

Abstract
The regulation of dietary cholesterol absorption was examined in C57BL/6 and transgenic mice with liver overexpression of the scavenger receptor BI (SR-BI Tg). In C57BL/6 animals, feeding 0.02 to 1% (wt/wt) dietary cholesterol resulted in a dose-dependent decrease in the percentage of dietary cholesterol absorbed. A plot of total daily mass of dietary cholesterol absorbed versus the percentage by weight of cholesterol in the diet yielded a curve suggesting a saturable process with a Km of 0.4% (wt/wt) and a Vmax of 0.65 mg cholesterol/g body weight per day. Dietary cholesterol suppressed hepatic 3-hydroxy-3-methylglutaryl CoA reductase activity, stimulated cholesterol 7α-hydroxylase activity, and enhanced fecal excretion of bile acids, but none of these changes correlated with the percentage of dietary cholesterol absorption. Dietary cholesterol also caused an increase in biliary cholesterol concentration, and in this case the concentration of biliary cholesterol was strongly and inversely correlated with the percentage dietary cholesterol absorption (r = −0.63, P < 0.0001). Biliary cholesterol concentration was also directly correlated with daily cholesterol intake, dietary cholesterol mass absorption, and liver cholesterol ester content. Transgene-induced overexpression of SR-BI resulted in a stimulation of excretion of cholesterol into the bile and suppressed percentage dietary cholesterol absorption. Furthermore, biliary cholesterol levels in SR-BI Tg mice were strongly and inversely correlated with the percentage of dietary cholesterol absorbed (r = −0.99, P < 0.0008). In summary, these results suggest that the excretion of cholesterol into the bile plays an important role in regulating the percentage absorption of dietary cholesterol.
Numerous epidemiological studies have indicated that diets high in cholesterol and saturated fats are associated with increased levels of low density lipoprotein (LDL) cholesterol and increased susceptibility to coronary heart disease (CHD) (1). These studies provide the basis for recommending a low-cholesterol, low-saturated-fat diet for the prevention of CHD (2). Clinical studies in humans indicate independent effects of dietary cholesterol and saturated fats on lipoprotein levels and, therefore, presumably on CHD susceptibility (3, 4). Although in humans dietary saturated fat appears to play a more major role quantitatively than dietary cholesterol in the regulation of lipoprotein levels, in some animal models the effects of dietary cholesterol predominate over dietary saturated fat (5) and there is also evidence for dietary cholesterol– saturated fat interaction (6, 7). In animal studies it also has been shown that raising dietary cholesterol alone can increase atherosclerosis susceptibility (5). Thus, it is clear that dietary cholesterol is important and the study of the pathways through which it exerts its metabolic effects is crucial to understanding how dietary cholesterol influences CHD susceptibility.
The absorption of dietary cholesterol from the intestine is the first step that allows it to exert its metabolic effects, yet the regulation of this process is only partially understood. Dietary cholesterol, which is mostly unesterified, is presented in micellar form to the intestinal mucosa, where it is passes through the brush border, is partially esterified by intestinal acyl-CoA:cholesterol acyltransferase, packaged into chylomicrons, and then, after triglyceride hydrolysis, chylomicron remnants are delivered to hepatic or extrahepatic tissues through remnant receptors. Studies in mice (8, 9), monkeys (10), and humans (11) have shown that feeding increasing amounts of dietary cholesterol results in a decrease in the percentage of dietary cholesterol absorbed; however, the mechanisms responsible have not been determined. In one clinical study the dietary cholesterol-induced decrease in percentage of dietary cholesterol absorption was attributed to increased cholesterol saturation of the intestinal mucosa (11, 12), but this interpretation of the data was challenged by subsequent studies from the same group (13). It has also been suggested that dietary cholesterol absorption is mediated by an intestinally derived brush border protein, but the protein has not been isolated and the theory remains unproven (14). In other experiments it was suggested that dietary cholesterol absorption is mediated by carboxyl ester lipase, a pancreas-derived protein that can attach to the intestinal brush border and was proposed to serve as a cholesterol transporter into the epithelial cell (15), or by intestinal acyl-CoA:cholesterol acyltransferase, the epithelial cell cholesterol-esterifying enzyme. Both of these theories have been challenged by finding apparently normal cholesterol absorption in knockout mice for the respective genes (16, 17). Thus, based on studies to date, the mechanism of regulation of dietary cholesterol absorption remains unresolved.
