

Abstract
Mucopolysaccharidosis type I (MPS-I or Hurler syndrome) is an inherited deficiency of the lysosomal glycosaminoglycan (GAG)-degrading enzyme α-l-iduronidase (IDUA) in which GAG accumulation causes progressive multi-system dysfunction and death. Early allogeneic hematopoietic stem cell transplantation (HSCT) ameliorates clinical features and extends life but is not available to all patients, and inadequately corrects its most devastating features including mental retardation and skeletal deformities. To test novel therapies, we characterized an immunodeficient MPS-I mouse model less likely to develop immune reactions to transplanted human or gene-corrected cells or secreted IDUA. In the liver, spleen, heart, lung, kidney and brain of NOD/SCID/MPS-I mice IDUA was undetectable, and reduced to half in heterozygotes. MPS-I mice developed marked GAG accumulation (3-38 fold) in these organs. Neuropathological examination showed GM3 ganglioside accumulation in the striatum, cerebral peduncles, cerebellum and ventral brainstem of MPS-I mice. Urinary GAG excretion (6.5-fold higher in MPS-I mice) provided a non-invasive and reliable method suitable for serially following the biochemical efficacy of therapeutic interventions. We identified and validated using rigorous biostatistical methods, a highly reproducible method for evaluating sensorimotor function and motor skills development. This Rotarod test revealed marked abnormalities in sensorimotor integration involving the cerebellum, striatum, proprioceptive pathways, motor cortex, and in acquisition of motor coordination. NOD/SCID/MPS-I mice exhibit many of the clinical, skeletal, pathological and behavioral abnormalities of human MPS-I, and provide an extremely suitable animal model for assessing the systemic and neurological effects of human stem cell transplantation and gene therapeutic approaches, using the above techniques to measure efficacy.
Keywords: Stem cell, mice, mucopolysaccharidosis I, animal model, glycosaminoglycans, iduronidase, Rotarod performance test, ganglioside
Introduction
MPS-I (Hurler syndrome) is an inherited metabolic disorder due to lack of the lysosomal GAG-degrading enzyme α-l-iduronidase (IDUA) [25]. The resulting GAG accumulation causes multi-system dysfunction and death within the first decade. One of the most devastating manifestations of Hurler syndrome occurs in the central nervous system (CNS), resulting in progressive neuropsychological deterioration [21,37].
Novel approaches such as gene therapy and stem cell-based therapy hold great potential for development into clinically useful interventions. Pre-clinical testing of these modalities requires the availability of animal models that consistently and accurately represent the human disease. Several such animal models include the MPS-I mouse [5,27,29,38], domestic cat [13,14] and Plott hound dog [32,33,35], A major limitation of the above models is that they are not immunodeficient, and can develop immune reactions against transplanted human or gene-corrected cells and/or the secreted enzyme [6,10,17,20,24,31]. Furthermore, evaluation of novel CNS-based approaches will require a test of neurological function that is easy to perform serially and consistently measures neurological deficits relevant to the human disease.
The two currently available murine models of MPS-I [5,27,29,38] were generated by insertion of a neomycin resistance gene in exon 6 of the IDUA gene in the background of the C57BL/6 strain, and both are immunocompetent.
IDUAnull mice developed by Clarke et al [5,29] lack detectable tissue enzyme activity, exhibit a 2-5 fold increase in urinary GAG excretion, progressive lysosomal accumulation of GAGs in various organs including the brain and progressive skeletal histopathology and dysostosis with visible broadening of facial features and digits [5]. These mice demonstrate abnormal growth and shortened life span (average survival 48 weeks), progressive Purkinje cell loss in the cerebellum, and increased accumulation of GM2 and GM3 gangliosides in the brain [29]. Cardiovascular abnormalities include aortic root dilatation, aortic valve insufficiency, increased left ventricular size and decreased left ventricular function [3]. Neurological function has not been reported on for this model.
The IDUAnull mice developed by Neufeld et al [27,38] have a similar but not identical phenotype as the Clarke model. Cardiovascular abnormalities include enlargement of the heart with thickening of the septal and posterior walls and reduced left ventricular function, thickening and distortion of the valves with insufficiency of the aortic and mitral valves, electrocardiographic abnormalities, disturbed circadian rhythm and aortic dilatation [16,19]. Distinct craniofacial skeletal abnormalities have been identified by computed tomographic scanning [11]. These mice have reduced hearing and photoreceptor function and fewer cells in the outer nuclear photoreceptor layer of the retina [19,22]. Behavioral abnormalities detected include abnormal habituation in the open field test [12,28] and impairment of long-term (but not short-term) memory for inhibitory avoidance training, whereas novel object recognition memory and nociception are normal [28].
We have characterized an immunodeficient mouse model of MPS-I [the NOD.129(B6)-PrkdcscidIduatm1Clk mouse] that breeds well under the conditions described here and demonstrates neurological and systemic manifestations of the disease. Further, we have validated that the Rotarod test represents an easily performed and consistent measure of impaired sensorimotor function and development of motor coordination in this mouse model. The NOD/SCID/MPS-I mouse thus provides a suitable model for evaluating the potential benefits of human stem cell, gene or enzyme-based therapies.
Methods
Animals
All studies were approved by the University of Minnesota's Institutional Animal Care and Use Committee (IACUC). Breeder pairs of NOD.129(B6)-PrkdcscidIduatm1Clk mice heterozygous for the IDUA mutation were obtained from The Jackson Laboratory (Bar Harbor, ME), housed under specific-pathogen-free (SPF) conditions and provided with chow (TEKLAD # 2018), sterile/acidified water and sulfmethoxazole-trimethoprim antibiotics biweekly. A total of 24 breeder pairs were set during the characterization of the colony (different combinations of the same mice within the colony). On average, a WT × WT mating produced 9 offspring whereas a heterozygous × heterozygous mating produced an average of 7 offspring. Attempts to mate MPS-I females with heterozygous males were unsuccessful. The colony was therefore propagated by heterozygous × heterozygous mating. This produced approximately 25% homozygous NOD-SCID-MPS-I (IDUAnull: MPS-I) mice, 50% heterozygous (IDUAheterozygous: Het) carriers and 25% wild type NOD-SCID mice (IDUAnormal: WT). The average litter size was 7, with 53% female and 47% male offspring produced.
