

Abstract
Acinar-to-ductal metaplasia in the pancreas is associated with an increased risk for tumorigenesis. Molecular dissection of this process in vitro has shown that primary acinar cells, in response to EGF receptor ligands, can transdifferentiate into duct-like epithelia, passing through a nestin-positive intermediate, in a Notch pathway-dependent manner. Here, we show that in vitro acinar transdifferentiation depends on matrix metalloproteinase 7 (MMP-7), a proteinase expressed in most metaplastic epithelia in vivo. MMP-7 was found to be required for Notch activation, which leads to dedifferentiation of acinar cells to the nestin-positive transitional cell. Besides being necessary for acinar transdifferentiation, it was found that MMP-7 activity was sufficient to induce the process, indicating that molecular signals capable of initiating MMP-7 expression also have the potential to induce formation of metaplastic epithelia in the pancreas.
Keywords: metaplasia, nestin, pancreatic cancer, matrilysin
Pancreatic ductal adenocarcinoma (PDA) is one of the most fatal human cancers. Pathological evidence strongly suggests that PDA arises from premalignant lesions known as pancreatic intraepithelial neoplasia (PanIN) (1). Although there is evidence that PanINs arise from a preexisting stem cell population (2), a second possible preneoplastic lesion is the metaplastic duct lesion (MDL), which is found predominantly associated with PDA as well as with chronic pancreatitis (CP), a risk factor for PDA (3). MDLs appear to arise from acinar-to-ductal metaplasia (ADM), in which acinar cells are progressively replaced by duct-like epithelia that are hypothesized to possess progenitor cell-like properties (4).
The matrix metalloproteinases (MMPs) comprise a family of extracellular, zinc-dependent proteinases frequently expressed in cancer (5). MMPs can collectively degrade all components of the extracellular matrix (ECM), suggesting a role in tumor invasion. However, evidence derived from mouse genetics has shown that MMPs contribute to multiple stages of tumor progression, including tumorigenesis and tumor growth, often by cleaving non-ECM substrates (6).
The MMP family member MMP-7 is expressed by the tumor cells of many adenomas, adenocarcinomas, and by associated metaplasia, including in colon (7), stomach (7), and the majority of PDAs, at all PanIN stages, and in 100% of tumor-associated MDLs (8). Animal models have supported a role for MMP-7 in tumorigenesis and early tumor growth. In ApcMin mice, MMP-7 deficiency inhibits intestinal tumor formation (9). Conversely, overexpression of MMP-7 in the mammary gland in the MMTV-Neu transgenic background accelerates tumor formation (10). Together, these data suggest that MMP-7 acts early in adenoma progression, perhaps in tumorigenesis itself. Although its role in PDA has not been directly addressed, in a mouse model of CP, all aspects of disease progression are inhibited in MMP-7-deficient (MMP-7−/−) mice, including MDL formation (8).
Many MMP-7 substrates have been linked to aspects of PDA progression, including syndecan-1 (11, 12), E-cadherin (13, 14), and TNF-α (15, 16). In mouse CP, FasL was determined to be a relevant in vivo MMP-7 substrate that controls acinar cell apoptosis (8). However, these data do not provide a satisfactory explanation as to why MDL formation is inhibited in MMP-7−/− mice. Indeed, given the inhibition of all aspects of CP progression in this model, including inflammation, fibrosis, apoptosis, and metaplasia, it was impossible to determine whether the processes are independent of one another.
ADM can be simulated in vitro by treating primary acinar cell explants embedded in three-dimensional fibrillar collagen with TGF-α (17). In this system, acinar cell differentiation is progressively lost with a reciprocal increase in duct cell differentiation, as determined by expression of cell-specific molecular markers. Recent studies have shown that this in vitro ADM represents a true cellular transdifferentiation, in which cells pass through a nestin-positive intermediate (18), and requires activation of the Notch receptor family (19).
Typically, Notch receptors (Notch 1–4) are activated upon interaction with one of their ligands (Jagged or Delta-like) on an adjacent cell (for review, see 20). Upon ligation, the extracellular portion of Notch is cleaved by a metalloproteinase followed by intramembrane cleavage by a presenilin-dependent γ-secretase. The newly liberated Notch intracellular domain (Notch-ICD) translocates to the nucleus where it interacts with the DNA-binding protein CSL [CBF-1/RBP-Jκ, Su(H), Lag-1], converting it from transcriptional repressor to transcriptional activator. Notch-ICD–CSL complexes activate transcription of genes such as Hairy Enhancer of Split (hes1), which function to block cellular differentiation. Notch blocks pancreatic acinar cell differentiation (21, 22) through the inactivation of PTF1, a transcription factor complex required for acinar differentiation, by Notch-ICD displacing Ptf1a from CSL (23), as well as by HES1 directly interacting with and blocking Ptf1a function (24).
