

Abstract
The nerve axon is a good model system for studying the molecular mechanism of organelle transport in cells. Recently, the new kinesin superfamily proteins (KIFs) have been identified as candidate motor proteins involved in organelle transport. Among them KIF1A, a murine homologue of unc-104 gene of Caenorhabditis elegans, is a unique monomeric neuron– specific microtubule plus end–directed motor and has been proposed as a transporter of synaptic vesicle precursors (Okada, Y., H. Yamazaki, Y. Sekine-Aizawa, and N. Hirokawa. 1995. Cell. 81:769–780). To elucidate the function of KIF1A in vivo, we disrupted the KIF1A gene in mice. KIF1A mutants died mostly within a day after birth showing motor and sensory disturbances. In the nervous systems of these mutants, the transport of synaptic vesicle precursors showed a specific and significant decrease. Consequently, synaptic vesicle density decreased dramatically, and clusters of clear small vesicles accumulated in the cell bodies. Furthermore, marked neuronal degeneration and death occurred both in KIF1A mutant mice and in cultures of mutant neurons. The neuronal death in cultures was blocked by coculture with wild-type neurons or exposure to a low concentration of glutamate. These results in cultures suggested that the mutant neurons might not sufficiently receive afferent stimulation, such as neuronal contacts or neurotransmission, resulting in cell death. Thus, our results demonstrate that KIF1A transports a synaptic vesicle precursor and that KIF1A-mediated axonal transport plays a critical role in viability, maintenance, and function of neurons, particularly mature neurons.
Organelle transport plays an important role in cellular morphogenesis and function, conveying and targeting important materials to correct destinations. Because of the lack of the protein synthesis machinery in the nerve axon, which can be up to 1 m long, all the proteins required in the axon and synapses have to be transported down the axon after synthesis in the cell body. Thus, organelle transport is fundamental to neuronal morphogenesis and function (Grafstein and Forman, 1980; Hirokawa, 1993, 1998). The nerve axon is a good model system for investigating the molecular mechanisms of organelle transport occurring also in other cells.
The motor proteins are key molecules conveying organelles along cytoskeletal filaments. Various types of membranous organelles are transported bidirectionally at varying velocities; those moving anterogradely include mitochondria and tubulovesicular structures, including precursors of axonal plasma membranes, synaptic vesicles, and synaptic plasma membranes, while those transported retrogradely include prelysosomal organelles, mitochondria, and endosomes. Kinesin and brain dynein are obvious candidates for motor proteins involved in this transport (Brady, 1985; Vale et al., 1985; Lye et al., 1987; Paschal et al., 1987).
In Caenorhabditis elegans, the kinesin-related gene unc-104 has been identified from a genetic approach (Hall and Hedgecock, 1991). In unc-104, null mutant axons have few synaptic vesicles, and neuronal cell bodies have surfeits of similar vesicles tethered together within the cytoplasm (Otsuka et al., 1991). This evidence suggested that UNC-104 is an axonal motor used for anterograde translocation of synaptic vesicles. On the other hand, we have identified the new kinesin superfamily proteins (KIFs)1 as microtubule-based motors involved in this transport (Aizawa et al., 1992; Hirokawa, 1993, 1996). Although some of the members of this superfamily of proteins have been characterized, their in vivo functions are largely unknown. Among the KIFs, KIF1A is a novel monomeric neuron-specific KIF. It exhibits the fastest reported anterograde motor activity in axons (from the cell body to synapses) and is a mammalian homologue of UNC-104 of C. elegans. Results of immunoprecipitation studies revealed that KIF1A in the axons of mature neurons associates with membranous organelles containing synaptic vesicle proteins such as synaptotagmin, synaptophysin, and Rab3A, but not others such as SV2 and presynaptic membrane proteins such as syntaxin 1A or SNAP-25 (Okada et al., 1995). Thus, these in vitro data suggest that KIF1A is a unique monomeric anterograde motor for transport of a subset of synaptic vesicle precursors.
To elucidate the in vivo function of KIF1A and the biological significance of the transport mediated by KIF1A, we disrupted the KIF1A gene using a gene targeting technique. Mice deficient in KIF1A mostly died within a day after birth, showing motor and sensory disturbances. A considerable reduction in the densities of nerve terminals and synaptic vesicles in the nerve terminals and an abnormal accumulation of small vesicles in neuronal cell bodies were observed on examination of the nervous system of these mice. This can be accounted for by the observed decrease of synaptic vesicle precursor transport. In addition, the deficiency in KIF1A resulted in marked neuronal degeneration and neuronal cell death both in vivo and in culture. The analyses in cultures of KIF1A mutant neurons suggested that the mutant neurons might not sufficiently receive afferent stimulation, such as neuronal contacts or neurotransmission, resulting in cell death. Thus, KIF1A mediates the transport of a synaptic vesicle precursor and is essential for the function, maintenance, and viability of neurons, particularly mature neurons.
Materials and Methods
Targeted Disruption of the KIF1A Gene
A 12.5-kb KIF1A genomic clone containing the first coding exon was isolated from the 129/Sv genomic mouse library using standard procedures (Sambrook et al., 1989). The targeting vector was constructed by subcloning the 1.5-kb AatII/Sma1 5′ fragment and the 6.5-kb HindIII/SalI 3′ fragment of KIF1A genomic clone into pBluescript (Stratagene, La Jolla, CA) and inserting the 1.8-kb PGK-neo cassette derived from pKJ1 (Li et al., 1992), the 3.2-kb lac Z gene fragment prepared from pMC1871 (Pharmacia Biotech, Piscataway, NJ), and the 1.0-kb diphtheria toxin A (DT-A) fragment cassette derived from pMC1 DT-A (Yagi et al., 1993). Hence, a 1.0-kb SmaI/HindIII region was deleted. The targeting vector was linearized by digestion with SalI and electroporated into J1 embryonic stem (ES) cells essentially as described previously (Thomas and Capecchi, 1987). After G418 selection, homologous recombination events were identified in 6/610 clones by Southern blot analyses after XbaI/EcoRV digestion of the clones. Of six ES clones carrying the disrupted KIF1A gene, four were injected into C57BL/6 blastocysts. Chimeric male mice were bred to C57BL/6 females, and agouti offsprings were generated from three of the clones, indicating germ line transmission of the ES genome. Heterozygous mice were interbred, and the genotype of the offspring was analyzed by PCR immediately after birth using tail DNA.
