

Abstract
Kinesin is a heterotetramer composed of two 115-kD heavy chains and two 58-kD light chains. The microtubule motor activity of kinesin is performed by the heavy chains, but the functions of the light chains are poorly understood. Mutations were generated in the Drosophila gene Kinesin light chain (Klc), and the phenotypic consequences of loss of Klc function were analyzed at the behavioral and cellular levels. Loss of Klc function results in progressive lethargy, crawling defects, and paralysis followed by death at the end of the second larval instar. Klc mutant axons contain large aggregates of membranous organelles in segmental nerve axons. These aggregates, or organelle jams (Hurd, D.D., and W.M. Saxton. 1996. Genetics. 144: 1075–1085), contain synaptic vesicle precursors as well as organelles that may be transported by kinesin, kinesin-like protein 68D, and cytoplasmic dynein, thus providing evidence that the loss of Klc function blocks multiple pathways of axonal transport. The similarity of the Klc and Khc (Saxton et al. 1991. Cell 64:1093–1102; Hurd, D.D., and W.M. Saxton. 1996. Genetics 144: 1075–1085) mutant phenotypes indicates that KLC is essential for kinesin function, perhaps by tethering KHC to intracellular cargos or by activating the kinesin motor.
Intracellular transport requires the action of molecular motors that bind cargo and generate movement coupled to ATP hydrolysis along cytoskeletal filaments (Gibbons et al., 1994; Bloom and Endow, 1995; Mooseker and Cheney, 1995). One type of motor is exemplified by kinesin (Vale et al., 1985; Brady, 1985), which plays an integral role in intracellular transport along microtubules in many cell types (Goldstein, 1993; Vale and Fletterick, 1997). Native kinesin is a heterotetramer composed of two copies each of two polypeptide chains: kinesin heavy chain (KHC)1 and kinesin light chain (KLC; Bloom et al., 1988; Kuznetsov et al., 1988; Johnson et al., 1990). KHC contains the motor domain (Penningroth et al., 1987; Bloom et al., 1988; Hirokawa et al., 1989; Scholey et al., 1989; Yang et al., 1989), which is sufficient to generate ATP-dependent forces along microtubules (Kuznetsov et al., 1989; Yang et al., 1990), whereas KLC is located in the non-motor tail domain of kinesin (Hirokawa et al., 1989; Gauger and Goldstein, 1990). Because KLC is located in the presumptive cargo-binding domain of kinesin, it has been suggested to play some role in mediating the interactions of the kinesin motor with its intended cargo. To date, however, little evidence to support this view has been obtained.
Secondary structure analyses predict that KLC participates in diverse protein–protein interactions. The amino terminal region of KLC is predicted to form an alpha-helical coiled coil that links KLC to KHC (Cyr et al., 1991; Gauger and Goldstein, 1993). The carboxy-terminal region of KLC is largely made up of six repeated units that are predicted to form tetratrico peptide repeat (TPR) domains (Gindhart and Goldstein, 1996) that are protein– protein interaction motifs identified in a diverse group of proteins (Lamb et al., 1995; Sikorski et al., 1990). Other proteins to which KLC might bind are not known, but a potential kinesin-binding protein is kinectin, the proposed kinesin receptor (Toyoshima et al., 1992; Futterer et al., 1995; Kumar et al., 1995; Yu et al., 1995).
Experimental evidence suggests that KLC may play one of two roles. One possibility is that KLC may be a positive factor necessary for kinesin–cargo binding (Cyr et al., 1991; Gauger and Goldstein, 1993; Stenoien and Brady, 1997). Alternatively, KLC might play a role in the negative regulation of kinesin activity, such that kinesin is inactive in the presence of KLC, but active in its absence or when its function is attenuated by cargo binding (Hackney et al., 1991, 1992; Matthies et al., 1993; Jiang and Sheetz, 1995). One recent attempt to test these hypotheses supported the view that KLC might be needed for cargo attachment of kinesin (Stenoien and Brady, 1997); however, additional data are clearly needed. To gain a better understanding of KLC function, we generated mutations in the Drosophila gene encoding KLC, studied the phenotypic consequences of loss of KLC function at the organismal and cellular level, and tested the positive and negative hypotheses of KLC function. We demonstrate that KLC is essential for axonal transport in Drosophila larva, and that locomotion defects associated with the loss of KLC function are a consequence of the disruption of multiple axonal transport pathways.
Materials and Methods
Identification of Klc Mutants
Identification of the P[lacW] (Bier et al., 1989) insertion 59A in Klc intron 1 was described previously (Desai et al., 1996). The location of P[lacW] within Klc was determined by sequencing genomic DNA flanking the insertion site, examining the size of PCR-generated DNA fragments using combinations of primers hybridizing to P[lacW] and KLC cDNA sequences to amplify 59A genomic DNA template, and by fine-structure restriction fragment length polymorphism mapping of 59A genomic DNA using cloned KLC genomic sequences as probes. The lethal P-element insertion P[lacW]l(3)A5-3-42 (Hartenstein and Jan, 1992) used for insertional mutagenesis of Klc was still present on the 59A mutant chromosome. To remove P[lacW]l(3)A5-3-42 from this chromosome, w; 59A/TM3 males were crossed to w; Dr/TMS, Sb Δ2-3 females (Robertson et al., 1988). Dysgenic F1 female progeny were crossed to w; TM3, Sb/TM6B Hu Tb males, and flies harboring the P[lacW] element in Klc, but not P[lacW]l(3)A5-3-42, were detected by alteration of the patterned 59A-specific eye color. Putative l(3)A5-3-42 revertants were crossed to P[lacW]l(3)A5-3-42/TM3, Sb to determine whether the P[lacW] element present in the l(3)A5-3-42 complementation group had excised precisely. A 59A derivative, Klc 1, lacks l(3)A5-3-42, but contains the P[lacW] insertion in Klc.