Here, we have used the acinar cell explant model of ADM to test the direct contribution of MMP-7 to acinar transdifferentiation. We have determined that MMP-7 was expressed in response to TGF-α and that primary acinar cells derived from MMP-7−/− pancreata did not convert to duct cells. Experiments to determine the hierarchy of MMP-7 among other molecules that contribute to acinar transdifferentiation supported a model in which MMP-7 cleaves and activates Notch to induce the transition to the nestin-positive intermediate and thus for ADM.
Results
MMP-7 Is Necessary for Acinar Transdifferentiation.
We have demonstrated that MMP-7 is necessary for most aspects of CP progression in vivo, including formation of MDLs (8). However, the complexity of the in vivo model makes it difficult to determine how MMP-7 affects MDL formation. To test the hypothesis that MMP-7 directly controls ADM, we used a simplified in vitro system in which pancreatic tissue explants enriched in acinar, centroacinar, and associated terminal duct epithelium, and depleted of stromal and islet cells, are embedded in a three-dimensional collagen matrix and treated with TGF-α. The ensuing transdifferentiation, which passes through a nestin-positive intermediate (18), concludes with the appearance of a cytokeratin-19 (CK-19)-positive ductal phenotype within 5 days in culture (19).
To test the appropriateness of this system for assessing MMP-7 function in acinar transdifferentiation, we first determined whether MMP-7 was expressed under these culture conditions (Fig. 1). Acinar explants were prepared from pancreata of wild-type or MMP-7−/− mice, and the explants were embedded in collagen and treated with TGF-α under low-serum conditions. Duplicate cultures were fixed on consecutive days, and MMP-7 expression was determined by immunofluorescence, with MMP-7−/− explants as a negative control [supporting information (SI) Fig. 9]. At day 0, MMP-7 was not detected, with strong expression found in 5.2% of acinar clusters on day 1, increasing to 54.5% on day 2, and to 69.7% on day 3 (Fig. 1). Expression of MMP-7 in these cultures was confirmed by semiquantitative RT-PCR (see SI Fig. 9).
Fig. 1.

MMP-7 is expressed in primary acinar cells treated with TGF-α. Pancreatic explants from wild-type mice embedded in collagen were treated with TGF-α. MMP-7 expression (green) was confirmed by immunofluorescence 1 (A), 2 (B), and 3 days (C) after culture, respectively. Staining was evident in the majority of acini on day 2. (Scale bar, 50 μm.) Cells were costained for amylase (red) and DAPI (blue). Data are representative of five independent experiments.
We then tested whether MMP-7 was necessary for acinar transdifferentiation. Wild-type and MMP-7−/− cultures were treated as before, fixed on days 0, 3, and 5, and examined for the acinar and ductal markers amylase and CK-19, respectively, by coimmunofluorescence (Fig. 2). By day 5, wild-type explants developed an amylase-negative, CK-19-positive ductal phenotype in ≈80% of cell clusters (Fig. 2 A–C). In contrast, acinar cells from MMP-7−/− mice maintained a predominantly acinar phenotype in 85% of cell clusters, confirmed by the persistence of amylase and the absence of CK-19 expression throughout the culture period (Fig. 2 D–F). To confirm that the block in transdifferentiation in MMP-7−/− cultures was the result of the immediate lack of MMP-7, we added active recombinant MMP-7 (rMMP-7) to the culture medium, which restored CK-19 positivity to 68% of MMP-7−/− cell clusters (Fig. 2 G–I). We conclude that in vitro acinar transdifferentiation is highly dependent on MMP-7.
Fig. 2.

MMP-7 is required for acinar-to-ductal transdifferentiation. Primary acinar cells from wild-type (A–C) and MMP-7−/− (D–I) mice were embedded in collagen and treated with TGF-α. Coimmunofluorescence for the acinar cell marker amylase (red) and the duct cell marker cytokeratin-19 (green) was performed on cells fixed on day 0 (A, D, and G), day 3 (B, E, and H), and day 5 (C, F, and I). TGF-α treatment of wild-type cultures induced acinar-to-ductal transdifferentiation by day 5 (C). TGF-α treatment of MMP-7−/− acinar cells showed virtually no conversion to ductal structures (D–F). Addition of 200 ng/ml rMMP-7 with TGF-α to MMP-7−/− acinar cells (G–I) restored transdifferentiation. (Scale bar, 100 μm.) DAPI is shown in blue. Data are representative of >10 independent experiments.
