

Abstract
It is generally agreed that most colon cancers develop from adenomatous polyps, and it is this fact on which screening strategies are based. Although there is overwhelming evidence to link intrinsic genetic lesions with the formation of these preneoplastic lesions, recent data suggest that the tumor stromal environment also plays an essential role in this disease. In particular, it has been suggested that CD34+ immature myeloid precursor cells are required for tumor development and invasion. Here we have used mice conditional for the stabilization of β-catenin or defective for the adenomatous polyposis coli (APC) gene to reinvestigated the identity and importance of tumor-infiltrating hematopoietic cells in polyposis. We show that, from the onset, polyps are infiltrated with proinflammatory mast cells (MC) and their precursors. Depletion of MC either pharmacologically or through the generation of chimeric mice with genetic lesions in MC development leads to a profound remission of existing polyps. Our data suggest that MC are an essential hematopoietic component for preneoplastic polyp development and are a novel target for therapeutic intervention.
Keywords: cancer, inflammation, polyposis, TNFα
The genesis of hereditary colon cancer [familial adenomatous polyposis of the colon (FAPC)] as well as sporadic colorectal neoplasias are closely linked with mutations that result in loss of function of the adenomatous polyposis coli (APC) gene (1–3) and lead to the stabilization of β-catenin (4). These molecular events are necessary and sufficient for triggering the formation of adenomatous polyps, precursors to invasive colon cancer (5). Mutations of the TGF-β receptor/SMAD4-signaling cascade in human also has been strongly implicated in the subsequent progression to malignant disease. Recently, in an elegant mouse model of adenocarcinoma, mutations of SMAD4 in concert with APC mutations were shown to induce the formation of invasive tumors (6). Intriguingly, the ability of these tumors to invade the tissues was shown to critically depend on a population of immature myeloid cells (iMC) recruited to the invasion front. These cells expressed CD34, CCR1, and matrix metalloproteinase (MMP) types 2 and 9 and were recruited by tumor cells synthesizing the CCR1 ligand, CCL9. Thus, although their identity has not yet been fully delineated, there is compelling evidence for the requirement of a specific hematopoietic component in the progression, and possibly the genesis, of colorectal neoplasia.
Mast cells (MC) are potent immunomodulatory cells that have been shown to play a key role in a variety of inflammatory diseases, including allergic asthma, autoimmune arthritis, and multiple sclerosis-like syndromes. They elaborate a potent array of cytokines, proteases, and inflammatory mediators (including TNFα, IL-6, TGF-β, VEGF, MMP2, and MMP9), which are known to be involved in tissue remodeling and angiogenesis. These cells are responsive to CCL9, and we have recently shown that they require the sialomucin CD34 (and CD43) for efficient trafficking in vivo (7, 8). Although mastocytosis is frequently associated with the emergence of tumors (6–13), their role in tumor progression or rejection is only beginning to be studied.
Here we have reevaluated the association of human MC with invasive adenocarcinoma and exploited two independent murine models of polyposis and three inhibitors of MC function (two genetic and one pharmacological) to specifically address the role of MC in the early development of colon cancer. Our observations indicate that MC are causative agents in polyp formation, the initiating step of colon cancer.
Results
Adenoma-Associated Mastocytosis.
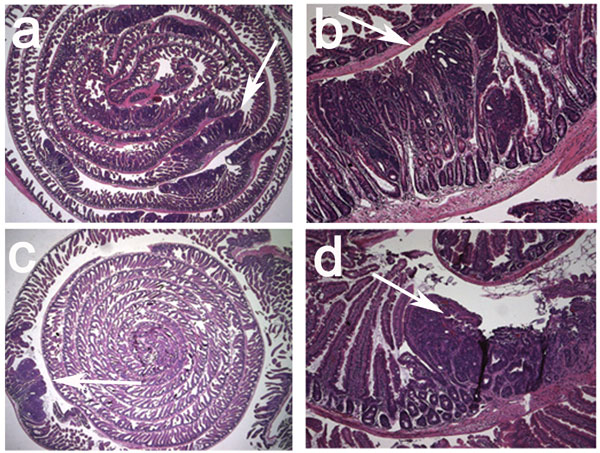
A screening of samples from 79 patients consisting of microinvasive adenocarcinomas, polyps, and adjacent normal tissue revealed an abundance of MC in the stroma and submucosa of the polyps and tumors. Quantitative analysis of the polyps (patients, n = 26) confirmed that MC were preferentially enriched in the polyp masses [supporting information (SI) Fig. 5]. To examine the kinetics of mastocytosis and to reproducibly observe early events in polyp growth, transgenic mice were generated that overexpress stabilized β-catenin in gut enterocytes in an inducible manner. CreERT2 mice express a tamoxifen-activatable Cre recombinase (9) driven by the intestine-specific TS4 promoter (10). These mice were crossed with Ctnnblox(ex3) mice (11), in which exon 3 of the β-catenin gene is flanked by loxP sequences. Serine and threonine residues encoded by this exon are targets of phosphorylation by glycogen synthase kinase 3β (GSK-3β), and excision of these sequences generates a stable dominant-acting mutant β-catenin (11). Feeding lactating female TS4-CreERT2 × Ctnnbllox(ex3) mice with tamoxifen (5 days, 1 mg per mouse per day) induced nascent crypts and polyps in the suckling pups and in the mother that were readily visible as early as 2.5 weeks after initiation of the treatment (Fig. 1 a and b). At the earliest time points evaluated, these lesions were consistently infiltrated with MC, as revealed by expression of CD34 (Fig. 1c) and chloracetate esterase (CAE) staining (Fig. 1d). Healthy neighboring tissue did not harbor significant numbers of MC (Fig. 1i).