Small deletions of Klc as well as Klc revertants were generated by remobilizing the Klc 1 insertion, then screening for loss of the w + phenotype encoded by P[lacW]. These flies were backcrossed to Klc 1/TM6B for complementation tests. Deletion breakpoints were identified by Southern hybridization of genomic DNA from deletion mutants to cloned fragments of Klc genomic DNA and cDNA. Approximately 70% of the Klc 1 excision events tested (74/104) failed to complement Klc 1.
Transgenic Rescue Constructs
The transgenic construct GEN-KLC is composed of a 12.4-kb EcoRI-NotI fragment from KLC cosmid 8.1 (Fig. 1) subcloned into pCaSPeR4 (Pirrotta, 1988) cut with EcoRI and NotI. This DNA fragment contains the Klc coding region, ∼5 kb of 5′ regulatory sequences, and 3 kb 3′ of Klc (see Fig. 1), but not Ptp69D. MYC-KLC was constructed by cutting pBS13a (Gauger and Goldstein, 1993) with DrdI and EcoRI, and then ligating a double-stranded linker DNA to the DrdI site at the 5′ end of the KLC cDNA (sequence of primer 1:5′AATTCCATGACGCAA3′; sequence of primer 2:3′GGTACTGCG5′). The linker DNA encodes amino acids 1–3 of KLC, and provides an EcoRI restriction site in the same translation frame as the EcoRI site in pWUM (Heck et al., 1993). The sequence tag amino-terminal of the KLC coding region in MYC-KLC is MEQKLISEEDLNS. This sequence is recognized by the anti-MYC antibody 9E10 (Evan et al., 1985). Fusion protein transcription is controlled by the Drosophila polyubiquitin promoter pUP2 (Lee et al., 1988), which ensures high-level expression in all cells. Rescue constructs were injected into y w 1118 embryos with helper plasmid pπ25.7wc (Rubin and Spradling, 1982) using conventional techniques (Robertson et al., 1988).
Figure 1.
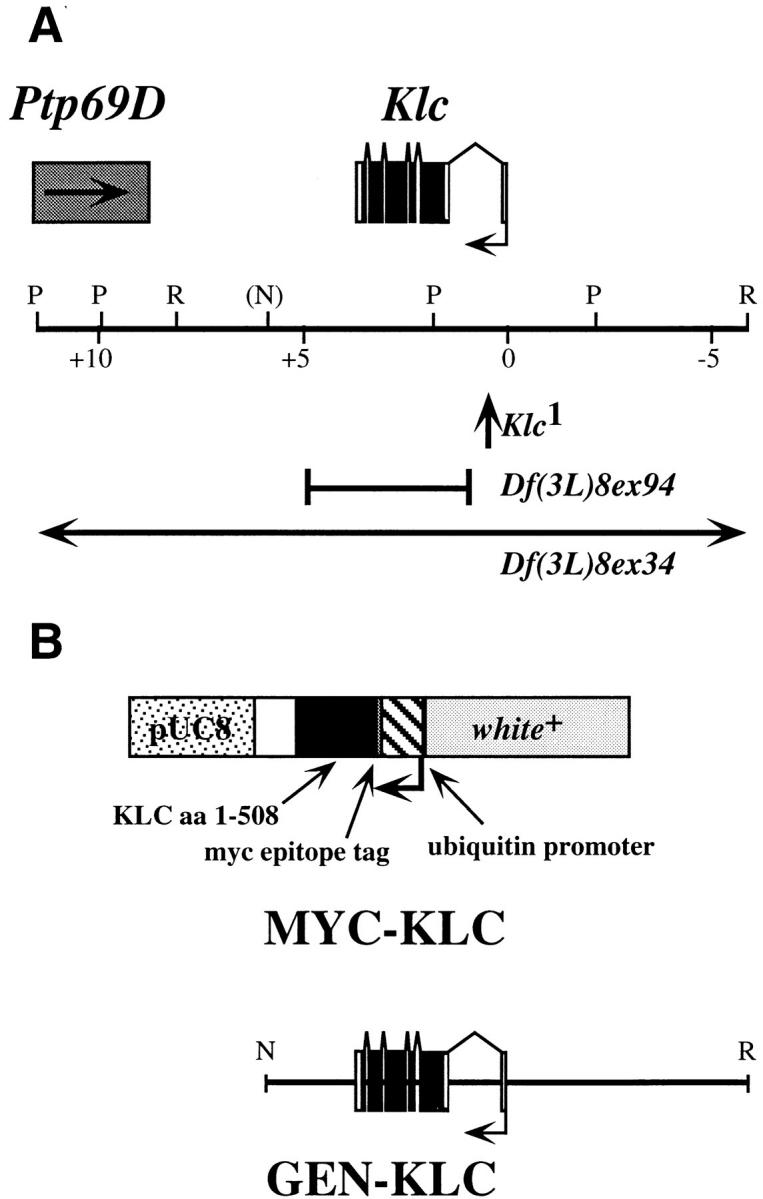
(A) Map of Klc genomic interval and location of Klc mutants used in this analysis. The Klc transcription unit is composed of six exons, and is transcribed from right to left in this diagram. Ptp69D (Desai et al., 1996) is located 3′ of Klc, and is transcribed toward Klc. Map coordinates are shown in kb. Restriction sites are shown as follows: P, PstI; R, EcoRI; N, the NotI site in cosmid 8.1 used for subcloning GEN-KLC (see Materials and Methods). The location of P[lacW] in Klc intron 1 is shown as an upward pointing arrow. Sequences deleted by the lesions Df(3L)8ex94 and Df(3L)8ex34 are shown as solid lines. The endpoints of Df(3L)8ex94 are shown; the endpoints of Df(3L)8ex34 are unknown. (B) Maps of KLC transgenic constructs MYC-KLC and GEN-KLC. The MYC-KLC transgene is composed of KLC amino acids 1–508 fused to a 13–amino acid epitope tag recognized by the anti-MYC monoclonal antibody 9E10 (Evan et al., 1985). This fusion protein is under the transcriptional control of the Drosophila polyubiquitin promoter, which ensures high levels of transgene expression in all tissues (Lee et al., 1988). The map position of the NotI-EcoRI genomic DNA fragment included in GEN-KLC is also shown.