In the present study we examined the mechanism of regulation of dietary cholesterol absorption by using C57BL/6 and liver expressing scavenger receptor BI (SR-BI) transgenic mice. SR-BI is a receptor shown to be involved in selective uptake of high density lipoprotein (HDL) cholesterol esters, and adenoviral expression of SR-BI was demonstrated to increase biliary cholesterol levels (18). In C57BL/6 mice we found that increasing amounts of dietary cholesterol decreased the percentage absorption of dietary cholesterol and increased the concentration of biliary cholesterol, and that these two events were strongly inversely correlated. In liver expressing SR-BI transgenic mice we found increased biliary cholesterol levels, decreased percentage absorption of dietary cholesterol, and a strong inverse relationship between biliary cholesterol levels and the percentage dietary cholesterol absorption. Taken together these results strongly suggest that biliary cholesterol excretion plays an important role in regulating the efficiency of dietary cholesterol absorption. This may occur by saturation of intestinal micelles with biliary cholesterol preventing proper presentation of dietary cholesterol to the intestinal mucosa for absorption.
METHODS
Animals and Diets.
C57BL/6 males were obtained from The Jackson Laboratory. SR-BI Tg animals were generated by cloning of 1.5 kb of the murine SR-BI cDNA fragment (19) into the HpaI site on pLVI-7 plasmid vector (20). A linearized fragment of the construct containing apolipoprotein E (apoE) promoter, first exon, first intron and part of the second exon of human apoE gene, murine SR-BI cDNA, poly(A)+ signal sequence, and the hepatic control region of the apoE/CI gene locus was isolated and purified after restriction digestion. The linear DNA fragment was injected into the male pronucleus of fertilized eggs from superovulated (C57BL/6 × CBA/J) F1 females that had been mated to males of the same genetic background. Founder animals were back-crossed two generations with C57BL/6 mice, and all the SR-BI Tg experiments were done with littermate controls. A manuscript describing the creation and characterization of the SR-BI Tg mice has been submitted for publication (N.W., Takeshi Arai, Yong Ji, Franz Rinninger, and A.R.T., unpublished data).
Animals were housed in a humidity- and temperature-controlled room with 12-h dark/12-h light cycle at the Laboratory Animal Research Center at The Rockefeller University. Unless otherwise indicated animals were fed for 3 weeks with Picolab (Bouncbrook, NJ) Rodent Chow 20 (5053) pellet containing 0.02–1% (wt/wt) cholesterol. The enrichment with cholesterol was achieved by dissolving crystallized cholesterol in ethyl ether, which was then mixed with chow diet (0.02% cholesterol) and served to animals in a pellet form.
Cholesterol Absorption Measurements.
Cholesterol absorption was determined in C57BL/6 and SR-BI Tg animals ages 10–13 weeks by using a modified form of the “dual isotope single-meal feeding” method (9, 21). Specifically, animals were placed for 24 h in metabolic cages, fasted for 4–6 h, and then given an intragastric bolus of 100 μl corn oil containing 1.67 μCi of [14C]cholesterol (DuPont) and 0.67 μCi of [3H]β-sitostanol (American Radiolabeled Chemicals, St. Louis). The animals were returned to metabolic cages, food intake was monitored, and feces were collected for 24 h unless otherwise specified. Collected feces were dried by overnight incubation at 55°C and homogenized, and a sample of 150 mg was extracted with 4 ml of chloroform/methanol (2:1, vol/vol). A 2-ml aliquot of the chloroform/methanol phase was thoroughly vortexed with 0.7 ml of 0.88% KCl, and the radioactive sterols in the feces were determined in 0.1 ml of the organic phase after evaporation to dryness and scintillation counting. Counting efficiency was calculated by the channel ratio method based on external standards. The efficiency of cholesterol absorption was expressed as percentage of administered dose absorbed by using the formula:
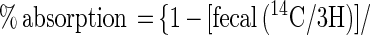 |
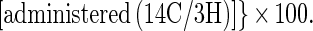 |
Measurements of Dietary Cholesterol Intake and Cholesterol Mass Absorption.
Food intake was measured by weighing the food before and 24 h after placing the animals in metabolic cages. Dietary cholesterol intake was calculated by multiplying the food intake by the percentage of cholesterol in the diet and expressed as mg cholesterol/g body weight per day. The mass of dietary cholesterol absorbed daily from the intestine was calculated by multiplying the value for dietary cholesterol intake by percentage of dietary cholesterol absorption and expressed as mg cholesterol/g body weight per day.
Bile Aspiration and Measurements of Liver Cholesterol Content.
Mice were fasted for 6 h and then anesthetized. The abdominal cavity was exposed through a ventral incision and gallbladder bile was aspirated. The bile was stored at 4°C and analyzed within 14 days for cholesterol, bile acid, and phospholipid content as described below. The liver was harvested, weighed, and homogenized in 2 ml ddH2O, and a predetermined amount of coprostanol was added as an internal standard for gas chromatography. After homogenization, 5 ml of chloroform/methanol (2:1, vol/vol) was added, the samples were vigorously vortexed and centrifuged, and the organic phase was transferred and split into two separate, fresh tubes. In one tube cholesterol ester hydrolysis was carried out by adding 2 ml of 1 M KOH in 95% ethanol and heating for 1 h at 85°C. Hydrolyzed and nonhydrolyzed samples were injected onto a packed silica capillary column in a Perkin– Elmer gas– liquid chromatograph for total and free cholesterol measurements, respectively. Cholesterol ester was calculated as the difference between total and free cholesterol. Data are expressed as mg cholesterol/g wet liver weight.