Quantitative Immunoblotting Analysis
Mice were killed about 20 h after birth, and then brain crude extract was prepared. Brain crude extract from six pairs of wild-type mice (+/+) and homozygous mutants (−/−) from four litters were analyzed. Immunoblotting was performed as described previously (Okada et al., 1995). The blot was probed with antibodies against KIF1A (Okada et al., 1995), KIF2 (Noda et al., 1995), KIF3 (Kondo et al., 1994), KIF4 (Sekine et al., 1994), H2 (Pfister et al., 1989; kind gift of Dr. G.S. Bloom, University of Texas, Dallas, TX), SUK4 (Ingold et al., 1988; kind gift of Dr. J.M. Scholey, University of California, Davis, CA), synaptophysin (Obata et al., 1986; kind gift of Dr. K. Obata, University of Gunma, Maebashi, Japan), and SV2 (Buckley and Kelly, 1985; kind gift of Dr. K.M. Buckley, Harvard Medical School, Boston, MA), and proteins reacting with the antibodies were quantified by a phosphor imaging analyzer (model BAS2000; Fujifilm, Tokyo, Japan)
Immunohistochemical Analysis
Monoclonal antisynaptophysin IgG and anti-SV2 IgG were purified from ascites fluid using E-Z-SEP (Pharmacia Biotech) and then labeled with a carbocyanine dye, Cy3.5 or Cy5 (Amersham International, Buckinghamshire, UK) respectively, at an [F/P] ratio of 1. Frozen cross sections of the spinal cord (thoracic level 7, T7) were stained with these antibodies. The thickness of the section and other factors that might affect the staining intensity were calibrated by staining the same sections with antitubulin antibody (data not shown). The samples were observed using a confocal laser scanning microscope (model MRC-1000; Bio-Rad Laboratories, Hercules, CA) in the photon counting mode. The number and the areas of the fluorescently labeled spots above a predetermined threshold were measured, and their density and mean area were calculated. As indices of the fluorescence intensity of these spots, upper tenth percentile values were used because the distributions of the fluorescence intensities were very skewed.
Electron Microscopic Analysis
Spinal cords (T7) were dissected out, fixed in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer overnight, fixed with 1% OsO4 for 1 h at 4°C, and embedded in Epon 812. Matching areas from the anterior horns were identified in semithin sections (1 μm) and ultrathin sections of similar thickness (100 nm) for comparison of the wild-type, and mutant mice were prepared and viewed under a transmission electron microscope (model 1200EX; JEOL, Tokyo, Japan) at 100 kV. For morphometric analysis, two sets of experiments were performed using two independent litters (Nos. 1 and 2). To determine the density of nerve terminals, defined areas of the anterior horns containing motor neurons were photographed, and accurate measurements of these areas were taken except for those of cell bodies, dendrites, and vessels in the wild-type (No. 1, 1,768.75 μm2; No. 2, 1,962.5 μm2) and mutant (No. 1, 1,884.38 μm2; No. 2, 1,784.38 μm2) mice. Nerve terminals were identified by the presence of postsynaptic density, and such nerve terminals were counted on the electron micrographs. To determine the density of synaptic vesicles, at least 26 nerve terminals with postsynaptic density were photographed randomly, and the numbers of synaptic vesicles in each nerve terminal were counted.
Sciatic Nerve Ligation Experiment
Sciatic nerve of wild-type or mutant mice was ligated with surgical thread within 12 h after birth. 3 h after ligation, the mice were fixed by perfusion with 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Frozen sections of the sciatic nerve were first blocked with Fab fragment of goat anti–mouse IgG (Jackson ImmunoResearch Inc., West Grove, PA) and then stained with antisynaptotagmin antibody (Wako Purechemical Inc., Tokyo, Japan) or anti–syntaxin 1A antibody (HPC-1; Sigma Chemical Co., St. Louis, MO). The samples were observed with a confocal laser scanning microscope (model MRC-1000; Bio-Rad Laboratories).
Analysis of Cultured Hippocampal Neurons
Cultures of hippocampal pyramidal neurons were prepared as described previously (Barlet and Banker, 1984; Harada et al., 1994). The embryos were genotyped using PCR. Wild-type neurons used for coculture were prepared from embryos of BALB/c mice. l-glutamate (15 μM) was added to the culture medium after 4 d of culture. Neuronal survival was assessed by counting of the viable neuronal cells using phase contrast microscopy. Cells showing cell body condensation and neurite fragmentation were judged as being dead. The numbers of the viable cells in five fields containing three selected regions and two random regions were counted and photographed. Three sets of independent experiments were carried out, and similar results were obtained. X-gal staining of cultured neurons was performed as previously described (Gossler and Zachgo, 1993). For immunoblot analysis of KIF1A in cultured neurons, the cells were prepared from embryos of wild-type littermates, equal amounts of cell lysate were analyzed by SDS-PAGE, and immunoblotting using an anti-KIF1A antibody was performed as previously described (Okada et al., 1995).
Results
Generation of KIF1A Mutant Mice
To disrupt the KIF1A gene in J1 ES cells, a lac Z/neor cassette was inserted into the first coding exon of the KIF1A gene (Fig. 1 a). The lac Z sequence was fused in-frame with the first 51 nucleotides of the KIF1A gene so that β-galactosidase expression reflected endogenous KIF1A gene expression. Six independent ES clones were identified as homologous recombinants by Southern blot analysis. Chimeric male mice derived from three targeted clones transmitted the mutated KIF1A gene through the germline. Mice heterozygous for the mutation showed no obvious morphological or behavioral abnormalities. These mice were interbred, and the DNA of 327 offspring was genotyped using PCR 1 d after birth. The resulting ratio of 77 wild-type, 176 heterozygous, and 74 homozygous mice represented the expected 1:2:1 Mendelian inheritance of the disrupted KIF1A gene. The disruption of the KIF1A gene in homozygous mice was confirmed by Southern blot analysis of the DNA of the offspring (Fig. 1 b). Homozygous mice derived from three targeted clones exhibited the same phenotype.
Figure 1.


Targeted disruption of the KIF1A gene. (a) Schematic representation of the targeting event. The diagrams depict the structure of the targeting vector (top), the KIF1A wild-type allele containing the first coding exon (middle), and the targeted allele (bottom). Restriction enzymes Xb, XbaI; Nc, NcoI; Hi, HindIII; Sm, SmaI; Sa, SalI; EV, EcoRV. A neomycin resistance gene driven by the pgk promoter (pPGK.Neo) and a diphtheria toxin A fragment gene controlled by the MC1 promoter (DT-A) were used as positive and negative selection markers in the targeting vector, respectively. The first coding exon (Ex. 1) is represented as a closed box. The lac Z sequence was fused in-frame with the first 51 nucleotides of the KIF1A gene to express functional β-galactosidase under the control of the KIF1A promoter. The 5′ genomic probe external to the targeting construct hybridized to a 9.6-kb XbaI/EcoRV fragment or a 10-kb NcoI fragment from the wild-type allele and to a 4.3-kb XbaI/EcoRV fragment or a 6.6-kb NcoI fragment from the targeted allele. (b) Southern blot analysis of tail DNA from homozygous (−/−), heterozygous (+/−), and wild-type (+/+) mice. Genomic DNA isolated from the newborn pups was digested with NcoI and hybridized with the 5′ probe. (c) Gross appearance of wild-type, heterozygous, and homozygous mice 1 d after birth. The homozygous mice were smaller than their wild-type and heterozygous littermates and failed to feed, leading to a lack of milk in their stomachs. Stomachs filled with milk in wild-type and heterozygous mice are marked with asterisks.