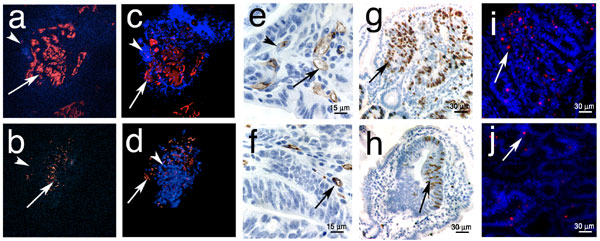
Fig. 1.

MC are enriched in adenomatous polyps. (a–h) Samples embedded in 6-μm paraffin sections or 2-μm plastic (where mentioned) sections. (a–d) TS4-CreERT2 × Ctnnblox(ex3) intestine 2.5 weeks after induction with tamoxifen. (a and b) H&E stain of nascent crypt (a) and aberrant crypt (b). (c) Polyp MC stained with antiCD34-HRP. (d) Polyp MC stained with CAE. Plastic sections are shown. (e and f) APCΔ468 (e) or Min-Rag−/− (f) intestine. Plastic sections were stained with CAE. (g and h) mMCP2 (g) and mMCP6 (h) stainings of APCΔ468 intestine. Arrows indicate MC. (i) MC per field in polyps of tamoxifen-induced CreERT2 × Catnblox(ex3) mice. Shown are the mean frequencies in polyps (17.5 ± 15.4; n = 357) and normal tissue (1.3 ± 1.6) (99% confidence, P < 0.001, ANOVA). (Magnification, ×200.) (j) MC per field in polyps in APCΔ468 (age, 5 months) or Min-Rag−/− (age, 3 months) mice. The mean frequencies of MC in the polyps of APCΔ468 mice (27.4 ± 1.7; n = 63) and Min-Rag−/− mice (28.3 ± 1.7; n = 52) were significantly higher than in the corresponding surrounding tissues (1.2 ± 0.3, and 0.3 ± 0.1, respectively; fields of view, n = 357) (99% confidence, P < 0.001; ANOVA).
To ensure that the observed mastocytosis was a characteristic event in an independent model of polyposis, we examined APCΔ468 mice, which bear a targeted insertion of truncating mutations after exon 10 of the APC gene (ref. 12 and unpublished data). In this hereditary model of polyposis, lesions were detected as early as 2 months after birth, due to loss of heterozygosis of the wild-type (wt) APC allele. Similar to the inducible model, polyps arising in this model were typically infiltrated with intraepithelial MC (Fig. 1e) restricted to the lesions (Fig. 1j). Thus, based on our evaluation of human polyps and using two independent mouse models, we find that mastocytosis is an early and persistent event associated with polyposis.
Lymphocyte Dependence and MC Subtypes.
Murine MC have historically been divided into two subpopulations, based on histochemical staining properties, expression of MC proteases, and localization to connective tissue mast cells (CTMC) versus mucosal mast cells (MMC) (13). Activation of MMC is antibody- and, consequently, B and T cell-dependent. We therefore examined the lymphocyte dependence of the polyp-specific mastocytosis. Min mice (14), which also bear a lesion in the APC gene, were bred to Rag2−/− mice that are T and B cell-deficient. These mice were then evaluated for polyps and MC infiltrates. Surprisingly, despite the lack of T and B cells in these mice, they rapidly developed polyps and harbored high levels of associated MC (Fig. 1 f and j). In mice, MMC express the murine MC proteases, mouse mast cell protease (mMCP)1 and mMCP2 (chymases) but rarely express tryptase. In contrast, CTMC typically express chymases mMCP4 and mMCP5 and tryptases mMCP6 and mMCP7 (13). We confirmed by immunohistochemistry, the presence of both MMC (mMCP2) and CTMC (mMCP6) in polyps (Fig. 1 g and h). In summary, the mixed mastocytosis observed in the intestine during colon carcinogenesis is, surprisingly, lymphocyte-independent.
Status of MC and Their Precursors in Regressing Polyps: A role for TNFα.