Analysis of Bile Composition.
Biliary cholesterol and bile acids were measured enzymatically by using Sigma Diagnostics commercial kits (352 and 450-A, respectively), and phospholipids were measured by using a Wako Commercial GmbH kit.
Measurements of Cholesterol 7α-Hydroxylase and Hepatic Hydroxy-3-Methylglutaryl CoA Reductase (HMGR) Activities.
Liver cholesterol 7α-hydroxylase activity in liver microsomes was assayed as described previously (22). [4-14C]Cholesterol (60 mCi/mmol, DuPont/New England Nuclear) was purified by chromatography on a silicic acid column (23) and mixed with purified, unlabeled cholesterol to a specific activity of 15 dpm/pmol. A 100-nmol aliquot of [4-14C]cholesterol was added to 0.75 mg of Triton X-100 (Sigma), and the acetone solvent was evaporated under nitrogen. The residue was dissolved in buffer (100 mM K2HPO4/0.1 mM DTT, pH 7.4) and preincubated for 2 min at 30°C with 2 units of NADPH-cytochrome c reductase and 50–200 μg microsomal protein in a final volume of 0.5 ml. The reaction was initiated with the addition of 0.6 μmol of NADPH. After a 10-min incubation in a shaking-water bath at 37°C, the reaction was stopped with 7.5 ml of dichloromethane/ethanol (5:1, vol/vol). After the addition of 3 ml of water, the sterols were extracted and separated by thin layer chromatography (TLC) as described previously (24). The amount of labeled 7α-hydroxycholesterol formed was determined by scintillation counting in 10 ml of EcoLume (ICN). All experiments were corrected with zero-incubation time controls. Enzyme activity was expressed as pmol of 7α-hydroxycholesterol formed per mg protein/min. Under the assay conditions used, the amounts of endogenous cholesterol, measured by capillary gas chromatography (25), was less than 5% of the exogenously added, labeled substrate. Hepatic microsomal HMGR activity was assayed by a method modified from a previously published procedure (26). Briefly, 50 and 100 μg of hepatic microsomal protein were preincubated for 2 min at 37°C with an NADPH-generating system (3.4 mM NADP+/30 mM glucose 6-phosphate/0.3 units of glucose-6-phosphate dehydrogenase) in a total final volume of 100 μl buffer (50 mM K2PHO4/70 mM KCl/10 mM DTT/30 mM EDTA, pH 7.4). The reaction was started with the addition of 15 μl 14C-labeled substrate ([14C]HMG CoA, purchased from Amersham, diluted with unlabeled HMG-CoA to a specific activity of 25 dpm/pmol and a final concentration of 300 μM). The mixture was incubated for 15 min at 37°C, and the reaction was stopped with 15 μl 6 M HCl. About 10,000 cpm of [3H]mevalonolactone and unlabeled mevalonolactone were added for recovery standard and product marker, respectively. After lactonization at 37°C for 15 min and the addition of 1 ml ether, 0.2 g sodium sulfate, and 100 μl water, the products were extracted twice with ether and separated by TLC on Silica Gel 60 plates (VWR Scientific) with benzene/acetone (1:1, vol/vol) as the solvent system. The immediate product (14C-labeled mevalonolactone) was quantitated by scintillation spectrometry in EcoLume as described above.
Fecal Bile Acid Analysis.
A mixture of 25 mg of freeze-dried feces and nor-deoxycholic acid (9.5 μg in 100 μl methanol) was added to 1 ml of 0.5 N sodium hydroxide and heated at 60°C for 1 h. After cooling to room temperature, the products were diluted with 1 ml water and extracted four times with 2 ml hexane. The aqueous phase was cooled in ice, acidified with 50% hydrochloric acid to pH 1, and extracted four times with 3 ml of ethyl acetate. The organic layer was washed with water to neutrality and evaporated at 55°C under nitrogen. The product was treated with 3% methanolic hydrochloric acid for 4 h at room temperature, solvent was evaporated at 55°C under nitrogen, and the residue was subjected to trimethyl silylation with 100 μl of Sil-Prep (Alltech Associates) for 30 min at 55°C. After evaporation under nitrogen, the trimethyl sily ether-methyl esters formed were dissolved in 200 μl of hexane, and 1–2 μl was used for gas– liquid chromatography (27). When needed, the mass spectra of the trimethyl sily ether-methyl esters of the bile acids were determined as described previously (28).