We initially observed that mothers often cannibalized their offspring during the first 1-4 days. Provision of a mix of peanut butter and oatmeal to the parents, and rubbing a small quantity of Vicks Vaporub® on the mothers' snouts and on the offspring were remarkably successful in reducing cannibalization.
Genotyping of mice
Tail clippings were digested overnight at 55°C in 200 μg/ml proteinase K in lysis buffer (1% SDS, 0.3M sodium acetate, 10 mM Tris and 1 mM EDTA). Samples were frozen at -80° C for at least 30 min, then clarified by centrifugation at 14,000 rpm at 4°C for 15 min. The supernatant was removed to a new tube, clarified again by centrifugation, and 2 μl was used for genotyping using PCR performed according to the protocol on The Jackson Laboratory website: (http://jaxmice.jax.org/pub-cgi/protocols/protocols.sh?objtype=protocol&objopr=query&protocol_id=466&no_rxns=20&button=Calculate). The following primers were used at a final conc. of 120 nM each:
Forward oIMR 1451: (5′ – GGAACTTTGAGACTTGGAATGAACCAG – 3′)
WT reverse oIMR 1452: (5′ – CATTGTAAATAGGGGTATCCTTGAACT – 3′)
MPS reverse oIMR 1453: (5′ – GGATTGGGAAGACAATAGCAGGCATGCT – 3′)
PCR products were resolved on a 1.5% agarose gel.
Photography of mice
Female and male mice at 6 months of age were sedated, placed on a black background and photographed using a 2.0 megapixel digital camera.
Skeletal radiographs
Eight month old mice were sedated and whole body radiographs obtained using a Faxitron (MNR 2000 Screen; exposure 7 sec/24 kV).
Rotarod test for sensorimotor and behavioral testing
Experimental method
A rotating rod apparatus (Model 8200 LSi Letica, PANLAB, Spain) was used to measure the ability of WT, heterozygous and MPS-I mice to remain balanced on a rotating rod (5 cm diameter) at different speeds. Mice used in this experiment were 20 weeks of age and naïve to this test. A total of 14 females (4 WT, 6 heterozygous and 4 MPS-I) and 21 males (6 WT, 9 heterozygous and 6 MPS-I) were tested. Mice were placed on the rod that was rotated at incremental speeds of 5, 15, 25 or 35 rpm 3 times daily for 5 consecutive days. Latency to fall (in secs) was scored for each animal at each speed in each trial. Animals rested for approximately 20 min between trials to prevent fatigue and exhaustion. This test requires a high degree of sensorimotor coordination and acquisition of motor skills [9,18]. The initial test thus served as a measure of baseline sensorimotor function, and impairment was indicative of damage to the basal ganglia and cerebellum. The ability to improve motor coordination was assessed by repeating the same test on 5 consecutive days to test for improvement in the balance time of individual mice.
Statistical considerations
We used a mixed model including both fixed and random effects to analyze the Rotarod data. The model that we fit estimates the effect of genotype, mouse gender, test number (i.e. time from baseline), and Rotarod rotation speed as fixed effects. We began by fitting a saturated model including the four-way interaction between all fixed effects. We used likelihood ratio tests to determine if the model could be simplified to a more parsimonious model with fewer fixed effects.
The experimental design resulted in many Rotarod duration measurements being recorded on each mouse. On each testing day, a mouse performed three trials at each of four different Rotarod RPMs. Given the repeated measurements made on each mouse, our statistical model accounts for correlation of measurements on the same mouse by using two sets of random effects. The first set of random effect is specific to each mouse and models the possibility that each mouse has an innate level of ability to run on the Rotarod that differs from other mice even after adjusting for the fixed effects in the model. This covariance matrix was also permitted to have a different level of variability for each genotype. The second random effect is specific to each mouse and test number pair. It models the possibility that measurements taken on the same day are likely to be more similar to one another than measurements taken on a different day and that testing days closer to one another may be more similar than testing days further apart in time. Due to uncertainty in how testing days might correlate with one another, we used an unstructured correlation matrix for the testing day random effects.
Mixed model parameters were estimated using the restricted maximum likelihood method as implemented in SAS 9.1. Type 3 F-tests with denominator degrees of freedom estimated using the Satterthwaite approximation were used to determine the overall significance of each fixed effect. We also tested for genotype effects within each combination of test day, gender, and Rotarod rotation speed. These tests were adjusted for multiple comparisons using the Tukey-Kramer method. All statistical tests were done at the 95% significance level.
Each combination of gender, genotype, rpm and test number (motor skills development) was modeled as having it's own distinct mean, without assumptions of additivity for any factor, since there was strong evidence of four-way interaction between the 4 items. The p-values were adjusted for multiple comparisons using the Tukey-Kramer method.
Urinary GAG excretion
Urine samples collected by placing the mice overnight in metabolic chambers (Metabolic Cage Model M-2100 from Lab Products Inc., Seaford, DE) was filtered and frozen at -80°C until analyzed. GAG concentration in the urine was determined by the 1,9-dimethylmethylene blue (DMMB; Sigma-Aldrich) method [4]. The dye was prepared by dissolving 16 mg DMMB in 5 ml of 95% ethanol and mixing it with a solution of formic acid (2 ml) and sodium formate (2 gm) in 1,000 ml distilled water. Serial dilutions of urine (25 μl) were made in 96 well plates, 25 μl of 0.48 M guanidine HCl and 200 μl of the DMMB dye were added to each well. The absorbance value was determined as: absorbance value at 550 nM minus the absorbance value at 650 nM. GAG concentration was calculated using a standard curve generated from chondroitin sulfate standards. Urinary creatinine concentration was determined using a picric acid based Creatinine Endpoint kit (Teco Diagnostics, Anaheim, CA). Urinary GAG excretion was expressed as μg GAG/mg creatinine in each urine sample.