KIF1A Mutant Mice Showed Motor and Sensory Disturbances
Mice homozygous for the mutant KIF1A gene were born alive. However, these mice showed several neurological abnormalities, and most of them (177 of 180) died within 24 h after birth and the rest within 72 h. The homozygous mice were significantly smaller and weighed 10% less (1.30 ± 0.08 g, n = 11) than their wild-type littermates (1.46 ± 0.08 g, n = 9) 1 d after birth (Fig. 1 c). The homozygous mice displayed reduced motor activity and abnormal limb movements with impaired balance, which could generally be referred to as ataxia. Since they could not position their extremities correctly when attempting to move, they failed to crawl about, in contrast to their wild-type and heterozygous littermates. They also showed an apparent inability to stretch their tails, and the movement of their hindlimbs was affected more severely than that of their forelimbs.
To examine responsiveness to pain stimulation, a pinching test was carried out (Table I). In pinching the dorsal portion of the neck skins or distal portion of the tails of mice with a forceps as intensely in all cases, responses were scored as strong if the mouse vocalized in response to the first pinch, and weak if any vocalization occurred during a series of three pinches. For both neck and tail pinches, all wild-type mice (n = 20) exhibited strong (neck, 85%, 17/20; tail, 100%, 20/20) or weak (neck, 15%, 3/20) responses. In contrast, no homozygous mice (n = 18) vocalized upon pinching of their tails, but approximately half of them exhibited a weak response (56%, 10/18) in the case of neck pinching. These observations indicate that homozygous mutant mice have motor and sensory disturbances, and their neurological defects are more severe in the caudal portion than in the rostral portion of the body.
Table I.
Numbers of Mice Responding to Pinching with Vocalization
| Pinching site | Response* | Wild-type (n = 20) | Heterozygote (n = 36) | Homozygote (n = 18) | ||||
|---|---|---|---|---|---|---|---|---|
| Neck | ||||||||
| Strong | 17 | 35 | 0 | |||||
| Weak | 3 | 1 | 10 | |||||
| None | 0 | 0 | 8 | |||||
| Tail | ||||||||
| Strong | 20 | 36 | 0 | |||||
| Weak | 0 | 0 | 0 | |||||
| None | 0 | 0 | 18 |
Responses were scored as strong if the mouse vocalized in response to the first pinch, and weak if any vocalization occurred during a series of three pinches.
Immunoblot Analysis of Synaptic Vesicle Proteins and KIFs
It has been previously proposed (Okada et al., 1995) that KIF1A functions as a motor protein for the transport of a subtype of synaptic vesicle precursor. Thus, we first measured the amounts of synaptic vesicle proteins in the crude extracts of total brain homogenates of wild-type and mutant mice. For the analysis, we selected synaptophysin and SV2 as markers because results of our previous study demonstrated that KIF1A is associated with synaptophysin-containing synaptic vesicle precursors but not with SV2-containing precursors. As shown in Fig. 2, the amounts of synaptophysin and SV2 in the homozygous mutant brain were 102 ± 8% (mean ± SEM, n = 6) and 109 ± 3% (n = 9) those of wild-type mice. Thus, the total amounts of these synaptic vesicle proteins are not significantly affected by the disruption of the KIF1A gene. One possible explanation for this is that some other motor protein might compensate for the loss of KIF1A. We therefore quantified the amount of other known brain KIFs by quantitative immunoblotting (Fig. 2). KIF2, KIF3, and KIF4 exhibited no significant increase (104 ± 8, 104 ± 6, and 100 ± 6%; n = 6), while kinesin heavy chain (KHC) increased (118 ± 2% with H2 antibody, 130 ± 10% with SUK4 antibody; n = 6). The difference between the values obtained using the two antibodies reflects differences in their reactivity to KHC isoforms. (There exist at least three isoforms in mouse brain: KIF5A [formerly KIF5; Aizawa et al., 1992], KIF5B [ubiquitous KHC; Gudkov et al., 1994], and KIF5C [Kato's KHC; Kato, 1991].) Unfortunately, we cannot quantify these three KHC isoforms at present, but the above result suggests that some isoform(s) of KHC might partially compensate for the function of KIF1A in the homozygous mutants. However, if this is the case, it is clear that KHC cannot compensate for the function of KIF1A fully because homozygous mutants developed severe neurological disorders and died shortly after birth.
Figure 2.

Quantitative immunoblot analysis. Three pairs of mice from two litters are shown here; #1 and #2, litter numbers. To the left of these lanes, calibration standards are shown. 10 to 1 μg of crude brain extract from a wild-type mouse was used as the standard. In the lane containing crude brain extract from homozygous mutants, a faint band corresponding to a molecular mass of 320 kD (**) was detected. The intensity of the signal was about 1% of that for the band of 200-kD KIF1A (*) from the brains of wild-type mice. This band was also detected with an anti–lac Z antibody (data not shown), suggesting that the band is a lac Z–KIF1A fusion protein. The concentrations of synaptic vesicle proteins (Synaptophysin and SV2) and most brain KIFs (KIF2, KIF3, KIF4) remained unchanged, while the KHC concentration was increased in the brains of the homozygous mutants.
Immunofluorescent Microscopy Demonstrated a Decrease of Synaptic Vesicle Protein Accumulation in the Synaptic Areas of KIF1A Mutant Mice
The amount of synaptic vesicle proteins in total brain homogenate does not directly reflect the function of motor proteins because it is possible that the total amount of synaptic vesicle proteins is unchanged from that of wild-type mice but that their localization in neurons is significantly affected. Our previous results on the function of KIF1A and the phenotype of unc104 mutant of C. elegans (Otsuka et al., 1991) suggest that the lack of KIF1A would decrease the transport of synaptic vesicle precursors in the axons. This would cause the decrease of synaptic vesicle proteins in the synaptic areas.
To test this possibility, we have measured the amount of synaptic vesicle proteins in synaptic areas using semiquantitative direct immunofluorescence microscopy. For the markers, we selected synaptophysin and SV2 as discussed in the previous section. Initially, we have anticipated from our previous result that synaptophysin, a KIF1A cargo protein, would decrease, while SV2, a non–KIF1A cargo protein, would remain unchanged.
However, as shown in Fig. 3, both synaptophysin and SV2 showed significant decrease. In wild-type and mutant mice, both synaptic vesicle proteins showed accumulation to numerous spots (arrows) outside the cell body (asterisks). Either with synaptophysin or with SV2, the density of these spots, their area, and their staining intensities were all significantly lower in the homozygous mutants than in the wild-type mice (summarized in Fig. 3 E). This supports the idea that the lack of KIF1A decreased the transport of synaptic vesicle proteins, which decreased the density of synapse and the number of synaptic vesicles. At the same time, this result suggests that both synaptophysin, a KIF1A cargo protein, and SV2, a non–KIF1A cargo protein, are equally affected.
Figure 3.