RESULTS
The first experiment was designed to examine the time course of excretion of fecal β-[3H]sitostanol and [14C]cholesterol in the mouse. In this experiment the animals were placed in metabolic cages and gavaged with labeled mixture, and feces were collected daily for 4 consecutive days and processed as described in Methods. As shown in Fig. 1, under these conditions more than 98% of the fecal β-[3H]sitostanol and 86% of [14C]cholesterol were recovered over the first 24 h of feces collection. This indicates that a 24-h collection is sufficient for the measurement of cholesterol absorption in the mouse, and this was used for all subsequent experiments.
Figure 1.

Daily fecal excretion of [3H]β-sitostanol and [14C]cholesterol. C57BL/6 males were gavaged with [3H]β-sitostanol and [14C]cholesterol and placed in metabolic cages, and feces was collected daily for 4 consecutive days. The percentage of daily [3H] and [14C] excretion out of the total of each label recovered in the feces over 4 days is plotted (mean ± SD, n = 5).
To examine the mechanism of regulation of dietary cholesterol absorption, C57BL/6 mice were fed increasing amounts of cholesterol and at each level of dietary cholesterol the percentage absorption was measured. As shown in Fig. 2A, as dietary cholesterol increased there was a stepwise decrease in the percentage of dietary cholesterol absorbed. Compared with the 0.02% cholesterol diet, the 1% cholesterol diet had a 50% decrease in the percentage of dietary cholesterol absorbed. Plotting the mass of dietary cholesterol absorbed per day versus the percent by weight of cholesterol in the diet gave a saturable curve characterized by a Km of 0.4% wt/wt and a Vmax of 0.65 mg cholesterol/g body weight per day as shown in Fig. 2B. Thus, in C57BL/6 mice dietary cholesterol absorption appears to be a saturable process.
Figure 2.

Effect of dietary cholesterol on cholesterol absorption. (A) Five different groups of C57BL/6 males were fed for 3 weeks with dietary 0.02–1% wt/wt cholesterol. Mice received a gastric bolus of 100 μl of corn oil containing 1.67 μCi [14C]cholesterol and 0.67 μCi [3H]β-sitostanol, they were placed in metabolic cages, and feces were collected for 24 h. The ratio of [14C]/[3H] labels in the fecal lipid extracts was determined and percentage absorption was calculated (mean ± SD, n = 5 animals in each group). ∗, P < 0.03 and ∗∗, P < 0.0001 vs. 0.02%. (B) The mass of absorbed cholesterol was calculated as the daily cholesterol intake divided by the body weight and multiplied by percentage cholesterol absorption (mean of 5–10 animals per group). Nonlinear regression shows saturable cholesterol absorption.
To rule out the possibility that the apparent dietary cholesterol-induced decrease in the percentage of dietary cholesterol absorption was because of exchange of labeled cholesterol with excess unlabeled dietary cholesterol within the intestinal lumen, the following experiment was performed. Mice were fed a 0.5% cholesterol diet, the absorption of dietary cholesterol was measured, and the mice were kept for an additional 3 weeks on the same diet. These mice were then given an additional gastric bolus of the radioactive mixture of cholesterol and sitostanol, switched to a 0.02% cholesterol diet, and, during the next 24 h, while the animals were consuming a 0.02% cholesterol diet, feces were collected and absorption was determined. As shown in Fig. 3, the acute switch to a low-cholesterol diet failed to change the apparent percentage of dietary cholesterol absorbed. This result indicates that the decrease in the percentage dietary cholesterol absorption in response to dietary cholesterol, observed in our experiments, is not attributable to nonspecific cholesterol exchange. This conclusion is supported further by our studies in the SR-BI Tg mice, as shown below. In another experiment we examined the effect of dietary cholesterol on cholesterol absorption in animals fed with a high-fat diet. In this experiment we found that, when added to high-fat diet (22% saturated fat), dietary cholesterol (0.5% wt/wt) efficiently suppresses the absorption rates (65 ± 6% vs. 44 ± 7% in high-fat- and high-fat, high-cholesterol-fed animals, respectively). These results strongly suggest that a decreased cholesterol solubility that may occur in cholesterol-enriched chow diet displayed minor, if any, effect on our results.
Figure 3.

Effect of an acute dietary cholesterol switch on percentage cholesterol absorption. C57BL/6 mice were preconditioned for 3 weeks with 0.5% cholesterol and cholesterol absorption was measured (solid bar). The same animals were fed for additional 3 weeks with the same diet, gavaged with radiolabeled mixture, and switched immediately afterwards into 0.02% cholesterol. During the next 24 h, while consuming a 0.02% cholesterol diet, feces were collected and cholesterol absorption was measured (open bar). (Mean ± SD, n = 5.)