Alpha-L-iduronidase (IDUA) activity assay
Sodium formate, formic acid, 4-methylumbelliferone, glycine, NaOH, Triton X-100 and sodium azide were obtained from Sigma (St. Louis, MO) and 4-methylumbelliferyl alpha-L-iduronide from Glycosynth (Warrington, Cheshire, UK).
After sacrifice using a ketamine/xylazine cocktail (10 μl/g), mice were perfused transcardially with 1x PBS. Samples from the brain, heart, kidney, liver, spleen and lungs were immediately harvested and flash-frozen for IDUA and GAG analysis. Harvested mouse tissues were placed in 1 ml PBS in an Eppendorf tube on ice and homogenized using a motorized pestle. Then 11 μl of 10% Triton X-100 in PBS was added and the homogenate kept on ice for 10 min. Protein concentration in the clarified supernatant was estimated by the Bradford colorimetric method [2].
IDUA activity was assayed essentially as described by Schuchman et al [30]. Briefly, 25 μl of a solution of 50 μM 4-methylumbelliferyl alpha-L-iduronide made in 0.4 M sodium formate buffer, pH 3.5, containing 0.2% Triton X-100 was added to 25 μl of tissue homogenate and incubated for 1 h at 37°C in the dark. The reaction was quenched by adding 200 μl of 0.5 M NaOH/glycine buffer, pH 10.3. Tubes were centrifuged for 1 min at 13,000 rpm at 4°C, the supernatant transferred to a 96 wells plate, and fluorescence read at 365 nm excitation wavelength and 450 nm emission wavelength using a Spectra Max Gemini XS fluorometric plate reader (Molecular Devices, Sunnyvale, CA).
IDUA activity in the tissue samples was calculated as: Activity in ng/h = (flourometric reading of the tissue sample × A) − B, where A and B were the values obtained from the curve fit equation of the standard curve generated using pure end product (4-methylumbelliferone). Specific activity of IDUA was expressed as nmol/h/mg protein in each sample.
Measurement of tissue GAGs
The remainder of the clarified supernatants of tissue homogenates were then processed to remove proteins using digestion with Proteinase K (equivalent to 3 times the amount of protein in the sample; incubation at 55°C for 24 h followed by boiling for 10 min to inactivate the enzyme), and nucleic acids by digestion with DNase + RNase (250 U DNase and 2.5 μg RNase; incubation at room temp. for 24 h followed by boiling for 10 min to inactivate the enzymes). All enzymes were from Sigma-Aldrich, St. Louis, MO. GAG concentration was then determined using the Blyscan Sulfated Glycosaminoglycan Assay (Accurate Chemical & Scientific Corp., Westbury, NY). Results were expressed as μg GAG/mg protein in each sample.
Statistical analysis of IDUA activity and tissue and urine GAG levels
Data were analyzed using GraphPad Prizm 4.0 software (GraphPad Software Inc., San Diego, CA). Results were expressed as mean +/- SE. The significance of differences between groups was examined using the non-parametric Mann-Whitney test to determine exact two-tailed P values.
Immunohistochemical assay for GM3 ganglioside accumulation in the brain
Mice were sacrificed and perfused with PBS and 4% paraformaldehyde (PFA). The brains were removed and fixed in 4% PFA for 24 h, then placed in 30% sucrose until the brains settled to the bottom of the vials. Sections (30 μM) were cut using a microtome, washed three times with PBS for 10 min each and blocked using 1% BSA with 1.5% normal goat serum and 0.05% Triton X-100 for 1 h at room temp. The sections were then incubated with primary antibody (Mouse anti-GM3, 1:100 dilution; Seikagaku Corporation Catalog # 370695) at 4°C for 24 h, washed with PBS and stained with the secondary antibody (biotinylated goat anti-mouse antibody; 1:200 dilution) for 1 h at room temp. Labeled cells were visualized by the avidin-biotin-peroxidase method (ABC Elite kit; Vector Laboratories, Burlingame, CA) with diaminobenzidine. Some sections were counterstained with neutral red or methyl green. Sections were examined by light microscopy.
Results
Genotyping
Determination of genotype was performed using DNA PCR. The WT allele produced a 550 bp band whereas the disrupted MPS-I allele produced a 350 bp band (Fig. 1). Genotype assignment of individual animals was confirmed by repeating the genotyping on tissue samples obtained at euthanasia, in order to exclude incorrect assignment due to sample labeling errors or technical problems with the PCR. No discrepancies were found between the initial and repeat genotyping (data not shown).
FIGURE 1. Genotyping by PCR.

The wild-type (WT) allele produced a 550 bp PCR product whereas the disrupted MPS-I allele produced a 350 bp PCR product. Heterozygous animals (Het) demonstrated both products. The last lane on the right shows a DNA size ladder.
Phenotypic features
Compared to WT mice, MPS-I animals had a broader head, more blunt nose, smaller ears, less prominent eyes and rough fur (Fig. 2A). These differences were more evident in female mice. The phenotypic features of heterozygous mice were intermediate between WT and MPS-I animals. Consistent with these facial features, plain radiographs showed a thickening of the zygomatic bone in MPS-I mice compared to WT or heterozygous mice (Fig. 2B).
FIGURE 2. Phenotypic features of wild type (WT), heterozygous and NOD/SCID/MPS-I (MPS) mice.

A. Facial features. M: males; F: females. MPS-I mice show coarsened fur and facial features, shortened snout, and broadened face resulting in lack of protrusion of eyes.