Lethal Phase Analysis
To determine the lethal phase of Klc mutant and control larvae, flies of the appropriate genotypes were placed in 8-oz. egg collection chambers. Egg collections (0–22h) were made on Karo-agar (9.5% Karo corn syrup; Best Foods, Englewood Cliffs, NJ, 3.4% agar) plates supplemented with yeast paste. All mutants were balanced by the chromosome TM6B, Hu Tb, which permits discrimination of short Tubby heterozygous larvae from longer non-Tubby larvae. 2 d after egg laying, mutant and control second instar larvae were placed in groups of 50 onto 60-mm Karo-agar plates supplemented with yeast paste. 2 d later, the numbers of surviving and dead larvae for each genotype were noted, and survivors were placed onto fresh plates to limit bacterial contamination. This procedure was repeated until pupation. Larvae were kept in a humid chamber at 25°C.
Antisera Production and Immunoblotting
Polyclonal rabbit antisera were generated against the peptide sequence CLTRAHEKEFGK (KLC-LG3), corresponding to amino acids 381–390 of Drosophila KLC (Gauger and Goldstein, 1993). This region is highly conserved among KLCs cloned from diverse species (Cyr et al., 1991; Gauger and Goldstein, 1993; Wedaman et al., 1993; Beushausen et al., 1993; Cabeza et al., 1993; Fan and Amos, 1994; Chernajovsky et al., 1996). The peptide was linked to BSA or keyhole limpet hemocyanin (KLH). Antisera production was provided by BAbCO (Richmond, CA). Affinity purification of antisera KLC-LG3-BSA and KLC-LG3-KLH was performed by linking the NH2-terminal cysteine of KLC-LG3 to a Sulfo-Link™ column (Pierce Chemical Co., Rockford, IL), and then purifying KLC-LG3-specific antisera according to the manufacturer's recommendations. Working dilutions of KLC-LG3 antisera are 1:100 for immunoblotting and immunohistochemistry (Fig. 2 c).
Figure 2.

(A–C). Klc mutant larvae have locomotion defects that are rescued by GEN-KLC. Anterior is to the left, and the dorsal surface of the larva is shown. (A) Wild-type and Df(3L)8ex94/+ (shown) larvae crawl using peristaltic waves of muscle contraction. Most if not all of the ventral surface remains on the medium. (B) Klc mutant larvae of the genotype Df(3L)8ex94/Klc 1 (shown) and other hypomorphic Klc mutant combinations flip their posterior end off the surface during crawling. This phenotype is quite similar to the tail flipping phenotype observed in certain Khc mutant combinations (Hurd and Saxton, 1996). (C) GEN-KLC rescues the tail-flipping phenotype. GEN-KLC; Df(3L)8ex94/Klc 1 larvae exhibit normal larval crawling behavior (compare wild-type [A] to [C] rescued larvae). The MYC-KLC transgene also rescues the Klc-dependent tail flipping phenotype (not shown). (D) Characterization of KLC antisera at different Drosophila life stages. Total protein was extracted from wild-type individuals at the following life stages: 0–6 h embryos (lane 1); third instar larvae (lane 2); adult females (lane 3); adult males (lane 4); and adult males transformed with MYC-KLC (lanes 5 and 6). Lanes 1–5 were incubated with affinity-purified KLC antisera, and lane 6 was incubated with an antibody recognizing the epitope tag of MYC-KLC. The epitope tag causes MYC-KLC to have an apparent molecular weight slightly larger than 58 kD, the molecular weight of Drosophila KLC (Gauger and Goldstein, 1993). Lane 6 shows that the epitope-specific antibody recognizes a protein of the same apparent molecular weight as KLC (lane 5) in MYC–KLC transformants. The adult-specific 91-kD protein may not be a KLC isoform, as it is recognized by antisera specific to KLC-LG3. peptide coupled to KLH, but not other KLC antisera (see Materials and Methods). Molecular weight standards are shown in kD (right).
