fig. 9 D expresses the transfected membrin-GFP fusion protein at a moderate to low level. Hence, the fainter more peripheral spotty structures distal to the intermediate compartment are not readily observed. Interestingly, 100% of the observed membrin-FP– labeled organelles transit away from and back to the Golgi region (Fig. 8 A). The most intricate of these is illustrated in yellow where a membrin-FP particle originates in a region that may be within the Golgi complex, transits out and away from the Golgi complex, wanders, and then returns to the Golgi region. Other organelles (blue, violet, pink) display similar patterns, moving back and forth between the Golgi region and more peripheral sites. The average rate of movement is 0.7 ± 0.3 μm/s. Organelles can be observed to intersect and form a pair, or to dissociate from a previously existing pair.
We speculate that these movements are antero- and retrograde movements of vesicles, clusters of vesicles, or small tubules between the ER, IC, and the Golgi apparatus. Interestingly, as a cell enters mitosis the membrin-GFP becomes fragmented, appearing more evenly dispersed about the cell (data not shown). Furthermore, organelle movements cease or are dramatically reduced, which is consistent with the previously described change in secretory events associated with mitotic cells (Warren et al., 1995). GOS28-GFP shows few if any detectable events on the time scale we recorded from the transfected cells (data not shown). This behavior of GOS28 is consistent with our previous observations that the protein changes little upon a temperature shift to 15°C and that GOS28 appears to be in distinct complexes from the other ER to Golgi SNAREs (Hay et al., 1998).
We also studied the dynamics of two post-TGN SNARE proteins, syntaxin 6-GFP and syntaxin 13-GFP (Fig. 9, E and F). The movements recorded for syntaxin 6-GFP are very unique as they are all of the type illustrated in Fig. 8 A, that is, spherical organelles migrating within the cell. A single organelle was traced through 11 frames illustrating movements towards, away from, and parallel to the TGN (Fig. 9 E, pink solid line). Syntaxin 6-GFP–labeled organelles can also be seen to reverse direction abruptly. In contrast to syntaxin 6-GFP, syntaxin 13-GFP–labeled organelles exhibit a wider variety of movements. Movements of spherical organelles as well as tubules are documented in Fig. 9 F. In addition, we observe syntaxin 13-GFP organelles that approach the plasma membrane, possibly fusing, and events that have the appearance of endocytosis (Fig. 8, G and H). Directions of the syntaxin 6 and 13-GFP movements are much more random in orientation than those of rsec22b-GFP and membrin-GFP.
To follow the movements of multiple SNAREs in a single cell we again used the YFP, CFP combination of fluorescent proteins. rsec22b-CFP coexpressed with membrin-YFP exhibits a higher degree of colocalization when compared to rsec22b-CFP coexpressed with syntaxin 13-YFP. However in the rsec22b-CFP/membrin-YFP cotransfected cells, the ratios of the two SNARE-FPs are not always equal because some organelles appear to contain more of one SNARE while other organelles contain more of the other (Fig. 10, A–C). This observation is consistent with the different overall dynamics observed for the two SNAREs. With this technique we are further able to observe the following: some membrin and rsec22b movements are distinct, and in some cases the colabeled rsec22b and membrin sites move synchronously.
Figure 10.