B. Radiograph of the skull: the arrow indicates the thickened zygomatic bone in MPS-I mice.
Sensorimotor and behavioral abnormalities
The Rotarod test demonstrated reproducible, highly significant abnormalities in the sensorimotor function and motor coordination learning capabilities of MPS-I mice (Fig. 3). The performance of heterozygous mice was comparable to that of WT mice. Because of the 4-way interaction between gender, genotype, speed and test number, each of the factors had an impact that varied with the other three factors, as described below. A likelihood ratio test found that the removal of the four-way interaction between genotype, mouse gender, test number, and Rotarod rotation speed results in a model that is statistically significantly different from the saturated model. We adopt the saturated model for all subsequent analyses. The statistical methodology does not account for the censoring in the data that occurred when mice were removed from the Rotarod if 120 secs was reached. Standard errors and means are therefore smaller than they might have been with uncensored data.
FIGURE 3. Sensorimotor and learning assessment by the RotaRod test.

Balance times on the Rotarod for male and female animals are shown separately, for the first (pre-training) and fifth (post-training) tests. Wild-type (WT): grey columns; heterozygous: hatched columns; MPS-I: black columns. The columns show the balance times at each speed (5, 15, 25 and 35 rpm) expressed as the mean +/- SE. Comparison between MPS-I and the other (WT or heterozygous) genotypes: *P<0.02; **P<0.001.
MPS-I mice of both genders had impairment of motor coordination, evidenced by lower balance times at higher speeds (≥15 rpm) when compared to WT or heterozygous mice on the initial (pre-training) tests. The MPS-I mice also demonstrated a relative inability to acquire motor coordination skills even after 5 training tests, evidenced by their lower balance times at higher speeds (≥25 rpm) on the post-training tests. This inability to improve motor coordination was more severe in males than in female MPS-I mice, evidenced by the lack of any improvement between the pre- and post-training balance time at 35 rpm in MPS-I males.
Effect of genotype
There were no differences between WT and heterozygous mice. Heterozygous and WT were both different from MPS-I mice and were able to remain on the Rotarod for longer periods of time than the MPS-I mice (P<0.0001). Moreover, these differences tended to be larger as the Rotarod speed increased. The WT/heterozygous differences from MPS began at lower speeds for males than females.
Effect of speed
Rotation speed had a highly significant effect on balance time on the Rotarod (P<0.0001). Comparing the effect of velocities at 5 rpm to each of the higher rotational speeds, only male MPS mice on their first test had significantly different latency times for falling from 5 rpm to 15 rpm, likely due to other mice already having achieved the full 120 secs at both 5 and 15 rpm. At 5 versus 25 rpm, males of all genotypes did significantly better at 5 rpm than 25 rpm for their first two tests. Also, both male and female MPS mice had significantly better 5 versus 25 rpm times. At 5 versus 35 rpm, males of all genotypes had better 5 rpm times. The effect was more pronounced in MPS mice where females had significantly better 5 rpm times as well.
Effect of test number (learning of motor coordination skills)
A highly significant effect of improvement over the course of the 5 consecutive tests was seen on balance times (P<0.0001). Comparing test 5 to test 1, the effect was significant in WT and heterozygous males at 25 and 35 rpm, and in MPS-I males at 15 and 25 rpm. At 35 rpm, MPS-I males appeared unable to improve over the course of 5 tests. When first tested at 15-35 rpm, MPS-I males were able to balance on the Rotarod for only half as long as WT or heterozygous males. By the 5th test, WT and heterozygous males were able to remain balanced on the Rotarod for longer at the higher speeds. The performance of MPS-I males also improved but never became equal to that of WT or heterozygous males.
Motor coordination improvement in female MPS-I mice was most pronounced at 25 and 35 rpm. When first tested at 35 rpm, female MPS-I mice were able to balance on the Rotarod for only 1/3rd as long as WT or heterozygous mice. Balance time at lower speeds was not impaired. By the 5th test, both WT and heterozygous females were able to remain balanced on the Rotarod for the maximum time tested (120 secs) even at the highest speed (35 rpm). The performance of MPS-I females also improved by the 5th test, but never became equal to that of WT or heterozygous females.
In some cases, the lack of improved Rotarod time over multiple tests was explained by the mice having no room for improvement (already at maximum of 120 sec).
Effect of gender
Gender also had a significant effect on balance times (P <0.0005). The performance of MPS-I males was more severely impaired than that of MPS-I females, evidenced by shorter balance times at 15 rpm. Furthermore, females tended to have longer Rotarod balance times than males in situations that required the most improvement in motor coordination (at the highest speed of 35 rpm) in the WT and heterozygous genotypes. Unlike MPS-I females, MPS-I males were unable to improve sufficiently to remain balanced longer at the highest speed (35 rpm) even after 5 tests. There was a trend towards a gender effect in MPS-I mice at all speeds above 5 rpm (again situations that require improvement of motor coordination since all MPS-I mice were impaired).
Urinary GAG excretion
GAG excretion was measured in the urine of WT, heterozygous and MPS-I mice. There was no increase in urinary GAG excretion by heterozygous mice (Fig. 4). In contrast, urinary GAG excretion by MPS-I mice was 6.5-fold higher than by WT or heterozygous mice.
FIGURE 4. Urinary excretion of GAGs.

Total GAGs and creatinine were measured in a 24 h urine collection, to calculate the relative GAG excretion in μg GAG/mg creatinine. Urine samples were collected from 8 animals of each genotype. Data is shown as the mean +/- SE. Comparison between MPS-I and WT or heterozygous mice: P <0.0002. Excretion of GAGs by heterozygous mice was equivalent to that by WT mice, whereas excretion of GAGs by MPS-I mice was consistently higher.
Tissue IDUA activity
IDUA activity was estimated in the brain, heart, liver, kidneys, spleen and lungs of WT, heterozygous and MPS-I mice each. In the WT mice, IDUA activity varied between different organs. The highest activity was seen in the spleen and lowest activity in the brain (Fig. 5). Heterogeneity in the IDUA activity in the liver, kidney, lung and spleen of WT mice was observed. Compared to WT mice, enzyme activity was reduced by 40-60% in all the organs of heterozygous mice, as expected; these differences were statistically significant for all organs. IDUA activity was completely undetectable in all the organs of MPS-I mice.