MMP-7 and Notch Are Required for Transition to the Nestin-Positive Intermediate.
The Notch pathway has been shown to be responsible for maintaining stem and progenitor cell populations and blocks pancreatic acinar differentiation (22). In acinar transdifferentiation, acquisition of the ductal phenotype is controlled by the Notch pathway (19). To investigate whether Notch and MMP-7 functionally interact in this system, we set out to define where both act during the process. Initially, we examined the effects of γ-secretase inhibition, which blocks intracellular Notch processing, on the transition to the nestin-positive intermediate. In wild-type acinar cells, nestin expression was discernable by immunofluorescence by day 3 (Fig. 3A), as reported (18). In the presence of the γ-secretase inhibitor (GSI), WPE III-31C, nestin expression was not detected at day 3 (Fig. 3B) nor at any point in the 5 days of culture (data not shown). Because the GSI can block the intramembrane cleavage of many proteins, we tested whether activating the Notch pathway could rescue nestin expression in the presence of the GSI. Indeed, infection with an adenovirus encoding the intracellular domain of Notch-1 (Ad-N1ICD), restored nestin positivity in the GSI-treated cultures (Fig. 3C), indicating that Notch is the relevant γ-secretase substrate in the assay. In parallel experiments, we found that MMP-7−/− acinar cells failed to initiate nestin expression at day 3 (Fig. 3D) or at any point during the 5-day culture period (data not shown). Also, GSI treatment had no effect on MMP-7 expression itself (see SI Fig. 10). Thus, activity of both Notch and MMP-7 was required for transition to the nestin-positive intermediate, and MMP-7 did not appear to be a downstream target of Notch.
Fig. 3.

Notch and MMP-7 are required for transition to the nestin-positive intermediate. Nestin immunofluorescence (green) was detectable on day 3 in wild-type cultures treated with DMSO (A), but not in cultures treated with 20 μM the γ-secretase inhibitor, WPE III-31C (GSI) (B). Infection with an adenovirus encoding a constitutively active V5-tagged Notch-1 intracellular domain (Ad-N1ICD-V5) bypassed the GSI, allowing nestin expression (C). Day 3 MMP-7−/− cultures did not show nestin immunoreactivity (D). (Scale bar, 50 μm.) Shown are amylase in red and DAPI in blue. Data are representative of at least three independent experiments.
Notch-ICD Bypasses the Requirement for MMP-7.
Because Notch and MMP-7 appeared to be working in the same part of the transdifferentiation process, we set out to test whether they acted as components of the same pathway or independent essential pathways. To distinguish between these possibilities, we infected wild-type and MMP-7−/− acinar explants with Ad-N1ICD-V5 to induce transdifferentiation, instead of TGF-α, with a GFP-encoding adenovirus (Ad-GFP) as a negative control (Fig. 4). GFP and V5 immunofluorescence were used to confirm infection efficiency, consistently ≈90%. At 3 days after infection, cells were fixed and analyzed for CK-19 expression to test for acinar transdifferentiation. GFP controls showed negligible conversion to a ductal phenotype (Fig. 4 C and I), whereas Ad-N1ICD-V5-infected cultures showed conversion of a majority of cells, approximately equivalent to the infection efficiency, in both wild-type and MMP-7−/− cultures (Fig. 4 F and L). Thus, Notch activity bypassed the requirement for MMP-7, suggesting that Notch acts downstream of MMP-7 in a common pathway.
Fig. 4.

Constitutively active Notch bypasses the requirement for MMP-7 in acinar transdifferentiation. Primary acinar cells from wild-type (A–F) or MMP-7−/− mice (G–L) were infected either with Ad-GFP, an adenovirus encoding GFP (A–C and G–I) or Ad-N1ICD-V5 (D–F and J–L) and embedded in collagen. After 3 days in culture, transdifferentiation in N1ICD cultures was evident (D and J) and was coincident with successful infection, confirmed by V5 immunofluorescence (E and K). Transdifferentiation was confirmed by immunofluorescence for CK-19 (F and L). (Scale bar, 50 μm.) DAPI is shown in blue. Data are representative of three independent experiments.