To provide possible insights into how dietary cholesterol might be regulating the percentage of absorption of dietary cholesterol, a number of parameters reflecting hepatic and biliary cholesterol and bile acid metabolism were measured. Increasing dietary cholesterol from 0.02 to 0.5% increased the liver content of total cholesterol by 46%, from 1.26 ± 0.11 to 1.84 ± 0.39 mg/g, respectively (P < 0.02), and cholesterol ester by 168%, from 0.25 ± 0.05 to 0.67 ± 0.25 mg/g, respectively (P < 0.008). Dietary cholesterol failed to significantly increase the liver content of free cholesterol. As shown in Fig. 4, dietary cholesterol effectively suppressed hepatic cholesterol biosynthesis, as represented by the activity of HMGR, and stimulated bile acid biosynthesis, as represented by the activity of cholesterol 7α-hydroxylase. Dietary cholesterol also increased biliary cholesterol concentration (from 178 to 491 mg/dl in animals fed 0.02% and 0.5%, respectively; P < 0.008), without affecting the concentration of biliary bile acids (Fig. 4) or phospholipids (data not shown). The increase in hepatic bile acid synthesis without an increase in biliary bile acid concentration could be explained by the dietary cholesterol-induced increase in fecal bile acids (0.5 ± 0.2 vs. 2.0 ± 0.6 μg/mg dry feces per day in 0.02% and 0.5% cholesterol-fed animals, respectively; P < 0.001).
Figure 4.

Effect of dietary cholesterol on hepatic HMGR and 7α-hydroxylase activities and bile composition. C57BL/6 mice were fed with increasing amounts of cholesterol for 3 weeks. Gallbladder bile was aspirated and analyzed. The activities of HMGR (■) and cholesterol 7α-hydroxylase (○) were determined in isolated hepatic microsomes. Biliary cholesterol (•) and bile acids (▴) were measured as described in Methods (mean ± SD, n = 5).
Correlations were sought between the dietary cholesterol-induced changes in hepatic and biliary parameters and the dietary cholesterol-induced change in the percentage of dietary cholesterol absorption. The changes in HMGR and cholesterol 7α-hydroxylase activities did not correlate with the changes in the percentage of dietary cholesterol absorption (data not shown). However, as shown in Fig. 5, biliary cholesterol concentration correlated inversely with the percentage dietary cholesterol absorption, r = −0.63, P < 0.0001. Biliary cholesterol concentration also correlated with daily dietary cholesterol intake, r = 0.69, P = 0.0004, dietary cholesterol mass absorption, r = 0.66, P = 0.0008, and liver cholesterol ester concentration, r = 0.74, P < 0.0003 (data not shown). These results suggest that dietary cholesterol stimulates the excretion of cholesterol into the bile, which, in turn, increases biliary cholesterol concentration, and which, in turn, decreases the percentage of dietary cholesterol absorption.
Figure 5.

Relation of percentage of absorption to biliary cholesterol. C57BL/6 mice were fed for 3 weeks with 0.02–1% cholesterol. Cholesterol absorption was measured and gallbladder bile was aspirated and measured for cholesterol content.
This hypothesis was tested further by studying the absorption of cholesterol in SR-BI Tg animals. We postulated that the stimulation of HDL cholesterol excretion into the bile will suppress the absorption of dietary cholesterol in these animals. As shown in Table 1, SR-BI overexpression significantly increased the biliary concentrations of cholesterol (141 ± 7 and 195 ± 28 mg/dl in control and SR-BI Tg, respectively; P < 0.003) with no significant effects on biliary phospholipids and bile acids. As displayed in Table 1, in the SR-BI Tg, increased concentrations of biliary cholesterol were associated with significantly decreased dietary cholesterol absorption rates (69 ± 11% and 46 ± 9% in control and SR-BI Tg, respectively; P < 0.005). Moreover, once again, we found strong and inverse relationships between biliary cholesterol levels and percentage dietary cholesterol in the SR-BI Tg group (r = −0.99, P < 0.0008; data not shown). Finally, the compromised cholesterol absorption rates resulted in a significant decrease in daily cholesterol mass absorption from 0.025 ± 0.001 to 0.021 ± 0.002 mg/g body weight per day in control and SR-BI Tg, respectively (P = 0.023). These observations in the SR-BI Tg mice strongly support the notion that biliary cholesterol excretion regulates dietary cholesterol absorption.
Table 1.
Bile composition and cholesterol absorption in 0.02% cholesterol-fed control and SR-B1 Tg animals
| Component | Control (n = 5) | SR-BI Tg (n = 5) | P |
|---|---|---|---|
| Cholesterol, mg/dl | 141 ± 7 | 195 ± 28 | <0.003 |
| Phospholipids, mg/dl | 1,963 ± 146 | 2,124 ± 373 | NS |
| Bile acids, mg/dl | 10,749 ± 1,585 | 8,789 ± 1,605 | NS |
| Cholesterol absorption, % | 69 ± 11 | 46 ± 9 | <0.005 |
Results are mean ± SD. NS, none significant vs. control.