To follow the fate of MC on the stabilization or regression of the polyps, we exploited the recent observation that polyposis and colitis-associated colon cancer in Min mice and Min-Rag2−/− mice can be reversed through the administration of anti-TNFα antibodies (15). Accordingly, we treated immunocompetent APCΔ468 mice with 200 μg of anti-TNFα antibody every 2 days for 2 weeks and examined them 1 week later. For comparison, age-matched mice served as controls. Significantly fewer polyps were observed in mice treated with anti-TNFα antibody than in control, untreated mice (P < 0.001) (Fig. 2a). The polyps that persisted after completion of the course of antibody treatment had a more flattened and differentiated morphology (SI Fig. 6) and tended to be smaller than the polyps observed in age-matched untreated mice (Fig. 2b). To examine the role of MC and their products in angiogenesis (16), blood vessels irrigating the polyps were imaged by using a near-infrared fluorescent probe (AngioSense) and scanning confocal microscopy (see Materials and Methods). The mean vessel volume in the polyps of anti-TNFα treated mice was four times less than in polyps of age-matched APCΔ468 mice (APCΔ468 mean volume, 881,600 μm3 ± 251,100 μm3; n = 4; and anti-TNFα-treated, 174,200 μm3 ± 49,610 μm3; n = 4; P = 0.0163, unpaired t test) (Fig. 2c and SI Fig. 7 a–d). Similar results were obtained by calculating the surface area of CD34+ vessels in histological sections (Fig. 2d and SI Fig. 7 e and f). Treated mice also had diminished frequencies of mitotic cells (Fig. 2e and SI Fig. 7 g and h) and increased apoptotic cells in the remaining polyps (Fig. 2f and SI Fig. 7 i and j). In summary, anti-TNFα treatment had a potent suppressive effect on polyp growth, expansion, and associated angiogenesis.
Fig. 2.

Polyposis requires TNFα. (a) Number of polyps in APCΔ468 mice with (n = 8) and without (n = 8) anti-TNFα treatment (untreated, 66 ± 3.6; treated, 34 ± 4.5, P < 0.001). The results shown are those of at least two independent experiments. (b) Median diameters and 25 and 75 percentiles of polyps (treated, 1.5 mm ± 0.05; untreated, 1.8 mm ± 0.29). (c) Mean blood vessel volumes measured by near-infrared fluorescent imaging (in each case, n = 4). (d) Relative areas covered by CD34+ endothelial cells (blood vessels). Results are from 20 micrographs from each of three mice: wt, 0.03 ± 0.007%; untreated, 1.99 ± 024%; anti-TNFα, 0.40 ± 0.045% (P < 0.001). (Magnification, ×400.) (e) Mitotic Index of adenoma in untreated (mean, 13 ± 1.4%) and anti-TNFα-treated (mean 4.4 ± 0.85%) mice. Results are from eight samples from each of three mice (P < 0.001). (Magnification, ×200.) (f) Mean values of TUNEL activity per field. Results are from 20 microphotographs from each of three mice: untreated polyps, 0.91 ± 0.17; anti-TNFα, 4.5 ± 0.4 (P < 0.001). (Magnification, ×200.) (g) Frequency of MCp among total MNC prepared from the intestine, determined by limiting dilution assay. Mean of three experiments, each with one wt control: APCΔ468, n = 12; anti-TNFα, n = 11; wt, n = 3 (one-way ANOVA with Bonferroni's multiple comparison test).
To clarify the impact of anti-TNFα treatment on the frequency of resident intestinal MC, MC progenitor (MCp) assays (17, 18) were performed on total mononuclear cell (MNC) preparations from the intestine of these mice. The average frequency of MCp in the intestine of APCΔ468 mice (n = 12) was 0.09 ± 0.2%, (880 ± 160 colonies per 1 × 106 MNC) (Fig. 2g), at least 9-fold higher than the frequency of MCp in the intestine of age-matched wt mice (n = 3; 0.01 ± 0.002%) (Fig. 2g). Four- to 5-fold lower frequencies of MCp were observed in the intestine of age-matched APCΔ468 mice that had received anti-TNFα antibody (n = 11; 0.016 ± 0.005%) when compared with the untreated mice (99% confidence, P < 0.001) (Fig. 2g). Thus, anti-TNFα treatment causes a clear reduction in the local MCp frequency in the intestine. To test whether the effect of anti-TNFα antibodies was related to lymphocyte function, we treated immunocompromised Min-Rag−/− mice with a similar protocol. Anti-TNFα treatment resulted in a potent reduction in the number of polyps (SI Fig. 8a). Again, the fraction of MCp in the intestine of Min-Rag−/− mice was reduced by anti-TNFα treatment (from 0.15 ± 0.015% in untreated intestine to 0.01 ± 0.002% in treated mice; n = 4) (SI Fig. 8b). Thus, the suppression of polyposis and the associated decrease in MCp by anti-TNFα treatment is lymphocyte-independent.
Intriguingly, serum TNFα in polyp-bearing APCΔ468 mice reached levels 10-fold higher than those in control mice by 4 months but declined to 3-fold higher levels in late-stage polyposis (Fig. 3a). APCΔ468Rag−/− mice had reproducibly higher serum TNFα levels than age-matched APCΔ468 mice (Fig. 3a). Consistent with earlier reports (19, 20), MC were a rich source of TNFα (SI Fig. 9). It has been suggested that TNFα supports MC differentiation (21). To test this, we performed MCp assays with MNCs derived from the diseased intestines of APCΔ468 mice, in the presence or absence of exogenous TNFα. TNFα had a potent stimulatory effect on MC colony formation, whereas anti-TNFα antibodies severely suppressed MC colony formation (1,415 ± 52.7 per 106 MNC versus 198.2 ± 16.3 per 106 MNC) (Fig. 3c). Similar observations were made with MNC preparations from the polyps of Min-Rag−/− mice, where addition of anti-TNFα in the cultures reduced the apparent MCp frequency by one-third, whereas addition of TNFα increased it by 2-fold (Fig. 3c and SI Fig. 8b). In aggregate, these observations support a positive-feedback model where lymphocyte-independent, MC production of TNFα leads to enhanced MC precursor proliferation and expansion.