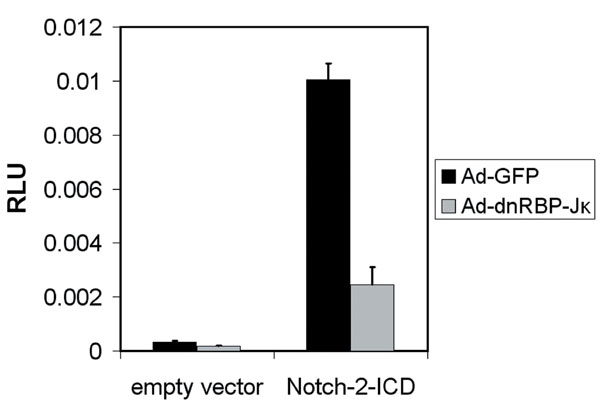
The ability of Notch to induce transdifferentiation in MMP-7−/− acinar cells suggested that MMP-7 acts upstream of Notch. To address this possibility directly, we tested whether MMP-7 was sufficient to induce Notch-dependent transdifferentiation. We performed the acinar transdifferentiation assay with wild-type acinar explants, replacing TGF-α with rMMP-7. Coimmuno fluorescence for amylase and CK-19 showed that by day 5, MMP-7 induced the ductal phenotype (Fig. 5A). This process was blocked by GSI (Fig. 5B) as well as by infection with an adenovirus encoding a dominant-negative form of RBP-Jκ (25) (Fig. 5 C and D), which blocked CSL-dependent reporter activity (see SI Fig. 11). Together, these data confirm that MMP-7 acts upstream of Notch to induce transdifferentiation.
Fig. 5.

MMP-7 activity is sufficient to induce Notch-dependent acinar-to-ductal transdifferentiation. (A and B) Primary acinar cells from wild-type mice were treated with 200 ng/ml rMMP-7 along with DMSO (A) or 20 μM WPE III-31C (GSI) (B). Coimmunofluorescence for amylase (red) and CK-19 (green) was performed on day 5. (C and D) Primary acinar cells infected with an adenovirus encoding a dominant-negative RBP-Jκ also blocked transdifferentiation induced by coinfection with Ad-N1ICD-V5 (amylase in red, V5 in green) (C) or by rMMP-7 (amylase in red, CK-19 in green) (D). (Scale bar, 50 μm.) DAPI is shown in blue. Data are representative of at least three independent experiments.
MMP-7 Activates Notch.
The most obvious model that fits the previous data is one where MMP-7 cleaves the extracellular domain of Notch, inducing subsequent γ-secretase intramembrane cleavage to form active Notch. To examine this possibility in detail, we moved to a system more amenable to biochemical analysis but with minimal background protease activity. We transfected COS-7 cells with a construct encoding full-length Notch-1 with a C-terminal V5 tag (Notch-1-V5) and treated them with molar equivalent amounts of recombinant pro-MMP-7 or active MMP-7. To analyze Notch processing by immunoblot, we used an antibody (Cleaved N1 Val-1744) that is specific for the neoepitope of Notch-1 exposed after γ-secretase cleavage. Notch cleavage was induced only by active MMP-7 (Fig. 6A). As a control for antibody specificity, cells were treated with WPE III-31C along with active rMMP-7 (Fig. 6A), which eliminated the appearance of the neoepitope. In a time course, we found that γ-secretase-cleaved Notch-1 was evident 2 h after the addition of rMMP-7 (Fig. 6B). Titration of rMMP-7 showed that as little as 2 ng/ml enzyme induced γ-secretase processing of Notch-1, with optimal cleavage occurring with 20 ng/ml rMMP-7 (Fig. 6C). For independent confirmation of MMP-7-induced Notch processing, we performed immunoblots for the C-terminal V5 tag (Fig. 6D), which revealed a size shift in the overexpressed protein consistent with P2 and P3 site processing (26).
Fig. 6.

MMP-7 activity induces γ-secretase cleavage of Notch-1. (A–C) Immunoblots for the γ-secretase-cleaved form of Notch-1 (Cleaved N1 Val-1744). (A) COS-7 cells expressing Notch-1 were treated with 100 ng/ml pro-MMP-7, 50 ng/ml active rMMP-7 with DMSO, or 20 μM WPE III-31C. (B) COS-7 cells expressing Notch-1 were treated with 200 ng/ml rMMP-7 for 0, 2, 4, 6, and 18 h. (C) COS-7 cells expressing Notch-1 were treated with varied rMMP-7 concentrations for 4 h. (D) COS-7 cells expressing Notch-1 with C-terminal V5 were treated with 200 ng/ml for 18 h. Data are representative of five independent experiments.