Rabbit polyclonal anti-KHC (Saxton et al., 1988) was affinity-purified (Olmsted, 1981) against bacterially expressed full-length KHC as follows: BL21(DE3) cells (Novagen, Inc., Madison, WI) containing pET-KIN were induced, harvested, lysed, and centrifuged according to Yang et al. (1990). A portion of the insoluble pellet containing high levels of KHC was analyzed by SDS-PAGE and then transferred to polyvinylidine difluoride membrane (Bio-Rad Laboratories, Hercules, CA). The band corresponding to KHC was excised from the blot, and then incubated with 1 ml of a 1:100 dilution of anti-KHC crude sera in TBST (15 mM Tris-Cl, 150 mM NaCl, 0.1% Tween-20, pH 7.5) for 2 h at room temperature. After washing, KHC-specific antibodies were eluted with 100 mM glycine, pH 2.5. The antibody solution was neutralized with 1 M Tris-Cl, pH 8.8, and FCS was added to 5%. The affinity-purified antibody was dialyzed overnight at 4°C with PBS in 50% glycerol. Affinity-purified anti-KHC was used at 1:10 for immunostaining.
Western blot analysis of KLC antisera on total fly protein (Fig. 2 c) was performed as follows: Drosophila embryos (10 μl), two third instar larvae, two adult females, three adult males, and three MYC-KLC transformant males were homogenized in 40 μl 5× protein gel loading buffer (Laemmli, 1970) and boiled for 3 min. Next, 10 μl of each homogenate were loaded onto a 10% SDS-PAGE gel, and then electrophoresed 4 h at 25 mA. Western transfer and detection were performed according to Barton et al. (1995). KLC antisera was used at a dilution of 1:100, and anti-MYC monoclonal antibody 9E10 (Evan et al., 1985) was used at a 1:1,000 dilution. Goat anti-mouse-HRP and goat anti-rabbit-HRP secondary antibodies (Cappel, Durham, NC) were used at a 1:20,000 dilution. The ECL kit (Nycomed Amersham Inc., Princeton, NJ) was used for secondary antibody detection.
Immunochemistry
Wild-type and mutant larvae were stained according to Hurd and Saxton (1996), with a few modifications. Larvae were dissected in dissection medium (64 mM NaCl, 2 mM CaCl2, 2 mM MgCl2, 1 mM KCl, 2.5 mM Hepes, pH 7.2, 18 mM sucrose) by pinning the anterior and posterior ends of the larvae with stainless steel pins onto a Sylgard™ (Precision Instruments, Sarasota, FL)-coated 35-mm petri plate, and then cutting along the dorsal surface with retinal scissors. Gut and fat body were removed, and the dorsal cuticle was pinned to the Sylgard plate. Larvae were fixed with 4% formaldehyde (Ted Pella, Inc., Redding, CA) in dissection buffer. Fixation conditions were 30 min at room temperature with five buffer changes. Fixed larvae were washed with antibody incubation buffer (PBS, 0.1% Triton X-100, 2% FCS) for 40–60 min at room temperature. All primary antibody incubations were performed overnight at 4°C. Secondary antibody incubations were 1–2 h at room temperature. Antibody washes were 40–60 min at room temperature in antibody incubation buffer. Primary antibodies used in this analysis include: affinity-purified rabbit polyclonal anti-KLC at 1:100, affinity-purified rabbit polyclonal anti-KHC at 1:10 (Saxton et al., 1988), rabbit polyclonal antisynaptotagmin at 1:500 (Littleton et al., 1993), mouse monoclonal anti-cysteine string protein at 1:20 (Zinsmaier et al., 1994), affinity-purified rabbit polyclonal anti-KLP68D at 1:20 (Pesavento et al., 1994), and mouse monoclonal anti–dynein heavy chain (DHC) at 1:1,000 (McGrail and Hays, 1997). FITC and Texas Red– conjugated goat anti–rabbit and goat anti–mouse secondary antibodies (Cappel) were used at a 1:200 dilution. All secondary antibodies were tested for cross-reactivity with fixed Drosophila tissue. Stained larvae were mounted in 90% glycerol, 100 mM Tris-Cl, pH 9.5, 2% N-propyl gallate.
Confocal and Video Microscopy
Samples were examined using a MRC 1000 confocal microscope (Bio-Rad Laboratories). Images were observed with a 40× oil immersion objective on an Optiphot™ (Nikon, Inc., Melville, NY) inverted microscope. The iris setting was 3, and the zoom setting was 3.5. Crawling behavior of wild-type and Klc mutant larvae was videotaped at 20× using a video camera mounted on a dissection microscope. Images were captured from videotape using a Snappy™ video frame grabber (Minolta, Ramsey, NJ).
Image files were prepared using Photoshop v.3.0 and 4.0 (Adobe Systems, San Jose, CA) and Canvas v.3.5 (Deneba Software, Miami, FL) on various Apple Macintosh systems. Images were printed on a Phaser IISDX™ printer (Tektronix. Inc., Beaverton, OR).