FIGURE 5. Tissue IDUA enzyme activity.

Samples of each of the indicated tissues were obtained from 10 animals of each genotype, except 9 animals each for MPS-I liver and spleen. Data is shown as the mean +/- SE. WT: grey columns; heterozygous: hatched columns; MPS: *no detectable activity. Comparison between MPS-I and WT or heterozygous mice: P <0.0001 for all tissues. Comparison between WT and heterozygous mice: brain: P <0.0001; heart: P <0.001; liver: P <0.005; kidney: P <0.009; lung: P <0.04 and spleen: P <0.002. IDUA activity was undetectable in all tissues from MPS-I mice, and was about ½ of WT in all examined tissues of heterozygous mice.
GAG accumulation in tissues
GAG concentration was measured in the brain, heart, liver, kidneys, spleen and lungs of WT, heterozygous and MPS-I mice. Marked accumulation of GAGs was seen in all tissues of MPS-I mice, being highest in the liver and lowest in the brain (Fig. 6). In MPS-I mice, the respective GAG content of different tissues was 3 to 38-fold higher than in WT or heterozygous mice; all differences were statistically significant.
FIGURE 6. Tissue accumulation of GAGs.

Total GAGs and protein were measured in each tissue sample, to calculate the GAG content in μg GAG/mg protein. Samples of each tissue were obtained from 8-13 animals of each genotype. Data is shown as the mean +/- SE. WT: grey columns; heterozygous: hatched columns; MPS: black columns. Comparison between MPS-I and WT or heterozygous mice: P <0.0003 for all tissues except MPS-I brain vs WT brain: P <0.008. The GAG content of different tissues from WT mice was about 5-15 μg/mg protein. In contrast, there were marked differences in the GAG content of different tissues from MPS-I mice, being lowest in the brain (3-fold higher than WT) and highest in the liver (38-fold higher than WT).
Ganglioside accumulation in the brain
Sections of the cerebral cortex, cerebellum, ventral brainstem, hippocampus and striatum of WT, heterozygous and MPS-I mice were stained for the presence of GM3 gangliosides. Detectable accumulation of GM3 gangliosides was seen in only rare cells in the brains of WT and heterozygous mice (Fig. 7). In contrast, considerable secondary accumulation of GM3 gangliosides was seen in several regions of the brains of MPS-I mice. This was most marked in the brainstem and striatum.
FIGURE 7. GM3 ganglioside accumulation in the brain.
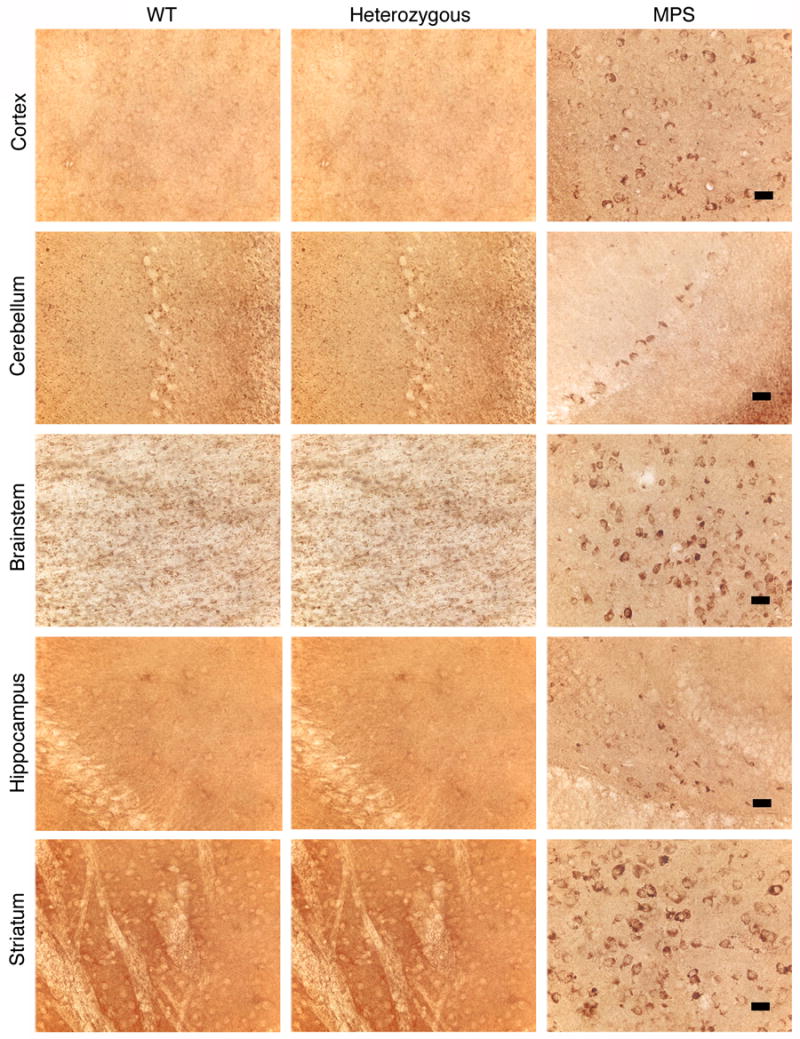
Immunohistochemical staining showing accumulation of GM3 gangliosides (brown staining cells) in all the examined regions of the brains of MPS-I mice, most notably in the brainstem and striatum. There was no detectable accumulation in the brains of WT or heterozygous mice. Magnification: 400x; bar: 25 μM.