Continuing along the Notch pathway, we examined nuclear translocation of the Notch-ICD. COS-7 cells transfected with the full-length Notch-1-V5 expression vector were treated with medium alone or with medium containing active rMMP-7. After 4 h of treatment, cells were fixed and analyzed by coimmunofluorescence (Fig. 7 A and B). V5 reactivity indicated transfected cells, and Cleaved N1 Val-1744 reactivity indicated Notch activation. With MMP-7 treatment, 34% of expressing cells exhibited cleaved nuclear Notch-1 compared with 10% of cells without MMP-7 (Fig. 7C).
Fig. 7.

MMP-7 activity leads to nuclear translocation of N1ICD and expression of hes-1. (A and B) COS-7 cells expressing full-length Notch-1 with a C-terminal V5 tag were treated for 4 h with MMP-7. Shown is immunofluorescence for Cleaved N1 Val-1744 antibody (green) and V5 antibody (red). (Scale bar, 20 μm.) Cells were treated for 4 h with medium alone (A) or with rMMP-7 (B). Arrows indicate cells with nuclear cleaved Notch-1. DAPI is shown in blue. (C) Quantification of Notch-1 nuclear translocation. Percentage represents cells with nuclear Notch-1 divided by total cells expressing Notch-1. (D) RT-PCR for hes1 from COS-7 cells expressing Notch-1 treated with MMP-7 or EDTA. Numbers represent fold expression relative to medium alone. Data are representative of five independent experiments.
Hes1, a Notch target gene, is often analyzed as an indication of Notch activity (19). Using semiquantitative RT-PCR, we assayed for hes1 transcript after MMP-7 treatment. COS-7 cells were transfected with Notch-1-V5 and treated with medium alone or medium containing active rMMP-7 for 4 h. EDTA-induced Notch activation (27) was used as a positive control. Relative to medium alone, hes1 was up-regulated 4.4-fold in response to rMMP-7 and 6.2-fold in response to EDTA (Fig. 7D). Collectively, these data indicate that MMP-7 activity can lead to Notch-1 γ-secretase cleavage, nuclear translocation, and target gene expression.
MMP-7 Activates Notch in Acinar Cell Explants.
With the observation that MMP-7 is necessary and sufficient to induce Notch-dependent acinar transdifferentiation and can induce Notch processing in COS-7 cells, we wanted to confirm that MMP-7 was required to activate endogenous Notch in acinar cells. First, we tested whether Notch-1 was expressed in wild-type and MMP-7−/− acinar cells by immunofluorescence and RT-PCR. We found that both wild-type and MMP-7−/− acinar explants expressed Notch-1 protein and transcript at day 0 (see SI Fig. 12). Thus, neither TGF-α nor MMP-7 was required for Notch-1 expression. We then used Cleaved N1 Val-1744 immunofluorescence to determine the status of Notch-1 activation in these cells. Wild-type acinar cells exhibited cleaved Notch-1 by day 3 in the majority of clusters, whereas MMP-7−/− acinar cells did not show immunoreactivity at day 3 (Fig. 8 A and B), or at any time during the assay. Cleaved Notch-1 immunofluorescence could be restored by including rMMP-7 in the cultures (Fig. 8C). As a final confirmation of MMP-7-dependent Notch activation, we tested for the expression of Notch target genes hes1 and hey1 in the explants. We found that MMP-7−/− explants expressed 2.1- and 2.3-fold lower hes1 and hey1, respectively, and that expression was restored when the cultures were supplemented with rMMP-7 (Fig. 8D). We conclude that MMP-7 is necessary for appropriate Notch processing and activation in primary acinar cells.
Fig. 8.

MMP-7 is required for Notch activation in acinar explants. (A–C) Acinar explants immunostained for Cleaved N1 Val-1744 (green) and amylase (red) on day 3. Wild-type acinar cells exhibited Notch cleavage (A), whereas MMP-7−/− cells did not (B). Inclusion of 200 ng/ml rMMP-7 rescued Notch cleavage in MMP-7−/− acinar cells (C). (Scale bar, 50 μm.) DAPI is shown in blue. (D) RT-PCR for hes1 and hey1 showed increased Notch activation in wild-type acinar cells and MMP-7−/− cells treated with rMMP-7, relative to MMP-7−/− cells. Data are representative of five independent experiments.