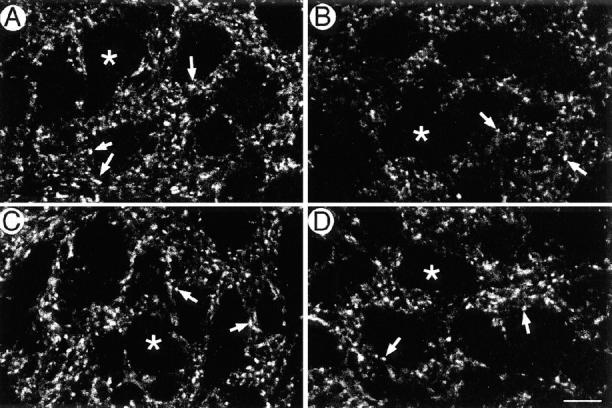

Immunohistochemical analysis of spinal anterior horn regions. (A–D) Semiquantitative direct immunofluorescence analysis of the localization of synaptic vesicle proteins in the spinal cord of wild-type mice (A and C) and homozygous mutants (B and D). High-magnification views of anterior horn of the T7 spinal cord stained with Cy5-labeled antisynaptophysin antibody (A and B) and Cy3.5-labeled anti-SV2 antibody (C and D). Some neuronal cell bodies are indicated with an asterisk, and some synaptic terminals are indicated with arrows. (E) Summary of the results of the quantitative analysis. Density, the density of the fluorescently labeled spots. Area, the mean area of the spots. Fluorescence, the upper tenth percentile value of the fluorescence intensity. Each value is shown in the format of mean ± SEM (n = 36 for wild-type mice, and 25 for mutant mice). Bar, 10 μm.
Decreased Densities of Synaptic Terminals and Synaptic Vesicles in the Terminals and Abnormal Clustering of Small Vesicles in the Nerve Cell Bodies of KIF1A Mutant Mice
Because the immunohistochemical data suggested some structural changes in the synaptic terminals, anterior horn regions of the spinal cords (thoracic level 7, T7) from the wild-type and homozygous littermates were examined by electron microscopy. In the wild-type mice, there was a high density of synaptic vesicles in the nerve terminals (Fig. 4, A and C). However, in the nerve terminals of the homozygous mutants, clear and small vesicles were reduced in number, and they were sparsely distributed in a large number of terminals (Fig. 4, B and D). However, no marked differences in the number of dense-cored vesicles or mitochondria in nerve terminals were observed between the wild-type and mutant mice. Results of morphometric analysis of two independent litters (Nos. 1 and 2) showed that the density of synaptic vesicles (Fig. 4 E) in the homozygous mutants was reduced over 40% compared with that of the wild-type littermates (P < 0.00001). KIF1A is believed to be an anterograde axonal motor (Okada et al., 1995). Therefore, a decrease in the number of synaptic vesicles in nerve terminals of homozygous mutants is likely to result from blockage of the anterograde transport of synaptic vesicle precursors by KIF1A in axons. To determine whether the deficiency in KIF1A in the mutant mice affected the number or area of nerve terminals, we measured the density and area of nerve terminals in the wild-type and mutant mice. The density of nerve terminals was decreased by >35% in the homozygous mutants, compared with the wild-type littermates (Fig. 4 F). However, no significant differences in presynaptic areas between the wild-type and homozygous mice were detected (data not shown). This observation indicates that KIF1A may play an essential role in the maintenance and function of nerve terminals. Overall, the results of the electron microscopic analysis suggested that the observed decrease in the number of synaptic vesicles and nerve terminals in the homozygous mutants may correlate with the reduced immunoreactivities of the spinal cord sections stained with antisynaptophysin and anti-SV2 antibodies as shown above.
Figure 4.
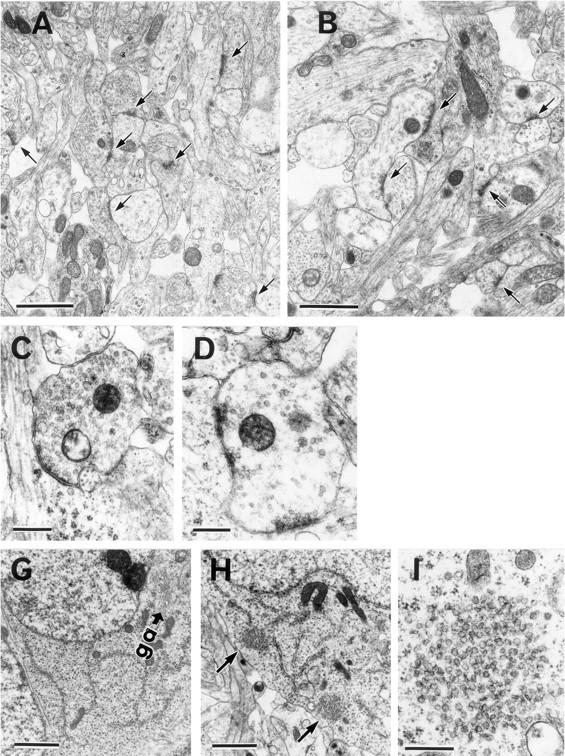

Electron microscopic analysis of spinal anterior horn regions. (A and B) Electron micrographs of cross sections of the anterior horn regions of the spinal cords (thoracic level 7, T7) of wild-type (A) and homozygous mutant (B) mice. The densities of nerve terminals and synaptic vesicles were decreased in the mutant mice. Arrows indicate nerve terminals. (C and D) Nerve terminals of wild-type (C) and homozygous mutant (D) mice. (E and F) Morphometric analysis of densities of synaptic vesicles (E) and nerve terminals (F) from two independent litters (#1, #2). Bar heights represent the mean ± SD (E) or the mean (F). T test comparisons of the density of synaptic vesicles between the wild-type and mutant mice reveal a statistically significant decrease in the density (*P < 0.00001) (E). (G and H) Neuronal cell bodies in the wild-type (G) and mutant mice (H). Neuronal cell bodies containing clusters of small vesicles were observed in the mutant mice (H) but not in the wild-type mice (G). Arrows, clusters of small vesicles (H). (I) A higher-magnification view of one of these clusters. ga, the Golgi apparatus. Bars: (A and B) 1.4 μm; (C and D) 400 nm; (G and H) 2 μm ; (I) 400 nm.
Very interestingly, in neuronal cell bodies accumulation of clusters of small and clear vesicles was observed in the homozygous mutants (Fig. 4, G–I). The abnormal clusters of vesicles were up to ∼1 μm in size distinct from vesicles located around the Golgi apparatus and mostly localized near the plasma membranes. Examination of more than 20 neurons in the anterior horn of both wild-type and mutant mice using serial sections showed that in all the mutant neurons examined (n = 23), up to three clusters were present in the cell bodies (three clusters, 4/23, 17%; two, 8/23, 35%; one, 11/23, 48%). However, no clusters were detected in the cell bodies of the wild-type neurons examined (n = 20). Abnormal clustering of vesicles was also observed in the pyramidal neurons of the hippocampus of the mutants (data not shown). For biogenesis of synaptic vesicles, it is proposed that proteins and membranes that are necessary for the formation of synaptic vesicles are sorted into several classes of vesicles in the cell body and transported as precursor forms to nerve terminals, where mature synaptic vesicles are assembled via exo/endocytosis (Bauerfeind and Huttner, 1993; Okada et al., 1995). The clustered vesicles observed in the cell bodies of the homozygous mutants may represent vesicles in an impaired state of vesicular transport.