Discussion
We demonstrate here that NOD/SCID/MPS-I mice exhibit the phenotypic, biochemical and neuropathological features that are representative of Hurler syndrome. In this model, IDUA activity levels in all tissues tested faithfully reflected the genotype, being reduced by about 50% in heterozygotes and being completely undetectable in MPS-I animals. IDUA enzyme activity in the livers of WT, heterozygous and MPS-I mice of the Clarke model [5,29] and in the liver, spleen, kidney and brain of WT and MPS-I mice of the Neufeld model [38] was similar to IDUA activity in corresponding tissues in the current model. Clarke et al [5,29] did not provide data on IDUA activity in the brain and kidney, and enzyme activity was not reported for the spleen, lungs or heart.
The modified method of measuring GAGs used here appears to be able to demonstrate increased levels of GAGs in the brains of NOD/SCID/MPS-I mice. Modifications included (a) not using a GAG precipitation step, in order to preclude the possibility of small un-precipitatable oligosaccharides in MPS-I tissues being lost in the supernatant, and (b) enzymatically digesting proteins and nucleic acids which may interfere with the GAG assay. The 3-fold difference in GAG concentration between WT and MPS-I brains (7.6 +/- 1.5 vs 21.8 +/- 6.5 μg GAG/mg protein, respectively) although smaller than in other organs, was nevertheless statistically significant. This is an important aspect of the method used, since it will allow direct assessment of biochemical response in the brain to cellular, gene-based or enzyme therapies. Overall, tissue GAG levels in the Neufeld model [19,22] were lower than in the current model. Nevertheless, as in the Neufeld model [38], the liver was the organ with the highest GAG content in NOD/SCID/MPS-I mice. Further investigations will be required to determine if differences between GAG levels in the various mouse models may be due to interference in the GAG assay by residual proteins and/or nucleic acids. No quantitative data was provided on tissue GAG accumulation in the Clarke model [5,29].
Urinary GAG excretion was 6.5-fold higher in NOD/SCID/MPS-I mice compared to WT animals, as seen in other mouse models [5]. However, the mean levels of GAG excretion by both WT and NOD/SCID/MPS-I mice were lower than those reported for the Clarke model [5]. We additionally show that GAG excretion by heterozygous animals is completely equivalent to that of WT animals. As there was no overlap between the values seen in NOD/SCID/MPS-I mice vs those in WT or heterozygous animals, and because urinary GAG excretion can be repeatedly measured in live animals over their life-span, it represents an extremely useful method of assessing the systemic biochemical response of the animal over time, to therapeutic interventions.
Thickening of the zygomatic arch of the skull is a feature common to all three MPS-I mouse models. However, the severity and widespread extent of skeletal abnormalities in the Clarke model [5,29] appears to be greater than that observed by us in the current model (data not shown).
One advantage of the current mouse model is that it is relatively easy to breed, although this may be at least partly a consequence of the possibly more favorable conditions in SPF rooms compared to rooms where immunocompetent mice (such as the Clarke and Neufeld models) are housed. Using the techniques described in Methods, we have bred approximately 300 animals over a 3 yr period. Robust breeding in this model makes a large number of MPS-I progeny and their WT and heterozygous littermates available for study.
The Clarke model of MPS-I demonstrates progressive accumulation of PAS+ material in the cerebellum and caudate nucleus and progressive Purkinje cell loss from the cerebellum [29]. However, baseline motor coordination, the ability to improve motor coordination skills, and differences in neurological functioning between males and females have not been reported for either the Clarke or Neufeld MPS-I mouse models. Our studies show that the Rotarod test is an easy, sensitive and consistent assay for evaluating sensorimotor function and improvement of motor coordination skills in NOD/SCID/MPS-I mice. These mice showed defective sensorimotor integration involving the cerebellum, striatal and proprioceptive pathways and cerebral cortex, and impaired acquisition of motor coordination. These abnormalities correlated with the location of neuropathological changes, demonstrated by GM3 accumulation, in the cerebellum and striatum. Impaired performance of MPS-I mice on the initial Rotarod test may in part be due to skeletal abnormalities, corneal or retinal abnormalities impairing visual cues or vestibular dysfunction. However, impairment on the Rotarod test due to these factors would not change on serial testing. The observed improvement in performance therefore indicates that this test does serve as a measure of neurological function. The serial Rotarod test therefore will be able to evaluate the functioning of relevant areas of the brain in MPS-I mice.
The NOD/SCID mouse is the immunodeficient mouse that has been used most commonly and extensively for testing engraftment, survival, migration and differentiation of various types of human and murine stem cells [1,7,8,15,23,36]. This mouse lacks functional B and T cells and complement activity and has impaired macrophage and antigen presenting cell functions [34]. Although NK cell activity is markedly attenuated in NOD/SCID mice [34], they still possess the capability to mount an NK-cell based rejection of transplanted cells. This can be prevented to a large extent by peri-transplant administration of an anti-mouse CD122 (IL-2 and IL-15 receptor β chain) blocking antibody [23].
The phenomenon of variable degrees of “leakiness” (tendency to produce some functional B and/or T cells) is common to diverse mutations in cell lines and animal models, and increases with age and housing in non-SPF conditions. The frequency of leakiness is considerably lower in NOD/SCID mice compared to SCID mice bred on other backgrounds [26]. In NOD/SCID mice, immunoglobulin levels >1 μg/ml have been found in only 3/41 (7%) mice [34]. T cell leakiness is also infrequent, evidenced by retention of MHC class-I disparate skin allografts by all NOD/SCID animals transplanted [34]. The NOD/SCID/MPS-I mouse is therefore not likely to develop humoral or T cell immune responses against implanted allogeneic or xenogenic cells or secreted IDUA enzyme, although this remains to be determined in prospective studies.
In conclusion, this immunodeficient mouse will likely be an excellent model for assessing the neurological as well as systemic effects of human stem cell transplantation and gene therapy. We have started using this model to assess the effect of intra-cerebro-ventricular implantation of various types of human stem cells on the biochemical, pathological and behavioral abnormalities in this mouse.