Discussion
Acinar-to-ductal metaplasia is strongly associated with pancreatic tumorigenesis (28, 29). Previous studies have shown that metaplasia in vivo is marked by expression of Notch target genes and, in vitro, Notch signaling is both necessary and sufficient for acinar-to-ductal transdifferentiation (19). Our previous in vivo studies have linked MMP-7 to the formation of MDLs in a duct obstruction model of CP (8). In the current work, we have shown that MMP-7 is both necessary and sufficient for activation of Notch signaling in acinar cells, which leads to dedifferentiation to a nestin-positive intermediate that precedes acquisition of a ductal phenotype. Consistent with this result, we find that nestin expression and Notch cleavage temporally coincide (see SI Fig. 13).
In this in vitro system, MMP-7 appears to function as a key mediator between TGF-α and Notch activation. MMP-7 expression requires TGF-α but not Notch activity. Notch-1 expression is constitutive, irrespective of TGF-α or MMP-7, but its cleavage by γ-secretase requires MMP-7. Dominant-active Notch-1 or Notch-2 can restore transdifferentiation in MMP-7−/− acinar cells, and MMP-7 can process both Notch-1 and Notch-2 (see SI Fig. 14) to generate the active ICD domain that can travel to the nucleus and initiate target gene transcription.
The dependence of Notch activation on MMP-7 in this system is surprising. During development, Notch signaling typically works by Notch binding to its ligand on an adjacent cell, which induces the ligand-binding domain to be transendocytosed into the ligand-expressing cell (20). This process exposes the transmembrane domain to cleavage by ADAM proteases (26, 30). Interestingly, ligand-dependent proteinases are clearly present in COS-7 cells because overexpression of Jagged-1 induced robust activation of a CSL reporter construct in the absence of rMMP-7 (data not shown). Also, using a Notch-2 construct tagged on N and C termini, we found that the ligand-binding domain was not transendocytosed but released into the medium in the presence of rMMP-7 (data not shown). Finally, rMMP-7 can directly cleave peptides containing the P2 cleavage site of the extracellular domain of Notch-1 (data not shown). We hypothesize that overexpressed, secreted MMP-7, unlike membrane-bound ADAMs, can access the P2 cleavage site without ligand, albeit inefficiently, to cleave Notch. It is also possible that MMP-7 may act indirectly by activating ADAMs; however, ADAM activation has been reported to occur intracellularly by a furin-like enzyme (31), making this possibility unlikely.
Despite the finding that Notch activation requires MMP-7 in this system, our data do not contradict what has been shown. We propose that Notch signaling in development is controlled by ligand-dependent cleavage by ADAMs. Consistent with this proposal, MMP-7−/− mice show no developmental defects attributable to impaired Notch activity, whereas ADAM-null mice do (32). However, we postulate that in disease, Notch processing can follow an alternate activation mechanism induced by an abundance of MMP-7 and perhaps other proteases that are hyperexpressed in a given pathological condition. In this context, it is interesting that another in vivo disease model has been shown to depend on both Notch and MMP-7: intestinal tumor formation in ApcMin mice. Blocking Notch activity with a GSI inhibits tumor progression in this system (33), whereas adenoma formation is suppressed in a MMP-7−/− background (9).
In conclusion, data from the current and previous studies (19) have supported a model in which Notch signaling is required for acinar cells to dedifferentiate and convert to MDLs. Recently, it has been shown that K-ras, the most commonly mutated oncogene in PDA, requires tissue damage to induce PDA when expression of the mutant K-ras is confined to adult acinar cells (34), lending support to the hypothesis that MDLs can act as preneoplastic lesions. Thus, Notch inhibition may prove useful in preventing pancreatic disease progression of both CP and PDA. Previous studies would suggest that ADAM-specific inhibitors would be one way to prevent Notch activation, but our data suggest that broad-spectrum MMP inhibitors may be required to block this pathway fully in pancreatic disease.
Materials and Methods
Mice.
The C57BL/6J MMP-7−/− mice were a gift from Lynn Matrisian (Vanderbilt University, Nashville, TN). Control C57BL/6J animals were purchased from The Jackson Laboratory. Animals were bred and maintained in a maximum isolation facility in the Division of Laboratory Animal Resources at Stony Brook University. All protocols herein were approved by the IACUC at Stony Brook University.
Primary Acinar Cell Cultures.
Primary acinar cell cultures were prepared by modifying published protocols (18). Transdifferentiation events were induced by addition of 50 ng/ml recombinant TGF-α (Chemicon International) or 200 ng/ml active rMMP-7 (Calbiochem) to medium. Where indicated, acinar cells were treated with 20 μM WPE III-31C (Calbiochem) in dimethyl sulfoxide. Cultures were maintained in a 37°C and 5% CO2 incubator for 5 days with medium replaced on days 1 and 3 (for details, see SI Materials and Methods).