Fig. 3.

Impact of TNFα on MCp frequencies. (a) Levels of TNFα in sera of wt (n = 4), APCΔ468 (n = 6 + 6), or APCΔ468Rag−/− (n = 8) mice were measured by ELISA. Ages shown above each bar are in months (m). Data are from two experiments. (b) Levels of TNFα in sera of mice reconstituted with wt or Cd34−/−Cd43−/− BM or wt mice (n = 3) were measured by ELISA (triplicate measurements). Mice were killed at 5 months of age, 6 weeks after BM reconstitution. (c) Impact of ex vivo addition or depletion of TNFα on the yield of MC colonies was determined by limiting dilution assay. TNFα neutralizing antibody (1 μg/ml; Bioexpress) and soluble TNFα (40 ng/ml; Cell Science) were included in the culture medium. For each treatment, percent increase or decrease relative to untreated mice from three to six assays is shown. Results are statistically significant to 99% confidence (P < 0.001, ANOVA).
Adenoma Formation Is MC-Dependent.
To provide a causative link between MC and polyposis, we generated hematopoietic chimeras by reconstituting polyp-prone APCΔ468 mice with bone marrow (BM) from either wt mice or mice with well characterized mutations in genes required for MC development: KitW-sh/Wsh (22, 23) and Cd34−/−Cd43−/− (24). c-kit is a well known MC growth factor receptor, and mice bearing an inversion of the promoter (KitWsh/Wsh) exhibit a selective defect in the ability to form mature MC. CD34 and CD43 are related cell surface sialomucins, and, although deletion of these genes has no effect on MCp formation in the BM, their loss leads to a profound impairment in the ability of MCp to traffic to peripheral tissues (8, 24). To generate chimeras, APCΔ468 mice were lethally irradiated and reconstituted with wt, KitWsh/Wsh or Cd34−/−Cd43−/− BM and analyzed 6 weeks later for polyps. Depletion of MC correlated with lowered levels of serum TNFα (Fig. 3b). The frequency and size of polyps showed a tight link with the frequency of both MC and MCp: wt BM recipients > KitW-sh/Wsh BM recipients > Cd34−/−Cd43−/− BM recipients (Fig. 4 a and b). Reconstitutions with Cd34−/−Cd43−/− BM resulted in a marked reduction in the frequency of polyps when compared with wt BM recipients (33 ± 10 or 47 ± 3.3 versus 72 ± 15) (Fig. 4a). Similarly, decreased blood vessel densities (Fig. 4e and SI Fig. 7 e and f) and decreased mitotic activity (Fig. 4f and SI Fig. 7 g and h) but increased apoptosis of aberrant epithelial cells (Fig. 4g and SI Fig. 7 i and j) were characteristically noted in MC-depleted mice. Morphological evidence for regression of polyps in Cd34−/−Cd43−/− BM recipient mice included microadenomas with villi appendages (SI Fig. 10a) and atypically large/highly apoptotic nascent crypts (SI Fig. 10 c, d, and f–h) poorly infiltrated by MC (SI Fig. 10 b and e). In summary, these data show that all major hallmarks of polyposis formation are strictly dependent on the ability of MC to provide an appropriate microenvironment and that depletion of MC adversely affects polyp integrity.
Fig. 4.

Polyposis requires MC. (a) Number of polyps in APCΔ468 mice, reconstituted with wt BM (60 ± 6.7, n = 10), Cd34−/−Cd43−/− BM (34 ± 4.9, n = 11), or KitW-sh/Wsh (SASH) BM (45 ± 1.5, n = 8). Data are from two independent experiments (P < 0.001 vs. wt). (b) Average diameter of polyps for wt BM (1.6 ± 0.06), Cd34−/−Cd43−/− BM (1.4 ± 0.04), or KitW-sh/Wsh (SASH) BM (1.5 ± 0.05). Median values and 25th and 75th percentiles are shown. (c) Frequencies of MCp determined by limiting dilution assay for wt BM (n = 9), Cd34−/−Cd43−/− BM (n = 9), and SASH BM (n = 8). (d) MC counts in polyps (n = 3 per group). Paraffin sections (6 μm) were stained with CAE and viewed at ×200 magnification. (e) Relative areas covered by CD34+ endothelial cells (blood vessels). Results are from 20 micrographs each of three mice. (Magnification, ×400.) The mean areas of vessels for wt BM and Cd34−/−Cd43−/− BM are 1.41 ± 0.093 and 0.055 ± 0.074, respectively (P < 0.001). (f) Mitotic index of polyps from Cd34−/−Cd43−/− BM (5.0 ± 0.61) and wt BM (9.6 ± 0.72) (P < 0.001). Data are from eight micrographs each of three mice. (Magnification, ×200.) (g) TUNEL activity of polyps for Cd34−/−Cd43−/− BM (3.9 ± 0.46) and wt BM (1.2 ± 0.27). The mean values of apoptotic cells for wt BM and CD34−/−CD43−/− BM are 1.2 ± 0.27 and 3.9 ± 0.46, respectively. Data are from 20 micrographs from each of three mice and are the result one-way ANOVA with Bonferroni's multiple comparison test (P < 0.001). (Magnification, ×200.)