Acknowledgments
The authors thank Dr. Elizabeth F. Neufeld, Professor and Chair, Department of Biological Chemistry, University of California Los Angeles, Los Angeles, CA, for help and advice with the biochemical assays used in this work. This work was supported by NIH R01-NS-48606 (P.G. and W.C.L.), the US Department of Veterans Affairs (P.G.) and the Childrens Cancer Research Fund (P.G.).
Footnotes
Conflict of Interest: There is no relevant conflict of interest for any of the authors of this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bhatia M, Bonnett D, Murdoch B, Gan OI, Dick JE. A newly discovered class of human hematopoietic cells with SCID-repopulating activity. Nat Med. 1998;4:1038–45. doi: 10.1038/2023. [DOI] [PubMed] [Google Scholar]
- 2.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 3.Braunlin E, Mackey-Bojack S, Panoskaltsis-Mortari A, Berry JM, McElmurry RT, Riddle M, Sun LY, Clarke LA, Tolar J, Blazar BR. Cardiac functional and histopathologic findings in humans and mice with mucopolysaccharidosis type I: implications for assessment of therapeutic interventions in hurler syndrome. Pediatr Res. 2006;59:27–32. doi: 10.1203/01.pdr.0000190579.24054.39. [DOI] [PubMed] [Google Scholar]
- 4.Chandrasekhar S, Esterman MA, Hoffman HA. Microdetermination of proteoglycans and glycosaminglycans in the presence of guanidine hydrochloride. Anal Biochem. 1987;161:103–8. doi: 10.1016/0003-2697(87)90658-0. [DOI] [PubMed] [Google Scholar]
- 5.Clarke LA, Russell CS, Pownall S, Warrington CL, Borowski A, Dimmick JE, Toone J, Jirik FR. Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum Mol Genet. 1997;6:503–11. doi: 10.1093/hmg/6.4.503. [DOI] [PubMed] [Google Scholar]
- 6.Di Domenico C, Villani GR, Di Napoli D, Reyero EG, Lombardo A, Naldini L, Di Natale P. Gene Therapy for a mucopolysaccharidosis type I murine model with lentiviral-IDUA vector. Hum Gene Ther. 2005;16:81–90. doi: 10.1089/hum.2005.16.81. [DOI] [PubMed] [Google Scholar]
- 7.Dick JE, Bhatia M, Gan OI, Kapp U, Wang JC. Assay of human stem cells by repopulation of NOD/SCID mice. Stem Cells. 1997;15:199–203. doi: 10.1002/stem.5530150826. [DOI] [PubMed] [Google Scholar]
- 8.Dick JE, Guenechea G, Gan OI, Dorrell C. In vivo dynamics of human stem cell repopulation in NOD/SCID mice. Ann N Y Acad Sci. 2001;938:184–90. doi: 10.1111/j.1749-6632.2001.tb03588.x. [DOI] [PubMed] [Google Scholar]
- 9.Fujimoto ST, Longhi L, Saatman KE, Conte V, Stocchetti N, McIntosh TK. Motor and cognitive function evaluation following experimental traumatic brain injury. Neurosci Biobehav Rev. 2004;28:365–78. doi: 10.1016/j.neubiorev.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Glaros EN, Turner CT, Parkinson EJ, Hopwood JJ, Brooks DA. Immune response to enzyme replacement therapy: single epitope control of antigen distribution from circulation. Mol Genet Metab. 2002;77:127–35. doi: 10.1016/s1096-7192(02)00148-8. [DOI] [PubMed] [Google Scholar]
- 11.Graupman P, Pan D, Konair B, Hartung S, McIvor S, Whitley C, Low W, Lam CH. Craniofacial abnormalities in a murine knock-out model of mucopolysaccharidosis I H: a computed tomography and anatomic study. J Craniofac Surg. 2004;15:392–8. doi: 10.1097/00001665-200405000-00009. [DOI] [PubMed] [Google Scholar]
- 12.Hartung SD, Frandsen JL, Pan D, Koniar BL, Graupman P, Gunther R, Low WC, Whitley CB, McIvor RS. Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human alpha-L-iduronidase gene. Mol Ther. 2004;9:866–75. doi: 10.1016/j.ymthe.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 13.Haskins ME, Aguirre GD, Jezyk PF, Desnick RJ, Patterson DF. The pathology of the feline model of mucopolysaccharidosis I. Am J Pathol. 1983;112:27–36. [PMC free article] [PubMed] [Google Scholar]
- 14.Haskins ME, Jezyk PF, Desnick RJ, McDonough SK, Patterson DF. Alpha-L-iduronidase deficiency in a cat: a model of mucopolysaccharidosis I. Pediatr Res. 1979;13:1294–7. doi: 10.1203/00006450-197911000-00018. [DOI] [PubMed] [Google Scholar]
- 15.Hogan CJ, Shpall EJ, McNulty O, McNiece I, Dick JE, Shultz LD, Keller G. Engraftment and development of human CD34+ enriched cells from umbilicl cord blood in NOD/LtSz-scid/scid mice. Blood. 1997;90:85–96. [PubMed] [Google Scholar]
- 16.Jordan MC, Zheng Y, Ryazantsev S, Rozengurt N, Roos KP, Neufeld EF. Cardiac manifestations in the mouse model of mucopolysaccharidosis I. Mol Genet Metab. 2005;86:233–43. doi: 10.1016/j.ymgme.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kakkis ED, Schuchman E, He X, Wan Q, Kania S, Wiemelt S, Hasson CW, O'Malley T, Weil MA, Aguirre GA, Brown DE, Haskins ME. Enzyme replacement therapy in feline mucopolysaccharidosis I. Mol Genet Metab. 2001;72:199–208. doi: 10.1006/mgme.2000.3140. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Chen J, Wang L, Zhang L, Lu M, Chopp M. Intracerebral transplantation of bone marrow stromal cells in a 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Neurosci Lett. 2001;316:67–70. doi: 10.1016/s0304-3940(01)02384-9. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Xu L, Hennig AK, Kovacs A, Fu A, Chung S, Lee D, Wang B, Herati RS, Ogilvie JM, Cai SR, Ponder KP. Liver-directed neonatal gene therapy prevents cardiac, bone, ear, and eye disease in mucopolysaccharidosis I mice. Mol Ther. 2005;11:35–47. doi: 10.1016/j.ymthe.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 20.Lutzko C, Kruth S, Abrams-Ogg AC, Lau K, Li L, Clark BR, Ruedy C, Nanji S, Foster R, Kohn D, Shull R, Dube ID. Genetically corrected autologous stem cells engraft, but host immune responses limit their utility in canine alpha-L-iduronidase deficiency. Blood. 1999;93:1895–905. [PubMed] [Google Scholar]