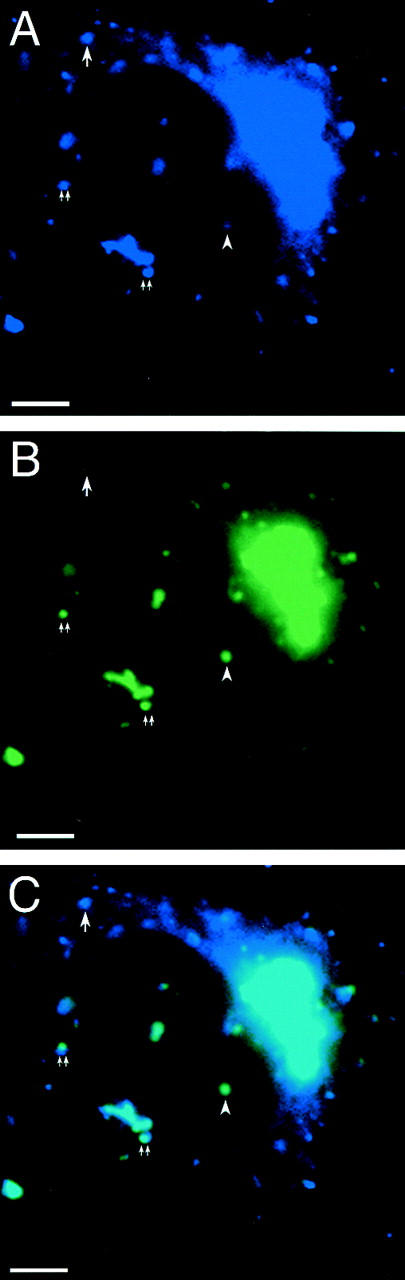
Overlapping and distinct SNARE trafficking pathways. Cotransfection of (A) rsec22b-CFP and (B) membrin-YFP reveals colocalizing structures (double arrow) and structures containing mostly membrin (arrowhead) or rsec22b (arrow). (C) A and B merged image. Moving structures that contain rsec22b and membrin appear as a double (yellow and cyan) spot because of the 1.25-s time difference between the exposures. Bars, 5 μm.
Discussion
We have studied the organization of the secretory pathway by fusing fluorescent proteins to VSV-G, a widely utilized cargo protein, and SNAREs, molecules important for membrane fusion. By following the dynamic movements of organelles labeled with these reagents we are able to better understand the mechanisms whereby proteins are translocated through the secretory pathway. Since SNARE proteins are critical determinants of an organelle's identity, these proteins serve as specific labels of functional compartments. Beginning at the ER, several interesting observations emerged from this work. At 40°C the ts-G-GFP is retained in the ER where the staining is similar to both calnexin and Bip. Many of the SNARE-labeled structures are closely juxtaposed to the ER, as if they lie alongside the tubules of the organelle. When the temperature of the transfected cells is lowered, allowing the VSV-G to flow through the secretory pathway, the protein accumulates in the 0.75–1-μm structures that precisely overlap with SNAREs at sites we believe to be the position of exit from the ER.
Double labeling with SNAREs and cargo reveals that rsec22b precedes cargo at these ER sites. The theory that these structures are ER exit sites is further confirmed by the dynamics of fluorescent SNARE proteins, particularly rsec22b-GFP. We observe streaks of fluorescence emanating from these sites that we interpret to be tubules or rows of closely spaced vesicles. Multiple streaks are commonly observed to emanate sequentially from a single exit site that suggests the ER exit sites are used multiple times. Their direction of transport is most often toward the Golgi region (suggesting anterograde trafficking) and sequential streaks follow similar paths along what are likely to be the same or closely aligned microtubules. This picture is somewhat different than that observed for the real-time dynamics of cargo, where the peripheral structures were noted to move toward the Golgi complex (Presley et al., 1997).
In contrast, our data support the view of a fixed ER exit site that serves to load transport organelles (vesicles or emerging tubules) with cargo while sorting ER proteins in a retrograde fashion back to the ER. When the transport vesicles or nascent tubules have matured, we propose that they translocate to the Golgi apparatus along microtubules and a new round of cargo loading and protein sorting initiates at the same site. Nevertheless, the regulation of membrane dynamics at these ER exit sites needs to be further investigated. For example, it would be important to learn what accounts for the apparently sporadic nature of the fluorescent streaks; perhaps they signify cargo export. In addition, it is of interest to know whether there is a functional difference between the SNARE-labeled sites that appear to colocalize with ER markers (perhaps in segments of ER tubules) and those puncta that exclude ER markers and lie directly alongside or some distance from ER tubules. Also, it is not yet known how SNAREs concentrate at certain sites while excluding resident ER proteins.