Decreased Transport of Synaptic Vesicle Precursors in the Axons
These immunohistochemical and electron microscopic results support the idea that the lack of KIF1A decreased the transport of synaptic vesicle precursors in the axons. For its direct demonstration, we tried a sciatic nerve ligation experiment. Actively transported proteins accumulate profoundly near the ligated site, and the degree of accumulation decreases when the transport is inhibited (Hirokawa et al., 1990, 1991; Okada et al., 1995). For the markers of synaptic vesicle proteins, we first used synaptophysin and SV2. However, we could not obtain reproducible results even with wild-type mice, probably because of the immaturity of neurons in these mice.
We next tried synaptotagmin, another KIF1A cargo synaptic vesicle protein, and syntaxin 1A, a non–KIF1A cargo synaptic plasma membrane protein. As shown in Fig. 5, both proteins accumulate significantly at the proximal region of ligation in wild-type mice. In mutant mice, however, no accumulation was observed with synaptotagmin. On the contrary, syntaxin 1A still showed significant accumulation, though its degree was slightly lower than in wild-type mice. This indicates that the transport of synaptic vesicle precursors decreased in KIF1A knockout mice, but the transport of synaptic plasma membrane precursors is not affected so much. These results support our previous biochemical results that KIF1A transports the synaptic vesicle precursors, but not the synaptic plasma membrane precursors.
Figure 5.

Sciatic nerve ligation experiment. Sciatic nerves of wild-type (a–d) and KIF1A knockout (e–h) littermate mice were ligated for 3 h and then stained with antisynaptotagmin (a and e) or with anti–syntaxin 1A (c and g). Both synaptotagmin and syntaxin accumulate significantly in the proximal regions of the ligated site (arrows) in wild-type mice (a and c). In mutant mice, synaptotagmin does not accumulate significantly (e), while syntaxin shows significant accumulation (g). b, d, f, and h show the negative control with nonspecific mouse IgG. Sections shown in a–d and e–h are from same nerves, respectively. Bar, 100 μm.
Neuronal Degeneration and Death in the Central Nervous System of KIF1A Mutant Mice
KIF1A homozygous mutants died shortly after birth displaying several neurological symptoms, indicating that KIF1A plays a critical role in the function of the nervous system around the time of birth. KIF1A was shown to be specifically expressed in neurons (Okada et al., 1995). X-gal staining of adult brains and spinal cords of heterozygous mice for detection of KIF1A-lacZ fusion expression also confirmed the widespread expression of KIF1A in neuronal tissues (data not shown). Thus, neuropathological examinations of neuronal tissues of the mutants were performed using light and electron microscopy. Extensive neuronal degeneration was found in the cerebrum, rhinencephalon, and amygdaloid areas of the mutants (Fig. 6 B). The degenerative neurons were characterized by severe swelling of the cytoplasm and nuclei (Fig. 6 D). In some neuronal cells, the nuclei became irregular in shape. Moreover, a large number of vacuoles were seen surrounding the degenerative neurons. Ultrastructurally, these vacuoles were bounded by a plasma membrane and contained a few degenerative membranous organelles. These could be extended dendrites at the progressive stage of degeneration (data not shown). But no degenerative lesions were detected in the wild-type mice (Fig. 6, A and C). Occasional degeneration of hippocampal pyramidal neurons was also observed in the mutants (data not shown).
Figure 6.
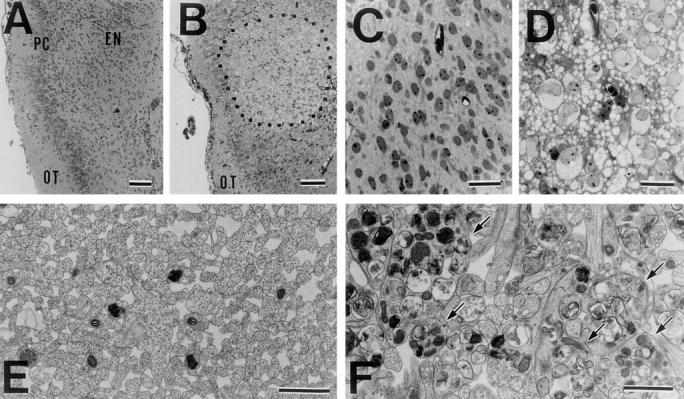
Neuropathological analysis. (A–D) Toluidine blue–stained cross sections of rhinencephalon areas of wild-type (A and C) and homozygous mutant mice (B and D). The focal lesion (dotted region) in the piriform cortex and endopiriform nucleus shows severe degeneration of neuronal cells and a large number of vacuoles surrounding the degenerative cells in the mutant mice. A higher-magnification view of the degenerative lesion (D). OT, lateral olfactory tract; PC, piriform cortex; EN, endopiriform nucleus. (E and F) Electron micrographs of cross sections of the fimbrias of hippocampi of wild-type (E) and mutant (F) mice. Massive degeneration of many axons (arrows) occurred in the mutant mice. The axons are very dilated and filled with membranous organelles. Bars: (A and B) 100 μm ; (C and D) 25 μm; (E and F) 1 μm.
Moreover, electron microscopic analysis revealed massive axonal degeneration in several areas such as the hippocampal fimbria, through which numerous axons of the hippocampal pyramidal neurons pass, and the lateral columns of the spinal cords in the homozygous mutants. In hippocampal fimbrias of the mutant, many axons were very dilated and filled with abnormally large autophagic membranous organelles (Fig. 6, E and F). Axonal degeneration observed in the mutants could be due to secondary changes in neurons of the mutants. These results indicate that mice deficient in KIF1A exhibit multiple neuropathological abnormalities in the nervous system and suggest that KIF1A plays an important role in the function and survival of neurons.
Massive Degeneration and Death of Cultured Hippocampal Neurons of KIF1A Mutant Mice
Because we observed massive neuronal degeneration in KIF1A-deficient homozygous mutants, we decided to examine the degenerative process in vitro in more detail. To characterize the abnormalities of developing neurons of mutants, cultured hippocampal neurons obtained from mouse embryos were analyzed. The axons and dendrites that develop in culture form synapses with one another and exhibit a normal neuronal polarity (Barlet and Banker, 1984; Dotti et al., 1988). Within the first 6 d in culture, the morphology and viability of wild-type cells were indistinguishable from those of mutant cells (Fig. 7, A and B). However, later than 7–8 d in culture, degeneration of mutant cells, as evidenced by cell body condensation or neurite fragmentation, was observed (Fig. 7 D), and the number of viable mutant cells, in comparison to wild-type cells (Fig. 7 C), decreased (Fig. 7 I). Finally, after 13 d in culture (D13), more than 90% of the mutant cells were dead (Fig. 7, F and I), while ∼74% of the wild-type cells were viable (Fig. 7, E and I).