Discussion
Colon cancer has arguably provided one of the best examples of an intrinsic genetic lesion that leads to tumor formation. Thus, it has been shown that loss of APC gene function and the resulting stabilization of β-catenin in epithelial precursors is necessary and sufficient for the genesis of polyps, the initiating step in this neoplasm. Despite the clear predisposition provided by these genetic lesions, mounting evidence suggests that tumor formation also strictly depends on a receptive stromal microenvironment for tumor expansion (reviewed in ref. 25). Although most studies have focused on stromal fibroblasts, myofibroblasts, and endothelial precursors (26–28), here we provide compelling evidence that at least one component of this microenvironment for gastrointestinal polyps is infiltrating MC.
MC are among commonly observed cancer-associated proinflammatory cells and decorate a variety of tumors, including hepatocarcinoma and cholangiocarcinoma (29), pancreatic (30), lung (31, 32), skin (33, 34), melanoma (35, 36), lymphoma (37) and neurofibroma (38). However, the contribution of MC to tumor growth or rejection is poorly understood. Here, through examination of a cohort of human tumors, as well as three independent conditional and hereditary murine models of polyposis (TS4CreER × Ctnnblox(ex3), Min, and APCΔ468), we have shown that MC are critical for epithelial tumorigenesis. MC appeared in early dysplatic tissue, and their expansion in polyps reflected elevated levels of MCp. Although reports suggest that masotcytosis is strictly lymphocyte-dependent (39), the absence of lymphocyte in APCΔ468Rag1−/− mice did not hinder the accumulation of MC in polyps. This is the first documentation of such local lymphocyte-independent expansion and maturation of MC and MCp in adenomatous polyps.
Because the contribution of MC to tumorigenesis is likely to be multifaceted, instead of targeting MC products we chose to inhibit the ability of these cells to populate tumor niches. We have previously shown that CD34 and the related antigen CD43 are essential for the recruitment of MC precursors to peripheral tissues (24). More recent studies using inflammatory models suggest that loss of CD34 alone in many cases is sufficient to impede recruitment of MC precursors to peripheral tissues (8). In the present study, knockout of Cd34 and Cd43 severely impeded migration of BM-derived MC to the intestine in APC-defective mice, leading to increased apoptosis, reduced mitotic activity, reduction of angiogenesis, and the loss of integrity of adenomatous polyps. MC requirement was confirmed by using mice reconstituted with c-kit-defective BM (KitW-sh/KitW-sh) (40, 41). Although human CD34 antigen expression ceases as MCp differentiate into mature MC in the peripheral tissues, migrating MCp express the antigen and, thus, should still be valid targets for therapy (7). Additionally, because deletion of the CD34-encoding gene has no obvious effects on murine development or steady-state immune responses, strategies to target this antigen in human MC are likely to be well tolerated.
Elevated levels of serum TNFα in diseased mice and suppression of cytokine levels in MC-depleted mice indicate a role for this cytokine in polyposis. TNFα is a major MC product (42) and is considered to act at the apex of proinflammatory pathways in inflammatory bowel disease (43–45) and to promote cancer (46). Here, we show that MC produce TNFα and require this cytokine for their expansion and differentiation ex vivo. TNFα was critically required for the progressive growth of adenomatous polyps. We propose that MC-synthesized TNFα functions as an autocrine factor to amplify the local MC pool at the sites of tumor formation, while driving a cascade of events to boost the incidence and progressive growth of adenomatous polyps, the immediate precursors to colon cancer. As noted in the next paragraph, because MC are generously endowed with a wide array of inflammatory modulators, it is likely that other MC-derived factors also will contribute to the promotion of polyposis.
MC are armed with a potent array of secreted inflammatory cytokines/mediators (e.g., TNFα, IL-1, IL-4, IL-5, IL-6, PGE2, PGD2, LTB4, LTC4, and PAF) as well as a variety of angiogenic and vascular permeability factors and proteases (VEGF, histamines, MMP2, MMP9, MCP1, MCP2, MCP4, MCP5, MCP7, and MCP8 among others), (reviewed in ref. 47). Thus, they are well positioned to influence tumor growth. VEGF, in particular, has a potent effect on angiogenesis and, therefore, tumor growth and metastasis (48, 49). Consistent with this notion, we have provided direct experimental evidence to show that impairment of MC infiltration negatively impacts polyp integrity in part through suppression of angiogenesis. The residual lesions that do develop in MC-depleted intestines are poorly supplied with blood vessels, are apoptotic, and may not develop beyond the stage of nascent crypts. These results are in agreement with the role of MC in regulating oncogene-promoted angiogenesis in a mouse model of skin cancer (50), although in this earlier study the use of the less viable W/Wv strain of mice (22) precluded the possibility of investigating impact of MC deficiency on tumor growth, because the mice died before developing tumors (L. Coussens, personal communication).