DISCUSSION
The results of the present study strongly suggest that dietary cholesterol absorption is a saturable process and shed light on the mechanism. In C57BL/6 mice increasing amounts of dietary cholesterol decrease the percentage of dietary cholesterol absorbed. This decrease correlates best with an increase in the cholesterol concentration in the bile, which, in turn, appears to be influenced by total daily dietary cholesterol intake, daily mass absorption of dietary cholesterol, and liver cholesterol ester content. In the SR-BI Tg mouse increased concentrations of biliary cholesterol resulted in a similar phenomena, e.g., decreased percentage of dietary cholesterol absorption. Taken together, these results in C57BL/6 and SR-BI Tg mice suggest a cause-and-effect relationship between increased biliary cholesterol content and the phenomenon of saturability of dietary cholesterol absorption.
After feeding increasing amounts of dietary cholesterol, a decrease in dietary cholesterol absorption efficiency has been reported by others in mice (8, 9), monkeys (10), and humans (11), but the mechanism has not been determined. Quintao et al. (11), in human studies, originally suggested that this phenomenon was because of an exchange of labeled with unlabeled cholesterol within the intestinal lumen, resulting in a decrease in absorption of labeled cholesterol on the high-cholesterol diet. However, in follow-up studies, Samuel and McNamara (13) showed that the contribution of the intestinal mucosa to luminal cholesterol, by either isotope exchange or cholesterol secretion, is minimal and therefore cannot explain the changes in the percentage of dietary cholesterol absorbed. Our experiments in which high dietary cholesterol-preconditioned animals were switched to low dietary cholesterol and failed to increase the percentage of absorption (Fig. 3) strongly argue against such a possibility.
The data presented in the present study strongly indicate that the suppression of the percentage of absorption of dietary cholesterol by feeding increasing amounts of dietary cholesterol is caused by changes in bile composition. Specifically, we could show in wild-type mice that, at a wide range of dietary intake, the efficiency of absorption of dietary cholesterol from the intestine is strongly inversely correlated with the concentration of cholesterol in the bile (Fig. 5). Our hypothesis is supported further by studies in the SR-BI Tg mouse. While on a chow diet these animals displayed severely diminished plasma HDL cholesterol levels and a 6- to 7-fold increase in liver HDL cholesteryl ester fractional catabolic rate (N.W., Takeshi Arai, Yong Ji, Franz Rinninger, and A.R.T., unpublished data), suggesting that HDL cholesterol represents the main source of biliary cholesterol in these animals. These observations provide strong evidence that cholesterol excretion into the bile, be it of dietary or endogenous (e.g., HDL cholesterol) sources, plays an important role in regulating the efficiency of dietary cholesterol absorption.
How does increased excretion of cholesterol into the bile decrease the percentage of dietary cholesterol absorbed? It is overlooked frequently that biliary cholesterol is the principal source of cholesterol within the intestinal lumen. For example, in humans, 800–1,200 mg of cholesterol is excreted daily from the biliary tract into the intestine. This cholesterol is almost exclusively in the form of nonesterified cholesterol and partitioned between the mixed micelles and phospholipid vesicles within the bile (29). In contrast, the dietary intake of cholesterol in Western countries is 300–500 mg per day. This cholesterol is mostly nonesterified and dissolved in the oil phase or the lammelar phase. The absorption of cholesterol from the oil and the lamellar phases is critically dependent on the availability of biliary-derived, intraluminal-mixed micelles that enable the absorption of dietary cholesterol by as yet poorly understood mechanisms (30). The distinctively different physicochemical properties of biliary and dietary cholesterol raise the possibility that the two cholesterol pools are subjected to absorption at different efficiencies. Studies in humans by Samuel and McNamara (13) suggested that biliary cholesterol is a preferable substrate for absorption from the intestine. We speculate that anything that stimulates cholesterol excretion into the bile (for example, overexpression of hepatic SR-BI), which then must be accommodated by the biliary vesicular and micellar phases, limits the amount of dietary cholesterol that can be transferred from the oil and lammelar phases to the micellar phase, thereby compromising dietary cholesterol absorption.