Interestingly, membrin, a SNARE proposed to function in concert with rsec22b through the formation of complexes (Hay et al., 1998) and whose still-life localization significantly overlaps that of rsec22b, appears to display significantly different dynamics than rsec22b. Membrin appears to reside on mobile ∼1-μm organelles that themselves move between the cis-Golgi region and more peripheral sites. On the other hand, while rsec22b is more apparent on stationary, peripheral, ∼1-μm structures from which steaks of fluorescence emanate, generally toward the Golgi region. One possibility is that rsec22b and rbet1 are more enriched in small transport vesicles/tubules that stream outward from larger foci. This would be consistent with the EM study that found a larger percentage of rsec22b and rbet1 vesicles also contained COPII when compared to syntaxin 5– or membrin-containing vesicles (Hay et al., 1998). The mobile 1-μm structures containing membrin may contain rsec22b and rbet1 as well, but these SNAREs may be more vigorously sorted away when these structures are translocated. This hypothesis might explain why the movements shown in Fig. 8 A appear rarer than those shown in Fig. 8 F, which are more common in the movies of rsec22b- and rbet1-FPs. What is the function of the mobile 1-μm structures? Perhaps they function as anterograde and/or retrograde transport organelles in conjunction with small ER-derived vesicles. The small vesicles/tubules may serve to fill the membrin-enriched larger structures which then act as carriers in and out of the Golgi area. These organelles could then be thought of as mobile extensions of the cis-Golgi network.
Since the fluorescence of syntaxin 5–transfected cells was not as bright as rsec22b, we may not have detected particular classes of movements. Given this caveat, syntaxin 5 dynamics were generally the same types as the other mobile ER and Golgi SNAREs; however, movements were less frequent, perhaps reflecting a less dynamic nature of this SNARE. The tubular extensions observed between organelles (see Fig. 8 I) may be conduits for the transfer of cargo between organelles. Alternatively, these structures may represent transport tubules that fuse with the acceptor compartment before fission from the donor compartment.
The dynamics of the two post-Golgi SNAREs, syntaxin 6 and syntaxin 13, were very different from the ER-to-Golgi proteins. Syntaxin 6 localizes largely to the TGN, and less so to endosomes. Syntaxin 6-FP appears on organelles that meander through the cytoplasm, some of which travel away from the Golgi region, reverse direction, and travel back toward this site again, perhaps reflecting cycling between the TGN and endosomes. The precise nature of any cargo contained in these organelles remains unknown. However, the localization and dynamics are consistent with a role in TGN to endosome trafficking. Syntaxin 13 is proposed to represent a SNARE in the plasma membrane/recycling endosome pathway (Prekeris et al., 1998; Tang et al., 1998). Consistent with this proposal, syntaxin 13-FP labels were the only organelles we were able to observe in potential plasma membrane fusion and recycling events. In addition, the dynamics appear to reveal tubules that extend from a central organelle. Perhaps these tubules, formed as proteins, are sorted within the early endosome.
The most remarkable feature of cellular dynamics reinforced by these studies is the specificity of the multitude of trafficking events. The direction and mode of movements, the size and shape of the organelles, and the destinations of the organelles are all precisely regulated. These events are likely to be the critical determinants of the specificity of vesicle trafficking and the membrane organization of cells. Clearly when an organelle arrives at its appropriate destination within the cell an adequate machinery, likely comprised of SNAREs, must catalyze the membrane fusion events. But how cargo and SNAREs are specifically coupled to the organelle translocation machinery, likely comprised of motors and cytoskeletal tracks, remains unclear. Recent reports of motor proteins binding specific Rab proteins may be part of the solution to the molecular recognition issues that underlie this problem. Perhaps purification of the SNARE-FP–tagged organelles will help lead to a solution to this problem.