MMPs, too, have been shown to be key players in colon tumor cell invasion and metastasis (51). In this regard, it is particularly intriguing that a recent study using a very similar model to the one described here showed that colon tumor invasion critically depends on the recruitment of CD34+, MMP9+, MMP2+, CCR1+ iMC to the invasion front (6). These cells were recruited to tumors by the chemokine CCL9. Importantly, we have previously shown that MC and their precursors are extremely difficult to distinguish from the earliest hematopoietic progenitors (52). In light of our observation that mature MC (i) express high levels of CD34 (24, 52), (ii) are well known sources of MMP2 and MMP9 (53, 54), and (iii) express CCR1 and chemotax in response to CCL9 (reviewed in ref. 55), it is likely that these iMC in fact represent mature MC, degranulated MC, or MCp. This notion is further strengthened by reports that, in human, iMC lack CD34 (6), because we previously have shown that, although the human Cd34 gene is expressed in trafficking MCp, it is shut off as they differentiate into mature MC in the periphery (reviewed in ref. 7). Our revelation that the iMC are MC should greatly enhance the development of additional interventional therapies.
Our observations provide further evidence in support of the paradigm that carcinogenesis is a process requiring tissue remodeling and collaboration between aberrant epithelium and cellular components of the tumor microenvironment. Our data suggests that in the absence of MC the genetically defective epithelium is unfit to propagate and progress into full tumors, providing experimental evidence for MC as a causative agent in colon cancer. Thus, we propose that MC deserve focused consideration as a therapeutic target in polyposis and colon cancer.
Materials and Methods
Human Colonic Tumor Specimens.
Representative examples of colonic adenocarcinomas (n = 53), colonic adenomas (n = 26), and corresponding normal colonic mucosa (120 sections in all) were identified by a retrospective search of the surgical pathology files of the Brigham and Women's Hospital Pathology Department. All procedures were approved by the Partners Human Research Committee. Sections (6 μm) were cut from paraffin blocks and used for toluidine blue and chloroacetate esterase staining as described below.
CAE Staining.
Paraffin sections were deparaffinized with xylene (two times, 5 min each) and rehydrated gradually with 100% ethanol, 95% ethanol, 70% ethanol, and water. The slides were stained with Naphthol-AS-D chloroacetate and counterstained with hematoxylin Gills II as described earlier (56, 57).
Preparation of Intestinal MNCs.
MNCs were prepared as described (18) by using 10 units of collagenase type 4 (WorthingtonBiochemical Corporation) in 25 ml of RPMI medium 1640 supplemented with 10% FCS (RPMI complete) and agitated at 37°C for 20 min. Cells were purified by 40–60% Percoll gradient centrifugation, washed, and resuspended in PBS plus 0.2% BSA for analysis.
MCp Assay.
Frequencies of MCp in MNCs was measured as described in ref. 18. Briefly, 10,000 MNCs per well were suspended in 100 ml of medium [RPMI complete with 20 ng/ml of SCF, 20 ng/ml of IL-3, and 100,000 irradiated spleen cells per well (3,000 rad)] in a single well of a 96-well plate and diluted serially in subsequent rows. After 10–15 days incubation at 37°C with 5% CO2, colonies of matured MC were scored.
CD34 Staining for Vessels and Anti-TNFα.
The CD34 rat monoclonal antibody (catalog no. Ab8158–100; Abcam) was used at a 1:100 dilution. The polyclonal rabbit anti-rat IgG (reference no. E0468; Dako) was diluted at 1:750, and a ready-to-use labeled polymer-HRP anti-rabbit from Dako was added (catalog no. K4011). Color was developed by using the DAB chromogenic substrate (catalog no. k4007; Dako), and sections were counterstained with hematoxylin. Anti-TNFα antibodies were derived from clone XT-3 (BioExpress).
Apoptosis Staining.
The ApopTag Red in situ apoptosis detection kit from Chemicon International (catalog no. S7185) was used according to the manufacturer's instructions. Products of the Tdt reactions were visualized by using digoxigenin-labeled NTPs and rhodamine conjugated anti-digoxigenin counterstained with DAPI.
BrdU Staining.
Mice were injected with 200 μl of 20 mg/ml BrdU (Sigma) 2 h before being killed. Paraffin sections (6 μm) were deparaffinized and stained for BrdU using the BD Pharmingen TM BrdU in situ detection kit (catalog no. 550803).
Near-Infrared Intravital Microscopy.
For imaging of blood vessels, mice were injected with AngioSense 750 (VisEn Medical), a high-molecular-weight (≈250,000 g·mol) fluorescent probe; they were then anesthetized, and their intestine were exposed through a skin incision and imaged (×4 dry lens) with the prototype Olympus IV100 laser scanning intravital scanning microscope. Z stacks were recorded and analyzed with ImageJ software (“USDC” plugins) in two separate channels. “Particle analysis” plugins and the command 3D objects counter were used to calculate mean vessel volumes, signal intensity, and the location of each vessel found in 525 × 525 × 80 μm cubes [threshold, 1,023; minimum size of particles, 100 voxels; maximum volume, 109 voxels (1 voxel = 1.04 μm3)]. Autofluorescence was detected at 505–510 nm.
Light Microscopic Data Images.
Microscopic images were collected with a Leica DCC camera. For multicolored images, the ImageJ plugin “threshold color” was used to identify cells of the color of interest, which were then counted with the plugin “nuclei counting.” CD34 stained cells were counted by using the command “analyze particles,” and only areas >60 μm2, including hollow structures, were recorded to exclude infiltrating hematopoietic cells (average area, ≈40 μm2).