Immunofluorescence.
Antibodies used for immunofluorescence analysis are listed in SI Materials and Methods. Immunofluorescent labeling of pancreatic explants was performed as described in ref. 18 (for details, see SI Materials and Methods).
Adenoviral Infections.
Before embedding explants in collagen, 1 ml of cell suspension was placed in 35-mm bacterial Petri dishes. Adenoviruses encoding GFP, Notch-1-ICD, Notch-2-ICD, or DN-RBP-Jκ [gifts from Lucy Liaw (Maine Medical Research Institute, Scarborough, ME) and Charles Eberhart (The Johns Hopkins University, Baltimore)] were added to the cell suspension at a moi of 20:1. Cell/virus mixtures were rocked every 15 min for 1 h at 37°C and remained in suspension overnight. Suspensions were mixed with collagen in medium without growth factors.
Notch Cleavage.
COS-7 cells were cultured in DMEM + 10% FBS at 37°C in a humidified 95% air/5% CO2 incubator. Cells (4 × 105) were transfected with 5 μg of pCDNA4V5-Notch-1 (gift from Deena Small, University of New Hampshire, Durham, NH), with Lipofectamine 2000 (Invitrogen). Forty-eight hours later, the medium was replaced with OptiMEM (Invitrogen), containing 100 ng/ml pro-MMP-7 (Calbiochem), 0–200 ng/ml active rMMP-7 for 2–18 h, or 20 μM WPE III-31C, as indicated. For EDTA activation, cells were washed in PBS and incubated in PBS + 10 mM EDTA for 15 min at 37°C. Cells recovered in DMEM for 4 h. For Western blotting, cells were washed in ice-cold PBS and lysed in ice-cold RIPA buffer (1% Nonidet P-40, 1% sodium deoxycholic acid, 0.1% SDS, 150 mM NaCl, 1 mM NaHPO4, 0.2 mM EDTA, plus protease inhibitors). Twenty micrograms of lysate was resolved on a 5% SDS/polyacrylamide gel, transferred to nitrocellulose, blocked in 5% milk/TBST, and probed with primary antibody overnight at 4°C, followed by secondary antibody, and visualized by chemiluminescence.
RT-PCR Analysis.
Total RNA was extracted by using the RNeasy mini kit (Qiagen). cDNA was synthesized by using SuperScript II reverse transcriptase (Invitrogen), and PCR was carried out for targets with 25, 30, and 35 cycles to confirm linear range (for primer sequences, see SI Materials and Methods). The signal was normalized by amplification of GAPDH for 22, 28, and 32 cycles.
Supplementary Material
Acknowledgments
We thank D. Small, C. Eberhart, and L. Liaw for expression vectors. Thanks also to L. Liaw, R. Gallagher, L. Bombardelli, and Y. Song for critiquing the manuscript. This work was supported by National Institutes of Health Grant R01CA100126 (to H.C.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705953104/DC1.
References
- 1.Klein WM, Hruban RH, Klein-Szanto AJ, Wilentz R. Mod Pathol. 2002;15:441–447. doi: 10.1038/modpathol.3880544. [DOI] [PubMed] [Google Scholar]
- 2.Carriere C, Seeley ES, Goetze T, Longnecker DS, Korc M. Proc Natl Acad Sci USA. 2007;104:4437–4442. doi: 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowenfels AB, Maisonneuve P. Best Pract Res Clin Gastroenterol. 2006;20:197–209. doi: 10.1016/j.bpg.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Song SY, Gannon M, Washington MK, Scoggins CR, Meszoely IM, Goldenring JR, Marino CR, Sandgren EP, Coffey RJ, Jr, Wright CV, et al. Gastroenterology. 1999;117:1416–1426. doi: 10.1016/s0016-5085(99)70292-1. [DOI] [PubMed] [Google Scholar]
- 5.Chambers AF, Matrisian LM. J Natl Cancer Inst. 1997;89:1260–1270. doi: 10.1093/jnci/89.17.1260. [DOI] [PubMed] [Google Scholar]
- 6.Matrisian LM, Wright J, Newell K, Witty J. Princess Takamatsu Symp. 1994;24:152–161. [PubMed] [Google Scholar]
- 7.McDonnell S, Navre M, Coffey RJ, Jr, Matrisian LM. Mol Carcinog. 1991;4:527–533. doi: 10.1002/mc.2940040617. [DOI] [PubMed] [Google Scholar]