Acknowledgments
The authors thank Dr. Suzanne R. Pfeffer for her efforts in establishing the Stanford University Cell Sciences Imaging Facility; Dr. Susan L. Palmieri for her excellent management of the facility; Dr. Carolyn Machamer (The Johns Hopkins University) for permission to use VSV-G-ts045; Dr. Suzie Scales for technical advice regarding VSV-G-ts045; and Paul Millman of Chroma Technology Corporation for technical expertise regarding fluorescent protein optical equipment. We also thank rotation students Mr. Igor Brodsky and Ms. Grace Park for help in making some of the constructs used in this work, and Viola Oorschot (Utrecht University) for preparing the ultrathin cryosections.
Abbreviations used in this paper
- B,C,G,Y FP
blue, cyan, green, yellow fluorescent protein
- IC
intermediate compartment
- NRK
normal rat kidney
- NSF
N-ethylmaleimide-sensitive factor
- SNAP-25
synaptosome-associated protein of 25 kD
- SNAP
soluble NSF attachment protein
- SNARE
soluble NSF attachment protein receptor
- VAMP
vesicle-associated membrane protein
- ts-G-GFP
temperature sensitive vesicular stomatitis virus-associated membrane protein
- VTC
vesicular tubular cluster
Footnotes
Address correspondence to Richard H. Scheller Howard Hughes Medical Institute, Department of Molecular and Cellular Physiology, Stanford, CA 94305-5428. Tel.: (650) 723-9075. Fax: (650) 725-4436. E-mail: scheller@cmgm.stanford.edu
J.C. Hay's present address is Department of Biology, University of Michigan, 830 North University, Room 3065C, Ann Arbor, MI 48109-1048.
References
- Advani RJ, Bae HR, Bock JB, Chao DS, Doung YC, Prekeris R, Yoo JS, Scheller RH. Seven novel mammalian SNARE proteins localize to distinct membrane compartments. J Biol Chem. 1998;273:10317–10324. doi: 10.1074/jbc.273.17.10317. [DOI] [PubMed] [Google Scholar]
- Alto MK, Ronne H, Keranen S. Yeast syntaxins Sso1p and Sso2p belong to a family of related membrane proteins that function in vesicular transport. EMBO (Eur Mol Biol Organ) J. 1993;12:4095–4104. doi: 10.1002/j.1460-2075.1993.tb06093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, McCafferey JM, Plunter H, Farquhar MG. Vesicular stomatitis virus glycoprotein is sorted and concentrated during export from the endoplasmic reticulum. Cell. 1994;76:841–852. doi: 10.1016/0092-8674(94)90359-x. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Scheller RH. The molecular machinery for secretion is conserved from yeast to neurons. Proc Natl Acad Sci USA. 1993;90:2559–2563. doi: 10.1073/pnas.90.7.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann JE. Using temperature-sensitive mutants of VSV to study membrane protein biogenesis. Methods Cell Biol. 1989;32:85–110. doi: 10.1016/s0091-679x(08)61168-1. [DOI] [PubMed] [Google Scholar]
- Brennwald P, Kearns B, Champion K, Keranen S, Bankaitis V, Novick P. Sec9 is a SNAP-25 like component of a yeast SNARE complex that may be the effector of Sec4 function in yeast. Cell. 1994;79:245–258. doi: 10.1016/0092-8674(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Cole NB, Ellenberg J, Song J, DiEuliis D, Lippincott-Schwartz J. Retrograde transport of Golgi-localized proteins to the ER. J Cell Biol. 1998;140:1–15. doi: 10.1083/jcb.140.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MS, Cornell-Bell AH, Chernjavsky A, Dani JW, Smith SJ. Tubulovesicular processes emerge from Trans-Golgi cisternae, extend along microtubules, and interlink adjacent Trans-Golgi elements into a reticulum. Cell. 1990;61:135–145. doi: 10.1016/0092-8674(90)90221-y. [DOI] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway-retention at the misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90:523–535. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- Hay JC, Klumperman J, Oorshot V, Steegmaier M, Kuo CS, Scheller RH. Localization, dynamics, and protein interactions reveal distinct roles for ER and Golgi SNAREs. J Cell Biol. 1998;141:1489–1502. doi: 10.1083/jcb.141.7.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RC, Scheller RH. Structural organization of the synaptic exocytosis core complex. Neuron. 1997;19:1087–1094. doi: 10.1016/s0896-6273(00)80399-2. [DOI] [PubMed] [Google Scholar]