Statistical Analysis.
The statistical analyses were preformed with the use of the Prism4 software. ANOVA one-way nonparametric with Bonenferroni post hoc test and 99% confidence intervals or unpaired one tailed t tests with Wells correction were used.
Supplementary Material
Acknowledgments
We thank Dr. Robert J. Mayer, Dr. Glenn Dranoff, and Dr. Harvey Cantor for scientific discussions, support, and encouragement. This work was supported by National Institutes of Health Grants R01-CA104547 (to K.K.), R01-AI059676 (to F.G.), and R01-CA108854 (to S.E.E.) and Canadian Institutes of Health Research Grant MOP-64278 (to K.M.M.). K.M.M. is a Michael Smith Foundation for Health Research Scholar.
Note.
While this manuscript was under review, Soucek et al. (58) reported that MC are required for the expansion of myc-induced pancreatic tumors, and Nonomura et al. (59) reported a positive correlation between the frequency of MC in human prostate cancer needle biopsy specimens and poor prognosis.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. L.C. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0704620104/DC1.
References
- 1.Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D, et al. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 2.Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Cancer Res. 1994;54:5953–5958. [PubMed] [Google Scholar]
- 3.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 4.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 5.Mayer RJ. Gastrointestinal Tract Cancer. New York: McGraw–Hill; 2001. [Google Scholar]
- 6.Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H, Aoki M, Oshima M, Hattori M, Takabayashi A, et al. Nat Genet. 2007;39:467–475. doi: 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- 7.Drew E, Huettner CS, Tenen DG, McNagny KM. Blood. 2005;106:1885–1887. doi: 10.1182/blood-2005-03-1291. [DOI] [PubMed] [Google Scholar]
- 8.Blanchet MR, Maltby S, Haddon DJ, Merkens H, Zbytnuik L, McNagny KM. Blood. 2007;110:2005–2012. doi: 10.1182/blood-2006-12-062448. [DOI] [PubMed] [Google Scholar]
- 9.Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D. Nucleic Acids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saam JR, Gordon JI. J Biol Chem. 1999;274:38071–38082. doi: 10.1074/jbc.274.53.38071. [DOI] [PubMed] [Google Scholar]
- 11.Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gounari F, Chang R, Cowan J, Guo Z, Dose M, Gounaris E, Khazaie K. Nat Immunol. 2005;6:800–809. doi: 10.1038/ni1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurish MF, Boyce JA. J Allergy Clin Immunol. 2006;117:1285–1291. doi: 10.1016/j.jaci.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Moser AR, Pitot HC, Dove WF. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 15.Rao VP, Poutahidis T, Ge Z, Nambiar PR, Boussahmain C, Wang YY, Horwitz BH, Fox JG, Erdman SE. Cancer Res. 2006;66:7395–7400. doi: 10.1158/0008-5472.CAN-06-0558. [DOI] [PubMed] [Google Scholar]
- 16.Heissig B, Rafii S, Akiyama H, Ohki Y, Sato Y, Rafael T, Zhu Z, Hicklin DJ, Okumura K, Ogawa H, Werb Z, Hattori K. J Exp Med. 2005;202:739–750. doi: 10.1084/jem.20050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sonoda T, Ohno T, Kitamura Y. J Cell Physiol. 1982;112:136–140. doi: 10.1002/jcp.1041120120. [DOI] [PubMed] [Google Scholar]
- 18.Gurish MF, Tao H, Abonia JP, Arya A, Friend DS, Parker CM, Austen KF. J Exp Med. 2001;194:1243–1252. doi: 10.1084/jem.194.9.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon JR, Galli SJ. Nature. 1990;346:274–276. doi: 10.1038/346274a0. [DOI] [PubMed] [Google Scholar]
- 20.McLachlan JB, Hart JP, Pizzo SV, Shelburne CP, Staats HF, Gunn MD, Abraham SN. Nat Immunol. 2003;4:1199–1205. doi: 10.1038/ni1005. [DOI] [PubMed] [Google Scholar]
- 21.Wright HV, Bailey D, Kashyap M, Kepley CL, Drutskaya MS, Nedospasov SA, Ryan JJ. J Immunol. 2006;176:2114–2121. doi: 10.4049/jimmunol.176.4.2114. [DOI] [PubMed] [Google Scholar]
- 22.Galli SJ, Kitamura Y. Am J Pathol. 1987;127:191–198. [PMC free article] [PubMed] [Google Scholar]