The relevance of the mouse model to human physiology deserves special attention. In a 70-kg human on a 2,500-kcal diet, ingestion of 200 mg cholesterol/day corresponds to 0.04% wt/wt dietary cholesterol, and ingestion of 500 mg cholesterol/day corresponds to 0.1% wt/wt dietary cholesterol. These intake values for dietary cholesterol are on the linear portion of the saturable dietary cholesterol absorption curve defined in this study for the mouse (Fig. 2B). Indeed, studies in humans fed comparable amounts of dietary cholesterol have suggested that the mass of dietary cholesterol absorbed from the intestine is linearly related to dietary cholesterol intake (11). It is also interesting to note that the Kd and Vmax values defined for dietary cholesterol absorption in the mouse suggest a low-affinity, high-capacity process supporting previous studies in humans that have concluded that dietary cholesterol absorption is a relatively inefficient process (11). Finally, in an as yet unpublished study, we have examined the mass of cholesterol absorbed in 18 subjects each fed diets containing 0.04% and 0.1% cholesterol. There was up to a 25% difference between people in the slope of the increase in dietary cholesterol absorption when individual subjects went from the 0.04% to the 0.1% cholesterol diet. This suggests that humans differ in the Km and/or Vmax for dietary cholesterol absorption even within the range of normal human dietary cholesterol consumption, thus supporting the notion that humans differ in the genetic and environmental factors that regulate the kinetics of dietary cholesterol absorption. From the mouse studies we speculate that these factors act by influencing biliary cholesterol concentration.
The capacity of dietary cholesterol to stimulate the biliary excretion of cholesterol in high-cholesterol-fed C57BL/6 animals raises questions about the pools of cholesterol that feeds into the bile. Previous studies suggest that HDL cholesterol is a preferable source for bile excretion (31). This view is strongly supported by the results of our studies in the SR-BI Tg animals (N.W., Takeshi Arai, Yong J., Franz Rinninger, and A.R.T., unpublished data) and by recently published studies in mice with adenoviral-induced overexpression of hepatic SR-BI (18). Yet, these studies do not exclude the possibility that dietary cholesterol may feed into the biliary tree through an HDL-independent pathways. For example, in our present studies in C57BL/6 males we could not detect significant effects of dietary cholesterol on HDL cholesterol levels. In addition, we examined the effect of dietary cholesterol on HDL cholesterol metabolism and found that dietary cholesterol did not affect the fractional catabolic rate of HDL cholesteryl ester (E.S. and J.L.B., unpublished data). Furthermore, in another set of experiments we examined the effect of dietary cholesterol on biliary cholesterol excretion in apolipoprotein E knockout animals. We found that, upon feeding with a high-cholesterol diet, the failure of the livers of these animals to dispose of dietary cholesterol in the form of chylomicron remnants resulted in a failure to stimulate the excretion of cholesterol into the bile (E.S. and J.L.B., unpublished data). We believe that these studies suggest that multiple plasma cholesterol sources may feed into the biliary tract including HDL and chylomicron remnant-derived cholesterol.
In summary, in the present study we used a mouse model to show that the efficiency of dietary cholesterol absorption is related to the control of cholesterol excretion into the bile. If similar mechanisms operate in humans, then the elucidation of the molecular events underlying cholesterol excretion into the biliary tract may lead to new insights into the understanding of responsiveness to dietary cholesterol and possibly new approaches to the prevention of atherosclerotic disease.
Acknowledgments
We are grateful to Dr. Paul Samuel for valuable discussions. This work was supported by National Institutes of Health Grants HL-32435-14 to J.L.B., HL-58033 to A.R.T., and DK 26756 to S.S. and by the Lipper Foundation Scholarship to E.S.
ABBREVIATIONS
- CHD
coronary heart disease
- HMGR
3-hydroxy-3-methylglutaryl CoA reductase
- SR-BI
scavenger receptor BI
- apoE
apolipoprotein E
- HDL
high density lipoprotein
References