Results
KLC Function is Essential for Drosophila Development
To generate mutations in the previously cloned Klc gene at chromosome band 69D (Gauger and Goldstein, 1993), we screened for the insertion of a nearby P transposable element into the Klc interval. This approach, known as P-element–directed mutagenesis (Cooley et al., 1988; Tower et al., 1993; Zhang and Spradling, 1993; Dalby et al., 1995), is a powerful tool for identifying mutations in a previously cloned gene without regard for its mutant phenotype. Two insertions were identified in a 15-kb region containing Klc and Ptp69D, a receptor tyrosine phosphatase required for motor axon guidance during embryonic development (Desai et al., 1996, Desai et al., 1997; Fig. 1). One of these insertions, 59A (subsequently renamed Klc 1), is located in the first intron of the Klc transcription unit (Fig. 1). Although this 12-kb insertion greatly reduces the amount of KLC protein synthesized within cells, some KLC protein can be detected by Western analysis (data not shown).
To generate small deletions that wholly or partially remove the Klc transcript, a second round of P-element mutagenesis was performed. Excision of a P-element, while sometimes causing mutant reversion by restoring the gene to its original structure, often results in deletion of sequences flanking the insertion site (O'Hare and Rubin, 1983; Searles et al., 1986; Tsubota and Schedl, 1986). Several imprecise excisions were identified; the most informative are shown in Fig. 1. Df(3L)8ex34 is one of several deletions that remove Klc and flanking DNA sequences, as detected by Southern hybridization of cloned DNA from the Klc interval to genomic DNA extracted from Klc mutant individuals (see Materials and Methods). In contrast, Df(3L)8ex94 appears to remove only the Klc transcription unit, as the 5′ breakpoint maps near the Klc transcription start site, and the 3′ breakpoint is between Klc and Ptp69D (Fig. 1; Desai et al., 1996). Both Klc 1 and Df(3L)8ex94 complement lethal mutations in Ptp69D, suggesting that these mutations do not affect the function of Ptp69D. Reduced levels of KLC accumulation are observed in Df(3L)8ex34 and Df(3L)8ex94 heterozygotes (data not shown), thereby confirming data suggesting that these deficiencies disrupt Klc.
The loss of Klc function causes lethality during the larval and pupal stages of development, depending upon the level of residual Klc activity in the allelic combinations tested. For example, null alleles such as Df(3L)8ex94 die at the boundary of the second and third larval instars, whereas trans-heterozygous combinations such as Klc 1/ Df(3L)8ex94 or Klc 1 homozygotes live until the late third larval instar and the pupal stages, respectively (Table I). Two lines of evidence suggest that the observed lethality is due to loss of Klc function. First, chromosomes from which the P[lacW] element has been precisely excised from Klc fully complement Klc mutations. Second, transgenic rescue constructs containing either Klc genomic DNA or cDNA sequences provide full or partial rescue of Klc mutations, depending on the allelic combinations used. The genomic rescue construct GEN-KLC contains 12.4 kb of genomic DNA, including 5 kb 5′ of the Klc transcription start site and 3 kb 3′ of Klc (Fig. 1). GEN-KLC rescues much of the lethality associated with Df(3L)8ex94 and Df(3L)8ex94/Klc 1, but rescues Klc 1 lethality only from midpupal stage to late pupal stage (Table I). The minimal rescue of Klc 1 by GEN-KLC is puzzling because we expected that Klc 1 would be rescued by GEN-KLC. The failure of GEN-KLC to rescue Klc 1 lethality suggests that other lethal mutations on the Klc 1 mutant chromosome may exist. However, additional data suggest that the lethality of Klc 1 is dependent upon P[lacW] insertion into Klc. First, Klc 1 revertants in which P[lacW] has been excised complement Klc 1. Second, the Klc 1 chromosome was generated multiple times by precise excision of a second lethal P[lacW] element from the 59A mutant chromosome; all isolates of Klc 1 have the same recessive lethal phenotype alone or in combination with other Klc 1 mutant chromosomes. Finally, attempts to separate secondary lethal mutations on the Klc 1 chromosome from Klc by meiotic recombination were unsuccessful (data not shown).
Table I.
Complementation and Lethal Phase Analysis of Klc Mutants
| Genotype* | Klc 1 | Df (3L)8ex94 | MYC-KLC, Klc 1 | MYC-KLC, Df (3L)8ex94 | GEN-KLC; Klc 1 | GEN-KLC, Df (3L)8ex94 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Klc 1 | 0% (315) | 0% (612) | 0% (494) | 24% (185) | 1.2% (956) | 40% (302) | ||||||
| MP | L3-EP | LP | ND | ND | ND | |||||||
| Df(3L)8ex94 | 0% (223) | 16% (281) | 0% (712) | 25% (471) | 40% (128) | |||||||
| L2-L3 | ND | L3 | ND | ND | ||||||||
| MYC-KLC, | 2.5% (364) | 45% (266) | ||||||||||
| Klc 1 | ND | ND | ||||||||||
| MYC-KLC, | 82% (761) | |||||||||||
| Df(3L)8ex94 | ND | |||||||||||
| GEN-KLC; | 2.8% (1579) | |||||||||||
| Klc 1 | ND |
Flies of the genotypes shown were mated, the numbers of mutant and nonmutant progeny were noted, and the stage of development at which mutant progeny died was determined. The percentages shown for each mating is the percentage of mutant1/mutant2 progeny observed relative to the expected number of mutant1/mutant2 progeny. The number in parentheses represents the total number of progeny scored for each mating. The lethal phase for each mating is shown as: L2, second larval instar; L3, third larval instar; EP, early (nonpigmented) pupa; MP, midpupa; i.e., approximately half the pupae are pigmented, whereas the other half are nonpigmented.