- 21.Lyon G, Adams RD, Kolodny EH. Neurology of hereditary metabolic diseases of children. McGraw Hill; New York: 1996. Late infantile progressive genetic encephalopathies; pp. 124–76. [Google Scholar]
- 22.Ma X, Liu Y, Tittiger M, Hennig A, Kovacs A, Popelka S, Wang B, Herati R, Bigg M, Ponder KP. Improvements in mucopolysaccharidosis I mice after adult retroviral vector-mediated gene therapy with immunomodulation. Mol Ther. 2007;15:889–902. doi: 10.1038/sj.mt.6300112. [DOI] [PubMed] [Google Scholar]
- 23.McKenzie JL, Gan OI, Doedens M, Dick JE. Human short-term repopulating stem cells are efficiently detected following intrafemoral transplant into NOD/SCID recipients depleted of CD122+ cells. Blood. 2005;106:1259–61. doi: 10.1182/blood-2005-03-1081. [DOI] [PubMed] [Google Scholar]
- 24.Meertens L, Zhao Y, Rosic-Kablar S, Li L, Chan K, Dobson H, Gartley C, Lutzko C, Hopwood JJ, Kohn DB, Kruth S, Hough MR, Dube ID. In utero injection of alpha-L-iduronidase carrying retrovirus in canine mucopolysaccharidosis type I: infection of multiple tissues and neonatal gene expression. Hum Gene Ther. 2002;13:1809–20. doi: 10.1089/104303402760372918. [DOI] [PubMed] [Google Scholar]
- 25.Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular basis of Inherited Disease. III. McGraw-Hill; New York, NY: 2001. pp. 3421–52. [Google Scholar]
- 26.Nonoyama S, Smith FO, Bernstein ID, Ochs HD. Strain-dependent leakiness of mice with severe combined immune deficiency. J Immunol. 1993;150:3817–24. [PubMed] [Google Scholar]
- 27.Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci USA. 2003;100:1902–07. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reolon GK, Braga LM, Camassola M, Luft T, Henriques JA, Nardi NB, Roesler R. Long-term memory for aversive training is impaired in Idua(-/-) mice, a genetic model of mucopolysaccharidosis type I. Brain Res. 2006;1076:225–30. doi: 10.1016/j.brainres.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 29.Russell C, Hendson G, Jevon G, Matlock T, Yu J, Aklujkar M, Ng KY, Clarke LA. Murine MPS I: insights into the pathogenesis of Hurler syndrome. Clin Genet. 1998;53:349–61. doi: 10.1111/j.1399-0004.1998.tb02745.x. [DOI] [PubMed] [Google Scholar]
- 30.Schuchman EH, Guzman NA, Desnick RJ. Human alpha-L-iduronidase. I. Purification and properties of the high uptake (higher molecular weight) and the low uptake (processed) forms. J Biol Chem. 1984;259:3132–40. [PubMed] [Google Scholar]
- 31.Shull R, Lu X, Dube I, Lutzko C, Kruth S, Abrams-Ogg A, Kiem HP, Goehle S, Schuening F, Millan C, Carter R. Humoral immune response limits gene therapy in canine MPS I. Blood. 1996;88:377–9. [PubMed] [Google Scholar]
- 32.Shull RM, Helman RG, Spellacy E, Constantopoulos G, Munger RJ, Neufeld EF. Morphologic and biochemical studies of canine mucopolysaccharidosis I. Am J Pathol. 1984;114:487–95. [PMC free article] [PubMed] [Google Scholar]
- 33.Shull RM, Munger RJ, Spellacy E, Hall CW, Constantopoulos G, Neufeld EF. Canine alpha-L-iduronidase deficiency. A model of mucopolysaccharidosis I. Am J Pathol. 1982;109:244–8. [PMC free article] [PubMed] [Google Scholar]
- 34.Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, McKenna S, Mobraaten L, Rajan TV, Greiner DL, Leiter EH. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. 1995;154:180–91. [PubMed] [Google Scholar]
- 35.Spellacy E, Shull RM, Constantopoulos G, Neufeld EF. A canine model of human alpha-L-iduronidase deficiency. Proc Natl Acad Sci U S A. 1983;80:6091–5. doi: 10.1073/pnas.80.19.6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang JC, Lapidot T, Cashman JD, Doedens M, Addy L, Sutherland DR, Nayar R, Laraya P, Minden M, Keating A, Eaves AC, Eaves CJ, Dick JE. High level engraftment of NOD/SCID mice by primitive normal and leukemic hematopoietic cells from patients with chronic myeloid leukemia in chronic phase. Blood. 1998;91:2406–14. [PubMed] [Google Scholar]
- 37.Whitley CB. The mucopolysaccharidoses. In: Moser HW, editor. Neurodystrophies and Neurolipidoses. 22. Vol. 66. Elsevier Science; 1996. pp. 281–328. [Google Scholar]
- 38.Zheng Y, Rozengurt N, Ryazantsev S, Kohn DB, Satake N, Neufeld EF. Treatment of the mouse model of mucopolysaccharidosis I with retrovirally transduced bone marrow. Mol Genet Metab. 2003;79:233–44. doi: 10.1016/s1096-7192(03)00116-1. [DOI] [PubMed] [Google Scholar]