Figure 7.
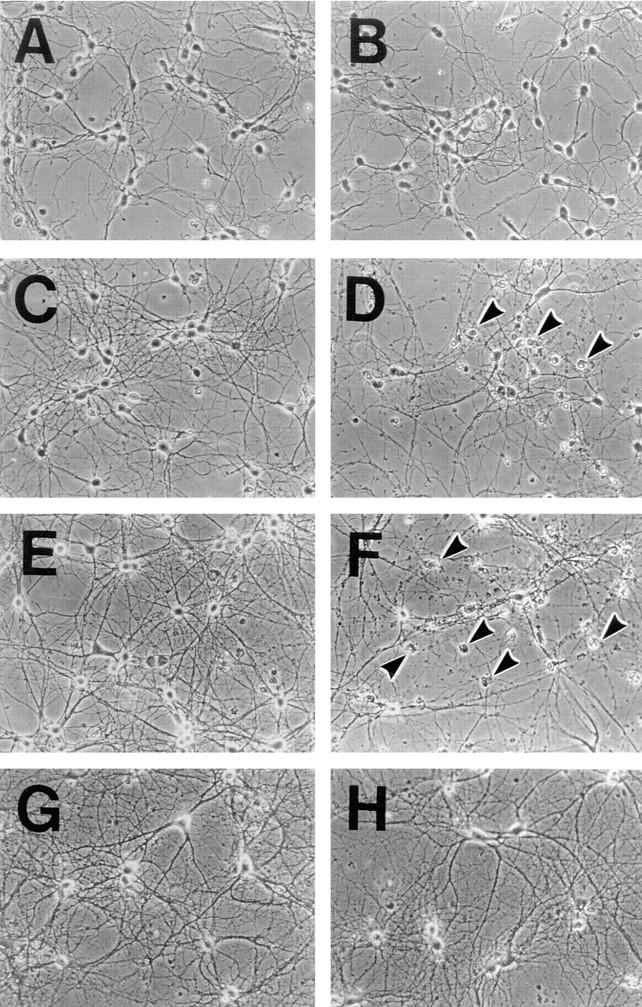

Analysis of cultured hippocampal neurons. (A–H) Phase contrast views of cultured hippocampal neurons from wild-type (A, C, E, and G) and homozygous mutant (B, D, F, and H) mice at 4 (A and B), 8 (C and D), 13 (E and F), and 18 d (G and H) in culture. Arrowheads indicate dying cells (D and F). Cultured neurons exposed to a low concentration of glutamate (15 μM) (G and H). (I) Survival curve for cultured hippocampal neurons from the wild-type (+/+) and mutant (−/−) mice. The exposure of the mutant cells to the low concentration of glutamate greatly promoted their survival (+/+Glu, −/−Glu). Each point represents the mean ± SD percentage of the cell numbers compared with that for each culture at D4. Inset shows the results of immunoblot analysis for determination of the level of KIF1A expression in the wild-type neurons at D4, 8, 10, and 13. (J) Coculture with wild-type cells prevented the mutant cells from dying. Equal numbers of the mutant and wild-type cells (prepared from embryos of wild-type BALB/c mice) were cocultured. Each point represents the mean ± SD percentage of the total cell numbers. WT, wild-type neurons derived from BALB/c mice. Inset shows the percentage of mutant cells at D18. To distinguish mutant cells from wild-type cells, we performed X-gal staining. We counted ∼2,000 β-gal–positive (homozygous or heterozygous mutant) cells and β-gal–negative (wild-type) cells. #1 and #2, litter numbers. Note that the numbers of homozygous cells and wild-type cells were almost equal.
To determine the level of expression of KIF1A in developing cultured neurons, immunoblot analyses were performed using extract from wild-type cells ranging from D4 to D13 (Fig. 7 I). The cells collected on D4 expressed a small amount of KIF1A, but the level of expression increased markedly in cells through D8, 10, and 13. Similar results were obtained from the analyses of cultured neurons derived from heterozygous embryos using X-gal staining (data not shown). Cultured hippocampal neurons developed well-differentiated axons and dendrites by the end of the first week, and then neurons will establish the maturation (Barlet and Banker, 1984; Dotti et al., 1988). Because no abnormalities of the mutant cells within the first 6 d in culture were observed and because only a low level of KIF1A expression in wild-type cells on D4 was detected, KIF1A may not be required in the early stages of development. However, when the cultured neurons matured, the level of KIF1A expression increased in the wild-type cells, and in contrast, the mutant cells had begun to degenerate. These observations indicate that KIF1A plays an essential role in mature neurons. Our results indicate that some molecules transported by KIF1A are indispensable for the viability and maintenance of mature neurons.
Afferent Neuronal Stimulation Prevented Neuronal Cell Death in Cultures of KIF1A Mutants
The formation of successful contacts between a presynaptic neurons and its target neuron is important for the survival of developing neurons. Removal of afferent input or pharmacological blockade of synaptic transmission can inhibit the survival of developing neurons (Lipton, 1986; Furber et al., 1987; Franklin and Johnson, 1992). In KIF1A mutant mice, considerable decrease in both densities of synaptic vesicles and synapses in vivo and neuronal degeneration and cell death both in vivo and in cultures suggested the cell death might be caused by a defect in synaptic function. To examine this possibility, we performed the coculture analyses of mutant and wild-type neurons. Interestingly, coculture of equal numbers of mutant and wild-type cells prevented the death of mutant cells through day 18 (Fig. 7 J). X-gal staining analysis of cocultured cells revealed that the numbers of homozygous mutant and wild-type cells were almost equal after 18 d in culture. These results suggest that afferent neuronal inputs or contacts from wild-type cells might be required for the survival of the mutant cells.
In addition, exposure of the mutant cells to the low concentration of glutamate (15 μM) greatly promoted the survival of the mutant cells, while the wild-type cells did not exhibit any change under the same culture conditions (Fig. 7, G–I). Cerebellar granule neurons maintained under nondepolarizing culture conditions, under which cell death is induced, are prevented from dying by exposure to depolarizing concentrations of K+ or a low concentration of glutamate (Yan et al., 1994). It is possible that the induction of neuronal depolarization by glutamate promotes survival in the mutant nerve cells.
These results in cultured neurons suggested that a lack of KIF1A may cause an impairment of afferent neuronal inputs, probably of synaptic transmission, leading to neuronal cell death.
Discussion
KIF1A knockout mice died within 24 h after birth. Before death, they exhibited motor and sensory disturbances. They also showed marked decrease in the transport of synaptic vesicle precursors but not in the transport of synaptic plasma membrane precursors. Consequently, the densities of the synaptic terminals and the numbers of synaptic vesicles decreased significantly. At the same time, the knockout mice showed neuronal degeneration, which was reproduced by hippocampal neuron culture in vitro. This in vitro experiment indicated the correlation between the time course of the neuronal death in mutant mice and the time course of KIF1A expression in wild-type mice. These results demonstrate that KIF1A plays essential roles in the function, maintenance, and survival of neurons.