Balancer chromosomes are omitted from the genotypes shown.
If Klc is nested within the regulatory region or transcription unit of a nearby gene, insertional mutations in Klc may alter the function of both Klc and its encompassing gene. Precise removal of the P[lacW] element from Klc would restore the function of both Klc and its nearby gene. Imprecise excisions such as Df(3L)8ex94, in contrast, restore the function of the nearby gene, but not Klc. This hypothesis predicts that Df(3L)8ex94/Klc 1 individuals can be rescued by GEN-KLC, because Df(3L)8ex94 lacks the secondary lethality associated with Klc 1. This prediction is indeed the case (Table I), as GEN-KLC rescues the heteroallelic combination Df(3L)8ex94/Klc 1 to adulthood. Therefore, we are able to conclude that the P[lacW] insertion associated with Klc 1 may affect the function of a nearby gene, but other Klc mutations such as Df(3L)8ex94 complement any secondary lethality that may be associated with Klc 1.
The cDNA rescue construct MYC-KLC encodes a myc-epitope tag-KLC fusion protein under control of the polyubiquitin promoter, which directs high levels of expression in all tissues (Figs. 1 and 2; Lee et al., 1988). The MYC-KLC transgene provides partial rescue of both the Klc insertion and deletion mutants (Table I). Some mutant combinations, such as Df(3L)8ex94/Klc 1, are rescued to adulthood, providing strong evidence that the phenotypes associated with this mutant combination are Klc-dependent. Like GEN-KLC, MYC-KLC incompletely rescues Klc 1, which again suggests that the P[lacW] insertion in Klc may also affect a nearby gene. Perhaps the most interesting MYC-KLC partial rescue phenotype is observed for the null mutant Df(3L)8ex94. Individuals completely lacking Klc function die at the boundary between the second and third larval instars; however, MYC-KLC facilitates survival of Klc null individuals to the end of the third larval instar, with apparently normal crawling behavior and activity levels. Intriguingly, larvae harboring MYC-KLC in a Klc null background live several days as late third instar larvae, but never enter the wandering third instar stage that foreshadows pupariation. A striking aspect of this phenotype is that MYC-KLC; Df(3L)8ex94 larvae remain alive in the food after all of their wild-type siblings become adults. The wanderer phenotype is rescued by GEN-KLC and is not observed in Df(3L)8ex94/Klc 1 individuals (Table I), suggesting that the wanderer phenotype is caused by defects in a gene deleted by Df(3L)8ex94, but present within GEN-KLC and unaffected by the Klc 1 insertion mutant. A second possibility is that MYC-KLC, because it encodes only one KLC isoform, may not encode alternate, minor KLC isoforms necessary for pupariation. Although the presence or absence of splice variants in Drosophila KLC has not been closely examined (Gauger et al., 1993), KLC homologs in other organisms undergo extensive alternative splicing to generate different KLC isoforms (Cyr et al., 1991; Beushausen et al., 1993; Wedaman et al., 1993). If other Drosophila KLC isoforms have unique functions, these functions will not be rescued by MYC-KLC, but may be rescued by GEN-KLC, which has the potential to encode multiple KLC isoforms, and by Klc 1, which synthesizes small amounts of wild-type protein. Nevertheless, MYC-KLC rescues many facets of the Klc mutant phenotype, thereby strengthening our assertion that phenotypes observed in Klc mutants result from loss of Klc function.
KLC Mutations Exhibit Phenotypes Reminiscent of KHC Mutations
The phenotypes of mutations in Drosophila Khc have been extensively studied (Saxton et al., 1991; Gho et al., 1992; Hurd and Saxton, 1996; Hurd et al., 1996). The Khc mutant phenotype is characterized by locomotion defects, progressive paralysis, and death during the larval stage of development. Paralysis is more severe at the distal (posterior) end of the larva, suggesting that the long motor axons innervating the posterior body wall muscles are more severely affected than the shorter axons of anterior segments. These phenotypes are caused by impairment of neuron function, as evidenced by electrophysiological defects including reduction of compound action potentials and the amplitude of excitatory junction currents, as well as reduction of the number of boutons at the neuromuscular junction. Light and electron microscopic analysis suggested that transport of a variety of cargos, both anterograde and retrograde, is blocked in Khc mutant larvae, leading to the formation of axonal swellings that accumulate diverse cargos.
The phenotype of null Klc mutations is quite similar to the Khc null mutant phenotype. KLC protein accumulation is observed at all stages of development; a single predominant species of 58 kD is observed (Fig. 2). The presence of KLC at all stages of development suggests that KLC function may be required for developmental processes. However, embryonic development appears normal, and first instar larvae hatch apparently unhindered by the loss of Klc function. The lack of zygotic KLC accumulation during embryogenesis and the first larval instar is presumably compensated by a maternally supplied pool of KLC protein. However, the second larval instar is characterized by a progressive loss of vigor as the larvae exhibit increasing amounts of paralysis, eventually resulting in complete paralysis and death near the end of the second larval instar. Occasionally a Klc null individual can proceed to the third larval instar, but these escapers often lack the strength to shed their second instar cuticle. Similar to Khc mutants, progressive paralysis associated with loss of Klc function begins at the posterior end of the larva and proceeds anteriorly. It is thus likely that paralysis of Klc mutants, like Khc, reflects the differential requirement for kinesin-based transport in the longer axons of the posterior segments relative to the shorter axons innervating the anterior segmental muscles.