- 1.Sonnenberg L M, Posner B M, Belanger A J, Cupples L A, D’Agostino R B. J Clin Epidemiol. 1992;45:413–418. doi: 10.1016/0895-4356(92)90042-l. [DOI] [PubMed] [Google Scholar]
- 2.Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. J Am Med Assoc. 1993;269:3015–3023. [PubMed] [Google Scholar]
- 3.Zanni E E, Zannis V I, Blum C B, Herbert P N, Breslow J L. J Lipid Res. 1987;28:518–527. [PubMed] [Google Scholar]
- 4.Denke M A, Breslow J L. J Lipid Res. 1988;29:963–969. [PubMed] [Google Scholar]
- 5.Kolodgie F D, Katocs A S, Jr, Largis E E, Wrenn S M, Cornhill J F, Herderick E E, Lee S J, Virmani R. Arterioscler Thromb Vasc Biol. 1996;16:1454–1464. doi: 10.1161/01.atv.16.12.1454. [DOI] [PubMed] [Google Scholar]
- 6.Katan M B, Berns M A, Glatz J F, Knuiman J T, Nobels A, de Vries J H. J Lipid Res. 1988;29:883–892. [PubMed] [Google Scholar]
- 7.Spady D K, Woollett L A, Dietschy J M. Annu Rev Nutr. 1993;13:355–381. doi: 10.1146/annurev.nu.13.070193.002035. [DOI] [PubMed] [Google Scholar]
- 8.Kirk E A, Moe G L, Caldwell M T, Lernmark J A, Wilson D L, LeBoeuf R C. J Lipid Res. 1995;36:1522– 1532. [PubMed] [Google Scholar]
- 9.Carter C P, Howles P N, Hui D Y. J Nutr. 1997;127:1344–1348. doi: 10.1093/jn/127.7.1344. [DOI] [PubMed] [Google Scholar]
- 10.Bhattacharyya A K, Eggen D A. Atherosclerosis. 1987;67:33–39. doi: 10.1016/0021-9150(87)90262-0. [DOI] [PubMed] [Google Scholar]
- 11.Quintao E, Grundy S M, Ahrens E H., Jr J Lipid Res. 1971;12:233–247. [PubMed] [Google Scholar]
- 12.Quintao E, Grundy S M, Ahrens E H., Jr J Lipid Res. 1971;12:221–232. [PubMed] [Google Scholar]
- 13.Samuel P, McNamara D J. J Lipid Res. 1983;24:265–276. [PubMed] [Google Scholar]
- 14.Thurnhofer H, Hauser H. Biochemistry. 1990;29:2142–2148. doi: 10.1021/bi00460a026. [DOI] [PubMed] [Google Scholar]
- 15.Lopez-Candales A, Grosjlos J, Sasser T, Buddhiraju C, Scherrer D, Lange L G, Kumar V B. Biochem Cell Biol. 1996;74:257–264. doi: 10.1139/o96-027. [DOI] [PubMed] [Google Scholar]
- 16.Howles P N, Carter C P, Hui D Y. J Biol Chem. 1996;271:7196–7202. doi: 10.1074/jbc.271.12.7196. [DOI] [PubMed] [Google Scholar]
- 17.Meiner V L, Cases S, Myers H M, Sande E R, Bellosta S, Schambelan M, Pitas R E, McGuire J, Herz J, Farese R V., Jr Proc Natl Acad Sci USA. 1996;93:14041–14046. doi: 10.1073/pnas.93.24.14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozarsky K F, Donahee M H, Rigotti A, Iqbal S N, Edelman E R, Krieger M. Nature (London) 1997;387:414–417. doi: 10.1038/387414a0. [DOI] [PubMed] [Google Scholar]
- 19.Ji Y, Jian B, Wang N, Sun Y, Moya M L, Phillips M C, Rothblat G H, Swaney J B, Tall A R. J Biol Chem. 1997;272:20982–20985. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- 20.Fan J, Wang J, Bensadoun A, Lauer S J, Dang Q, Mahley R W, Taylor J M. Proc Natl Acad Sci USA. 1994;91:8724–8728. doi: 10.1073/pnas.91.18.8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zilversmit D B, Hughes L B. J Lipid Res. 1974;15:465–473. [PubMed] [Google Scholar]
- 22.Shefer S, Salen G, Bullock J, Nguyen L B, Ness G C, Vhao Z, Belamarich P F, Chowdhary I, Lerner S, Batta A K, et al. Hepatology. 1994;20:213–219. doi: 10.1016/0270-9139(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 23.Nicolau G, Shefer S, Salen G, Mosbach E H. J Lipid Res. 1974;15:94–98. [PubMed] [Google Scholar]
- 24.Shefer S, Cheng F W, Hauser S, Batta A K, Salen G. J Lipid Res. 1981;22:532–536. [PubMed] [Google Scholar]
- 25.Nguyen L B, Shefer S, Salen G, Horak I, Tint G S, McNamara D J. Metabolism. 1988;37:346–351. doi: 10.1016/0026-0495(88)90134-5. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen L B, Shefer S, Salen G, Ness G C, Tint G S, Zaki F G, Rani I. J Clin Invest. 1990;86:923–931. doi: 10.1172/JCI114794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batta A K, Aggarwal S K, Mirchandani R, Shefer S, Salen G. J Lipid Res. 1992;33:1403–1407. [PubMed] [Google Scholar]
- 28.Batta A K, Arora R, Salen G, Tint G S, Eskreis D, Katz S. J Lipid Res. 1989;30:1953–1962. [PubMed] [Google Scholar]
- 29.Somjen G J, Gilat T. Gastroenterology. 1986;91:772–775. doi: 10.1016/0016-5085(86)90652-9. [DOI] [PubMed] [Google Scholar]
- 30.Wilson M D, Rudel L L. J Lipid Res. 1994;35:943–955. [PubMed] [Google Scholar]
- 31.Robins S J, Fasulo J M. J Clin Invest. 1997;99:380– 384. doi: 10.1172/JCI119170. [DOI] [PMC free article] [PubMed] [Google Scholar]