The decrease in the transport of synaptic vesicle precursors in the axons is consistent with our previous study (Okada et al., 1995). We have previously demonstrated with a mature peripheral nerve preparation that KIF1A associates with a class of synaptic vesicle precursors that contains synaptophysin, synaptotagmin, and Rab3A, but not SV2. KIF1A does not associate with synaptic plasma membrane precursors that contain syntaxin 1A, either. From these results, we have concluded that KIF1A transports a class of synaptic vesicle precursors but not synaptic plasma membrane precursors. This conclusion was clearly demonstrated by the sciatic nerve ligation experiment, which indicates the decreased transport of synaptotagmin and the almost normal transport of syntaxin 1A. Unfortunately, we were unable to assay the transport of SV2, a non–KIF1A-associated synaptic vesicle protein, mostly because of the technical problems. Therefore, we cannot definitely answer the question of whether SV2 transport is affected in the KIF1A knockout mice at present.
An interesting finding is that synaptic vesicles and synaptic vesicle proteins accumulate in the synapse, even when the transport of synaptic vesicle precursors are severely affected by the lack of KIF1A. One possibility is that other KIFs, unidentified KIF(s), or slightly increased conventional kinesin might partially compensate for the function of KIF1A. Another possibility is that synaptic vesicle precursors are transported by KIFs other than KIF1A in immature neurons. As shown in Fig. 7 I, inset, the expression of KIF1A increases after 8 days in culture, much later than the synapse formation and the accumulation of synaptic vesicle proteins (Fletcher et al., 1994). This suggests that synaptic vesicle precursors are transported by KIF(s) other than KIF1A in immature neurons and that the transport machinery switches to KIF1A after maturation. One candidate for this juvenile-type motor is KIF4, which is dominantly expressed in juvenile neurons and transports vesicles to the end of growing neurites (Sekine et al., 1994).
The second interesting finding is the accumulation of small clear vesicles in the cell body that are connected by fuzzy structures. These vesicles could be either precursors of synaptic vesicles that were not transported to the axon or ectopic synaptic vesicles formed most likely through ectopic exocytosis, endocytosis, and recycling underneath the plasma membrane of the cell body because precursors were not transported to the axon. In the nerve cell bodies of C. elegans unc-104 mutants, accumulation of clustered vesicles is observed, but the vesicles have denser cores and appear to be different from the vesicles observed in KIF1A mutants, although KIF1A could be the mammalian homologue of UNC-104 (Hall and Hedgecock, 1991). The reason for this difference is not clear but it may be due to differences in the types of neurons.
The third interesting finding is the neuronal death. In KIF1A mutants, axonal degeneration and degeneration of neuronal cell bodies occurred in areas of the central nervous system such as the rhinencephalon, amygdaloid area, and hippocampus. The results of in vitro culture of hippocampal neurons clearly indicated that neuronal cell death is correlated with the level of KIF1A expression in wild-type mice. In this culture system, hippocampal neurons mature and establish mature synapses after ∼8 d in culture, at which time the level of KIF1A expression in wild-type mice increases and neuronal cell death commences in KIF1A mutants. These observations clearly indicate that KIF1A is essential for the function and survival of mature neurons. Although paralyzed movement and defect of an anterograde translocation of synaptic vesicles that were observed in unc-104 mutants resemble the phenotypes of KIF1A mutant mice, the neuronal degeneration and death mentioned above were not reported for unc-104 mutants. The difference in viabilities of neurons between unc-104 and KIF1A mutants could correlate with differences in their expressions during development.
For understanding of the function of KIF1A in vivo it is important to determine the cause of neuronal cell death. One possibility is that based on the decrease in transport of precursors of mature synaptic vesicles in the KIF1A mutants and the fact that the cargoes transported by KIF1A contain some synaptic vesicle proteins involved in neurotransmitter release such as synaptotagmin and Rab3A (Okada et al., 1995), the neurotransmission at the nerve terminals is considerably impaired in the KIF1A mutants so that the electrical activities in the mutant neurons is significantly reduced, resulting in death of these neurons. In fact, coculture with wild-type cells and exposure to a low concentration of glutamate rescued the mutant cells from death in culture, consistent with this possibility.
On the other hand, the knockout mice lacking of synaptotagmin (Geppert et al., 1994a ), synaptophysin (McMahon et al., 1996), or Rab3A (Geppert et al., 1994b ), which cargoes transported by KIF1A were suggested to contain, and mice deficient in synapsin I or II (Rosahl et al., 1995; Takei et al., 1995), in which a marked decrease in synaptic vesicle density in the nerve terminals was observed, have been generated and reported. However, these mutant mice did not exhibit such neuronal degeneration and cell death as observed in KIF1A mutant mice. Thus, only the explanation mentioned above may not be enough to make clear about the cause of the neuronal cell death in KIF1A mutant mice. Another possibility is that KIF1A may have an additional unknown function. It may be conceivable that synaptic vesicle precursors transported by KIF1A may also contain some molecules that are essential for neuronal survival or neurotransmitter action, such as ion channel proteins, neurotrophic factors, or neurotrophic factor receptors.
Axonal degeneration similar to that observed in KIF1A mutants has also been reported for several neurodegenerative diseases such as senile dementia (Adams and Duchen, 1992). In some neurodegenerative diseases, neuronal cell death caused by defects in the transport of synaptic vesicle precursors by KIF1A may be involved. Future studies will elucidate the mechanism of neuronal degeneration and death in KIF1A mutant mice.
Acknowledgments
We thank Drs. G.S. Bloom, J.M. Scholey, K. Obata, and K.M. Buckley for kindly providing the antibodies. We are grateful to Ms. H. Sato and Mrs. H. Fukuda for technical assistance and members of the Hirokawa laboratory for their help and discussions.
Abbreviations used in this paper
- DT-A
diphtheria toxin A
- ES
embryonic stem
- KHC
kinesin heavy chain
- KIFs
kinesin superfamily proteins
Footnotes
The present study is supported by a grant for the Center of Excellence (COE) Research from the Ministry of Education, Science, and Culture of Japan to N. Hirokawa.
References
- Adams, J.H., and L.W. Duchen. 1992. Greenfield's Neuropathology. Edward Arnold, London. 1,557 pp.