Combinations of less severe Klc mutations live to the late third larval instar or pupal stage of development (Table I). Like the null Klc mutants, the terminal phenotype of Klc hypomorphic alleles is larval paralysis, with the exception of weak mutants that pupate but fail to eclose. In addition, partial loss of Klc function often results in unusual locomotion defects. Wild-type larvae move along a surface by rhythmic contractile waves that originate at the posterior end and move in a concerted fashion toward the anterior. These contractile waves are facilitated by subcuticular body wall muscles controlled by motor axons whose cell bodies are located in the larval CNS. Normally, the dorsal and ventral muscles of each segment contract in unison, ensuring that the larva maintains contact with the surface. However, Klc mutations that allow survival to late third larval instar cause the posterior end of the larva to lift its tail off the medium (Fig. 2). Severely affected individuals can be observed lifting the posterior 40% of their body. This tail-flipping phenotype is also observed in certain Khc mutant combinations (Hurd and Saxton, 1996). It has been proposed that the tail-flipping observed in Khc mutants is the result of a temporal gradient of paralysis such that the ventral body wall muscles lose muscle tone before dorsal body wall muscles. Contraction of the dorsal body wall muscles in the absence of counterbalancing ventral muscle contraction then causes the tail to flip upward. The tail-flipping and paralysis phenotypes observed in Klc mutant larvae are quantitatively rescued by the transgenic constructs GEN-KLC and MYC-KLC (Table I; Fig. 2), providing strong evidence that the neuromuscular defects of Klc mutants result from the loss of Klc function.
Loss of Klc Function Disrupts Multiple Pathways of Axonal Transport
Macromolecular structures required for synapse function, such as membranous vesicles, neurotransmitters, and the machinery controlling synaptic vesicle fusion and recycling, must be transported great distances from the point of synthesis in the cell body to the synapse. Similarly, for chemical signals received at the synapse to be acted upon by the neuron, the signals must be transported from the synapse to the cell body. Microtubule motors such as kinesin are necessary for these transport phenomena. Perhaps the paralytic and tail-flipping phenotypes associated with loss of Klc function are the result of a defect in axonal transport owing to mislocalization of kinesin cargoes. In fact, loss of Khc function is known to cause massive disruption of fast axonal transport, resulting in focal swellings packed with many different types of membrane-bound organelles, including mitochondria, prelysosomal mutivesicular bodies, and synaptic vesicle precursors. Loss of Khc function does not disrupt slow axonal transport, however, as molecules that undergo slow axonal transport, such as tubulin, are not observed in Khc organelle jams (Hurd and Saxton, 1996).
To determine if loss of Klc function results in formation of axonal organelle jams similar to those observed in Khc mutants, we studied the cellular phenotype of Klc mutant axons from tail-flipping Klc mutant larvae. Biochemical analyses in mammalian systems suggest that kinesin is associated with many different types of membranous vesicles, including synaptic vesicles and mitochondria (Hollenbeck, 1989; Pfister et al., 1989; Dahlstrom et al., 1991; Hirokawa et al., 1991; Leopold et al., 1992; Burkhardt et al., 1993; Vallee and Sheetz, 1996). Therefore, we studied the transport of proteins such as the synaptic vesicle components synaptotagmin (SYT) and cysteine string protein (CSP), which undergo high levels of microtubule-based axonal transport (Littleton et al., 1993; Zinsmaier et al., 1994; Parfitt et al., 1995). Segmental nerves from control Klc/+ larvae exhibit punctate but relatively uniform CSP and SYT staining in the segmental nerves (Fig. 3, a and b). However, large immunoreactive clusters of CSP and SYT are observed in the segmental nerves of Klc mutant larvae (Fig. 3, c and d). These clusters appear to represent organelle jams similar to those observed in Khc mutant larvae (Hurd and Saxton, 1996). Small clusters of immunoreactivity are sometimes observed in control larvae, but their size and frequency are greatly reduced in comparison with Klc mutant segmental nerves. Immunostaining of Khc mutant larvae exhibiting the tail-flipping behavioral phenotype reveals similar staining patterns for SYT and CSP (Hurd and Saxton, 1996; our unpublished results). These results suggest that, in addition to sharing behavioral phenotypes, Khc and Klc mutations appear to cause a similar disruption of fast axonal transport. Furthermore, the similar phenotype of Klc and Khc mutants suggests that both the light chains and heavy chains are necessary for kinesin function, and that the light chains may have a positive role in kinesin function (such as cargo binding) instead of being a negative regulator of kinesin activity.
Figure 3.

Loss of Klc function causes disruption of axonal transport. Larvae heterozygous for Klc (A, B) or Klc 1/Df(3L)8ex94 mutants (C, D) were simultaneously incubated with antibodies specific for cysteine string protein (A, C) and synaptotagmin (B, D) to compare the accumulation patterns of these synaptic vesicle components in wild-type and Klc mutant backgrounds. Although accumulation of CSP and SYT is punctate but fairly uniform in the segmental nerves of control larvae (A, B), large aggregates of CSP and SYT immunoreactivity are observed in Klc mutants (C, D), suggesting that normal axonal transport of these proteins are blocked. Furthermore, the pattern of CSP (C) accumulation completely overlaps SYT (D), suggesting that these proteins are found in the same subset of organelle jams. Individual organelle jams are presumed to be confined to a single axon (Hurd and Saxton, 1996). Bar, 25 μM.