- 23.Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Am J Pathol. 2005;167:835–848. doi: 10.1016/S0002-9440(10)62055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drew E, Merzaban JS, Seo W, Ziltener HJ, McNagny KM. Immunity. 2005;22:43–57. doi: 10.1016/j.immuni.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Mueller MM, Fusenig NE. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 26.Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 28.Bhowmick NA, Neilson EG, Moses HL. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terada T, Matsunaga Y. J Hepatol. 2000;33:961–966. doi: 10.1016/s0168-8278(00)80129-4. [DOI] [PubMed] [Google Scholar]
- 30.Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, Bevilacqua G, Campani D. J Clin Pathol. 2004;57:630–636. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imada A, Shijubo N, Kojima H, Abe S. Eur Respir J. 2000;15:1087–1093. doi: 10.1034/j.1399-3003.2000.01517.x. [DOI] [PubMed] [Google Scholar]
- 32.Takanami I, Takeuchi K, Naruke M. Cancer. 2000;88:2686–2692. [PubMed] [Google Scholar]
- 33.Farnoush A, Mackenzie IC. J Oral Pathol. 1984;13:359–365. doi: 10.1111/j.1600-0714.1984.tb01434.x. [DOI] [PubMed] [Google Scholar]
- 34.de Rey BM, Palmieri MA, Duran HA. Tumour Biol. 1994;15:166–174. doi: 10.1159/000217888. [DOI] [PubMed] [Google Scholar]
- 35.Toth-Jakatics R, Jimi S, Takebayashi S, Kawamoto N. Hum Pathol. 2000;31:955–960. doi: 10.1053/hupa.2000.16658. [DOI] [PubMed] [Google Scholar]
- 36.Ribatti D, Ennas MG, Vacca A, Ferreli F, Nico B, Orru S, Sirigu P. Eur J Clin Invest. 2003;33:420–425. doi: 10.1046/j.1365-2362.2003.01152.x. [DOI] [PubMed] [Google Scholar]
- 37.Ribatti D, Vacca A, Marzullo A, Nico B, Ria R, Roncali L, Dammacco F. Int J Cancer. 2000;85:171–175. [PubMed] [Google Scholar]
- 38.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Visser KE, Korets LV, Coussens LM. Cancer Cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Berrozpe G, Timokhina I, Yukl S, Tajima Y, Ono M, Zelenetz AD, Besmer P. Blood. 1999;94:2658–2666. [PubMed] [Google Scholar]
- 41.Duttlinger R, Manova K, Chu TY, Gyssler C, Zelenetz AD, Bachvarova RF, Besmer P. Development (Cambridge, UK) 1993;118:705–717. doi: 10.1242/dev.118.3.705. [DOI] [PubMed] [Google Scholar]
- 42.Suto H, Nakae S, Kakurai M, Sedgwick JD, Tsai M, Galli SJ. J Immunol. 2006;176:4102–4112. doi: 10.4049/jimmunol.176.7.4102. [DOI] [PubMed] [Google Scholar]
- 43.Mizoguchi E, Mizoguchi A, Takedatsu H, Cario E, de Jong YP, Ooi CJ, Xavier RJ, Terhorst C, Podolsky DK, Bhan AK. Gastroenterology. 2002;122:134–144. doi: 10.1053/gast.2002.30347. [DOI] [PubMed] [Google Scholar]
- 44.Kollias G. Semin Arthritis Rheum. 2005;34:3–6. doi: 10.1016/j.semarthrit.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Choo-Kang BS, Hutchison S, Nickdel MB, Bundick RV, Leishman AJ, Brewer JM, McInnes IB, Garside P. Trends Immunol. 2005;26:518–522. doi: 10.1016/j.it.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Szlosarek P, Charles KA, Balkwill FR. Eur J Cancer. 2006;42:745–750. doi: 10.1016/j.ejca.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Galli SJ, Nakae S, Tsai M. Nat Immunol. 2005;6:135–142. doi: 10.1038/ni1158. [DOI] [PubMed] [Google Scholar]
- 48.Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D. Science. 1999;284:808–812. doi: 10.1126/science.284.5415.808. [DOI] [PubMed] [Google Scholar]
- 49.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mook OR, Frederiks WM, Van Noorden CJ. Biochim Biophys Acta. 2004;1705:69–89. doi: 10.1016/j.bbcan.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 52.Drew E, Merkens H, Chelliah S, Doyonnas R, McNagny KM. Exp Hematol. 2002;30:1211. doi: 10.1016/s0301-472x(02)00890-1. [DOI] [PubMed] [Google Scholar]
- 53.Fang KC, Wolters PJ, Steinhoff M, Bidgol A, Blount JL, Caughey GH. J Immunol. 1999;162:5528–5535. [PubMed] [Google Scholar]
- 54.Tanaka A, Arai K, Kitamura Y, Matsuda H. Blood. 1999;94:2390–2395. [PubMed] [Google Scholar]
- 55.Juremalm M, Nilsson G. Chem Immunol Allergy. 2005;87:130–144. doi: 10.1159/000087640. [DOI] [PubMed] [Google Scholar]
- 56.Leder LD. Am J Dermatopathol. 1979;1:39–42. [PubMed] [Google Scholar]
- 57.Caughey GH, Viro NF, Calonico LD, McDonald DM, Lazarus SC, Gold WM. J Histochem Cytochem. 1988;36:1053–1060. doi: 10.1177/36.8.3134486. [DOI] [PubMed] [Google Scholar]
- 58.Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Nat Med. 2007;13:1211–1218. doi: 10.1038/nm1649. [DOI] [PubMed] [Google Scholar]
- 59.Nonomura N, Takayama H, Nishimura K, Oka D, Nakai Y, Shiba M, Tsujimura A, Nakayama M, Aozasa K, Okuyama A. Br J Cancer. 2007;97:952–956. doi: 10.1038/sj.bjc.6603962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}