- Liou W, Geuze HJ, Slot JW. Improving structure of cryosections for immunogold labeling. Histochem Cell Biol. 1996;106:41–48. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- Loyet KM, Kowalchyk JA, Chaudhary A, Chen J, Prestwich GD, Martin TFJ. Specific binding of phosphatidylinositol 4,5-bisphosphate to calcium-dependent activator protein for secretion (CAPS), a potential phosphoinositide effector protein for regulated exocytosis. J Biol Chem. 1998;273:8337–8343. doi: 10.1074/jbc.273.14.8337. [DOI] [PubMed] [Google Scholar]
- Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Nichols BJ, Ungermann C, Pelham HR, Wickner WT, Haas A. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 1997;387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- Novick P, Zerial M. The diversity of Rab proteins in vesicle transport. Curr Opin Cell Biol. 1997;4:496–504. doi: 10.1016/s0955-0674(97)80025-7. [DOI] [PubMed] [Google Scholar]
- Pevsner J, Hsu S-C, Braun JEA, Calakos N, Ting AE, Bennett MK, Scheller RH. Specificity and regulation of a synaptic vesicle docking complex. Neuron. 1994;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR. Transport vesicle docking: SNAREs and associates. Annu Rev Cell Dev Biol. 1996;12:441–461. doi: 10.1146/annurev.cellbio.12.1.441. [DOI] [PubMed] [Google Scholar]
- Plutner H, Davidson HW, Saraste J, Balch WE. Morphological analysis of protein transport from the ER to Golgi membranes in digitonin-permeabilized cells: role of the P58 containing compartment. J Cell Biol. 1992;119:1097–1116. doi: 10.1083/jcb.119.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekeris R, Klumperman J, Chen YA, Scheller RH. Syntaxin 13 mediates cycling of plasma membrane proteins via recycling endosomes. J Cell Biol. 1998;143:957–971. doi: 10.1083/jcb.143.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Presley JF, Smith C, Hirschberg K, Miller C, Cole NB, Zaal KJM, Lippincott-Schwartz J. Golgi membrane dynamics. Mol Biol Cell. 1998;9:1617–1626. doi: 10.1091/mbc.9.7.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE, Warren G. Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr Biol. 1994;4:220–223. doi: 10.1016/s0960-9822(00)00051-8. [DOI] [PubMed] [Google Scholar]
- Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action of COPII and COPI. Cell. 1997;90:1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Sciaky N, Presley J, Smith C, Zaal KJM, Cole N, Moreira JE, Terasaki M, Siggia E, Lippincott-Schwartz J. Golgi tubule traffic and the effects of Brefeldin A visualized in living cells. J Cell Biol. 1997;139:1137–1155. doi: 10.1083/jcb.139.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of rat. J Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993a;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps in synaptic vesicle docking, activation, and fusion. Cell. 1993b;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Tang BL, Tan AE, Lim LK, Lee SS, Low DY, Hong W. Syntaxin 12, a member of the syntaxin family localized to the endosome. J Biol Chem. 1998;273:6944–6950. doi: 10.1074/jbc.273.12.6944. [DOI] [PubMed] [Google Scholar]
- Wang H, Frelin L, Pevsner J. Human syntaxin 7: a pep12p/Vsp6p homologue implicated in vesicle trafficking to lysosomes. Gene. 1997;199:39–48. doi: 10.1016/s0378-1119(97)00343-0. [DOI] [PubMed] [Google Scholar]
- Warren G, Levine T, Misteli T. Mitotic disassembly of the mammalian Golgi apparatus. Trends Cell Biol. 1995;5:413–416. doi: 10.1016/s0962-8924(00)89094-7. [DOI] [PubMed] [Google Scholar]