- 8.Crawford HC, Scoggins CR, Washington MK, Matrisian LM, Leach SD. J Clin Invest. 2002;109:1437–1444. doi: 10.1172/JCI15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson CL, Heppner KJ, Labosky PA, Hogan BL, Matrisian LM. Proc Natl Acad Sci USA. 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rudolph-Owen LA, Chan R, Muller WJ, Matrisian LM. Cancer Res. 1998;58:5500–5506. [PubMed] [Google Scholar]
- 11.Li Q, Park PW, Wilson CL, Parks WC. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 12.Conejo JR, Kleeff J, Koliopanos A, Matsuda K, Zhu ZW, Goecke H, Bicheng N, Zimmermann A, Korc M, Friess H, et al. Int J Cancer. 2000;88:12–20. doi: 10.1002/1097-0215(20001001)88:1<12::aid-ijc3>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 13.McGuire JK, Li Q, Parks WC. Am J Pathol. 2003;162:1831–1843. doi: 10.1016/S0002-9440(10)64318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Aynati MM, Radulovich N, Riddell RH, Tsao MS. Clin Cancer Res. 2004;10:1235–1240. doi: 10.1158/1078-0432.ccr-03-0087. [DOI] [PubMed] [Google Scholar]
- 15.Haro H, Crawford HC, Fingleton B, Shinomiya K, Spengler DM, Matrisian LM. J Clin Invest. 2000;105:143–150. doi: 10.1172/JCI7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karayiannakis AJ, Syrigos KN, Polychronidis A, Pitiakoudis M, Bounovas A, Simopoulos K. Anticancer Res. 2001;21:1355–1358. [PubMed] [Google Scholar]
- 17.De Lisle RC, Logsdon CD. Eur J Cell Biol. 1990;51:64–75. [PubMed] [Google Scholar]
- 18.Means AL, Meszoely IM, Suzuki K, Miyamoto Y, Rustgi AK, Coffey RJ, Jr, Wright CV, Stoffers DA, Leach SD. Development (Cambridge, UK) 2005;132:3767–3776. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, Sriuranpong V, Iso T, Meszoely IM, Wolfe MS, et al. Cancer Cell. 2003;3:565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 20.Nichols JT, Miyamoto A, Olsen SL, D'Souza B, Yao C, Weinmaster G. J Cell Biol. 2007;176:445–458. doi: 10.1083/jcb.200609014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Proc Natl Acad Sci USA. 2003;100:14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esni F, Ghosh B, Biankin AV, Lin JW, Albert MA, Yu X, MacDonald RJ, Civin CI, Real FX, Pack MA, et al. Development (Cambridge, UK) 2004;131:4213–4224. doi: 10.1242/dev.01280. [DOI] [PubMed] [Google Scholar]
- 23.Beres TM, Masui T, Swift GH, Shi L, Henke RM, MacDonald RJ. Mol Cell Biol. 2006;26:117–130. doi: 10.1128/MCB.26.1.117-130.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh B, Leach SD. Biochem J. 2006;393:679–685. doi: 10.1042/BJ20051063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato H, Taniguchi Y, Kurooka H, Minoguchi S, Sakai T, Nomura-Okazaki S, Tamura K, Honjo T. Development (Cambridge, UK) 1997;124:4133–4141. doi: 10.1242/dev.124.20.4133. [DOI] [PubMed] [Google Scholar]
- 26.Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A. Mol Cell. 2000;5:207–216. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 27.Rand MD, Grimm LM, Artavanis-Tsakonas S, Patriub V, Blacklow SC, Sklar J, Aster J. Mol Cell Biol. 2000;20:1825–1835. doi: 10.1128/mcb.20.5.1825-1835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsa I, Longnecker DS, Scarpelli DG, Pour P, Reddy JK, Lefkowitz M. Cancer Res. 1985;45:1285–1290. [PubMed] [Google Scholar]
- 29.Wagner M, Luhrs H, Kloppel G, Adler G, Schmid RM. Gastroenterology. 1998;115:1254–1262. doi: 10.1016/s0016-5085(98)70098-8. [DOI] [PubMed] [Google Scholar]
- 30.Pan D, Rubin GM. Cell. 1997;90:271–280. doi: 10.1016/s0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- 31.Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, et al. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- 32.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lübke T, Lena Illert A, von Figura K, et al. Hum Mol Genet. 2002;11:2615–2624. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- 33.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, et al. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 34.Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}