- Aizawa H, Sekine Y, Takemura R, Zhang Z, Nangaku M, Hirokawa N. Kinesin family in murine central nervous system. J Cell Biol. 1992;119:1287–1296. doi: 10.1083/jcb.119.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlet WP, Banker GA. An electron microscopic study of the development of axons and dendrites of hippocampal neurons in culture. II. Synaptic relationships. J Neurosci. 1984;4:1954–1965. doi: 10.1523/JNEUROSCI.04-08-01954.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerfeind R, Huttner WB. Biogenesis of constitutive secretory vesicles, secretory granules and synaptic vesicles. Curr Opin Cell Biol. 1993;5:628–635. doi: 10.1016/0955-0674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- Brady ST. A novel brain ATPase with properties expected for the fast axonal transport motor. Nature. 1985;317:73–75. doi: 10.1038/317073a0. [DOI] [PubMed] [Google Scholar]
- Buckley K, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neuronal and endocrine cells. J Cell Biol. 1985;100:1284–1294. doi: 10.1083/jcb.100.4.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin JL, Johnson EM. Suppression of programmed neuronal cell death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992;15:501–508. doi: 10.1016/0166-2236(92)90103-f. [DOI] [PubMed] [Google Scholar]
- Fletcher TL, De Camilli P, Banker G. Synaptogenesis in hippocampal cultures: evidence indicating that axons and dendrites become competent to form synapses at different stages of neuronal development. J Neurosci. 1994;14:695–706. doi: 10.1523/JNEUROSCI.14-11-06695.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furber S, Oppenheim RW, Prevette D. Naturally-occurring neuron death in the ciliary ganglion of the chick embryo following removal of preganglionic input: evidence for the role of afferents in ganglion cell survival. J Neurosci. 1987;7:1817–1832. doi: 10.1523/JNEUROSCI.07-06-01816.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC. Synaptotagmin I: a major Ca2+sensor for transmitter release at a central synapse. Cell. 1994a;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Sudhof TC. The role of Rab3A in neurotransmitter release. Nature. 1994b;369:493–497. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- Gossler, A., and J. Zachgo. 1993. Gene and enhancer trap screens in ES chimeras. In Gene Targeting: A Practical Approach. A.L. Joyner, editors. IRL Press at Oxford University Press, New York. 207–213.
- Grafstein B, Forman DS. Intracellular transport in neurons. Physiol Rev. 1980;60:1167–1283. doi: 10.1152/physrev.1980.60.4.1167. [DOI] [PubMed] [Google Scholar]
- Gudkov AV, Kazarov AR, Thimmapaya R, Axenovich SA, Mazo IA, Roninson IB. Cloning mammalian genes by expression selection of genetic suppressor elements: association of kinesin with drug resistance and cell immortalization. Proc Natl Acad Sci USA. 1994;91:3744–3748. doi: 10.1073/pnas.91.9.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. . Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Axonal transport and the cytoskeleton. Curr Opin Neurobiol. 1993;3:724–731. doi: 10.1016/0959-4388(93)90144-n. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Organelle transport along microtubules—the role of KIFs. Trends Cell Biol. 1996;6:135–141. doi: 10.1016/0962-8924(96)10003-9. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Yoshida T, Sato-Yoshitake R, Kawashima T. Brain dynein localizes on both anterogradely and retrogradely transported membranous organelles. J Cell Biol. 1990;111:1027–1037. doi: 10.1083/jcb.111.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Kobayashi N, Phisfer KK, Bloom GS, Brady ST. Kinesin associates with anterogradely transported membranous organelles in vivo. J Cell Biol. 1991;114:295–302. doi: 10.1083/jcb.114.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingold AL, Cohn SA, Scholey JM. Inhibition of kinesin-driven microtubule motility by monoclonal antibodies to kinesin heavy chains. J Cell Biol. 1988;107:2657–2667. doi: 10.1083/jcb.107.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K. Sequential analysis of twenty mouse brain cDNA clones selected by specific expression patterns. Eur J Neurosci. 1991;2:704–711. doi: 10.1111/j.1460-9568.1990.tb00460.x. [DOI] [PubMed] [Google Scholar]
- Kondo S, Sato-Yoshitake R, Noda Y, Aizawa H, Nakata T, Matsuura Y, Hirokawa N. KIF3A is a new microtubule-based anterograde motor in the nerve axon. J Cell Biol. 1994;125:1095–1107. doi: 10.1083/jcb.125.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Bestor T, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Blockade of electrical activity promotes the death of mammalian retinal ganglion cells in culture. Proc Natl Acad Sci USA. 1986;83:9774–9778. doi: 10.1073/pnas.83.24.9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye RJ, Porter ME, Scholey JM, McIntosh JR. Identification of a microtubule-based cytoplasmic motor in the nematode C. elegans. . Cell. 1987;51:309–318. doi: 10.1016/0092-8674(87)90157-7. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Bolshakov VY, Janz R, Hammer RE, Siegelbaum SA, Sudhof TC. Synaptophysin, a major synaptic vesicle protein, is not essential for neurotransmitter release. Proc Natl Acad Sci USA. 1996;93:4760–4764. doi: 10.1073/pnas.93.10.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda Y, Sato-Yoshitake R, Kondo S, Nangaku M, Hirokawa N. KIF2 is a new microtubule-based anterograde motor that transports membranous organelles distinct from those carried by kinesin heavy chain or KIF3A/B. J Cell Biol. 1995;129:157–167. doi: 10.1083/jcb.129.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Nishiye H, Fujita SC, Shirao T, Inoue H, Uchizono K. Identification of a synaptic vesicle-specific 38,000 dalton protein by monoclonal antibody. Brain Res. 1986;375:37–48. doi: 10.1016/0006-8993(86)90956-x. [DOI] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Otsuka AJ, Jeyaprakash A, Garcia AJ, Tang LZ, Fisk G, Hartshorne T, Franco R, Born T. The C. elegans unc-104gene encodes a putative kinesin heavy chain-like protein. Neuron. 1991;6:113–122. doi: 10.1016/0896-6273(91)90126-k. [DOI] [PubMed] [Google Scholar]
- Paschal BM, Shpetner HS, Vallee RB. MAP1C is a microtubule-activated ATPase which translocates microtubules in vitro and has dynein-like properties. J Cell Biol. 1987;105:1273–1282. doi: 10.1083/jcb.105.3.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister KK, Wagner MC, Stenoien DL, Brady ST, Bloom GS. Monoclonal antibodies to kinesin heavy and light chains stain vesicle-like structures, but not microtubules, in cultured cells. J Cell Biol. 1989;108:1453–1463. doi: 10.1083/jcb.108.4.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Sudhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A laboratory Manual. Second edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sekine Y, Okada Y, Kondo S, Aizawa H, Takemura R, Hirokawa N. A novel microtubule-based motor protein (KIF4) for organelle transports, whose expression is regulated developmentally. J Cell Biol. 1994;127:187–202. doi: 10.1083/jcb.127.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Harada A, Takeda S, Kobayashi K, Terada S, Noda T, Takahashi T, Hirokawa N. Synapsin I deficiency results in the structural change in the presynaptic terminals in the murine nervous system. J Cell Biol. 1995;131:1789–1800. doi: 10.1083/jcb.131.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheets MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Nada S, Watanabe N, Tamemoto H, Kohmura N, Ikawa Y, Aizawa S. A novel negative selection for homologous recombinants using diphtheria toxin A fragment. Anal Biochem. 1993;214:77–86. doi: 10.1006/abio.1993.1459. [DOI] [PubMed] [Google Scholar]
- Yan G, Ni B, Weller M, Wood KA, Paul SM. Depolarization or glutamate receptor activation blocks apoptotic cell death of cultured cerebellar neurons. Brain Res. 1994;656:43–51. doi: 10.1016/0006-8993(94)91364-1. [DOI] [PubMed] [Google Scholar]