The observation that organelle jams from Khc mutant larvae contain a variety of membranous organelles, including both anterograde and retrograde cargoes (Hurd and Saxton, 1996), led us to test directly the hypothesis that failure of kinesin transport in Klc mutants interferes with the ability of other molecular motors to transport their cargoes. To determine if Klc mutations interfere with transport pathways of other motors, we examined immunolocalization of two other motor proteins that are likely to play important roles in fast axonal transport. The first of these motors is KLP68D (Pesavento et al., 1994), which is a plus-end directed neuronal motor homologous to members of the murine KIF3 family and the sea urchin KRP85/ 95 family (Scholey, 1996; Yamazaki et al., 1995). The second motor we examined is DHC, which is the heavy chain of the retrograde fast axonal transport motor cytoplasmic dynein (McGrail and Hays, 1997). KLP68D is obviously localized to large aggregates in Klc mutant larvae (Fig. 4 b), whereas its localization in Klc/+ control segmental nerves is fairly uniform (data not shown), suggesting that the presence of KLP68D aggregates is a direct consequence of loss of Klc function. Some of these aggregates colocalize with CSP (compare Fig. 4, a and b), demonstrating that KLP68D protein is present in organelle jams. Identification of organelle jams that contain KLP68D but not CSP suggests that the composition of the membranous organelles in jams is heterogeneous. This heterogeneity may reflect the stochastic nature of organelle jam formation, or could be due to the presence of both sensory and motor axons in segmental nerves, each accumulating different ratios of intracellular cargos. While the degree of impairment of KLP68D-dependent transport in Klc mutant axons cannot be directly measured, the presence of high levels of KLP68D immunoreactivity in organelle jams suggests that loss of kinesin function causes a pleiotropic block of anterograde fast axonal transport.
Figure 4.

KLP68D and cytoplasmic dynein are found in organelle jams. Segmental nerves of Df(3L)8ex94/Klc 1 larvae were incubated with antibodies to CSP (A) and KLP68D (B), or SYT (C) and DHC, the motor component of cytoplasmic dynein (D). Both KLP68D and DHC immunoreactivity are observed in organelle jams. Bar, 20 μM.
To test whether the organelle jams found in Klc mutant axons may impede retrograde fast axonal transport, immunolocalization of DHC in control and Klc mutant larvae was compared. DHC is observed in large aggregates in Klc mutants (Fig. 4 d), but not in Klc/+ control larvae, and the DHC aggregates sometimes colocalize with SYT (compare Fig. 4, c and d). However, many DHC-labeled organelle jams do not colocalize with SYT-labeled organelle jams, suggesting again that the distribution of intracellular cargos within organelle jams is heterogeneous. This result demonstrates that cytoplasmic dynein is also present in organelle jams, and supports the hypothesis that loss of kinesin function inhibits multiple pathways of motor-driven fast axonal transport. However, we cannot conclude that Klc mutations disrupt fast retrograde axonal transport. Cytoplasmic dynein is a fast retrograde motor in axons, but much cytoplasmic dynein in axons may be in an inactive state, transported by anterograde fast axonal transport to the synapse (Hirokawa et al., 1990; Dillman et al., 1996). Because we cannot determine whether the DHC in organelle jams is in an active state, additional experiments are required to determine unequivocally the relationship between loss of Klc function and disruption of cytoplasmic dynein-dependent transport pathways. In conclusion, these results strongly suggest that behavioral defects observed in Khc and Klc mutant larvae are not solely due to a failure to localize specific kinesin cargos, but result from a general failure of several pathways of motor-mediated axonal transport (Hurd and Saxton, 1996).
KLC May Be Required for KHC Attachment to Cargoes In vivo
An important question is whether the kinesin light chains are necessary for kinesin binding to its intracellular cargoes. Whereas some results suggest that the KHC tail domain is sufficient for kinesin–cargo interaction (Skoufias et al., 1994; Bi et al., 1997), other experiments demonstrate that KLC function may be necessary for a subset of kinesin binding to intracellular cargoes (Stenoien and Brady, 1997). We attempted to test directly the role of KLC in cargo binding by studying immunolocalization of KHC in Klc mutant axons. The absence of KHC from Klc mutant organelle jams would support a model in which KLC is necessary for cargo attachment. In contrast, the observation of KHC immunoreactivity in the absence of KLC would strongly suggest that some kinesin–cargo interactions are KLC-independent. Immunostaining of Klc 1/ Df(3L)8ex94 mutant larvae with antisera to KHC (Fig. 5 b) and CSP (Fig 5 a) demonstrates that KHC is present in Klc larval organelle jams. This result suggests that KLC is dispensable for kinesin–cargo interactions; however, it is possible that KHC is tethered to cargoes by residual light chains present in Klc mutant larvae. The allelic combination used for KHC immunolocalization, Klc 1/Df(3L)8ex94, makes low but detectable levels of KLC protein. Fig. 5 demonstrates that KLC protein, like KHC, is also observed in Klc mutant larvae, and that its immunoreactivity partially overlaps CSP (Fig. 5, c and d). Biochemical fractionation experiments also demonstrate that nearly all the residual KLC protein in light chain mutants is membrane-bound (our unpublished results), suggesting attachment to organelle cargoes. Although these results support a role for KLC in kinesin–cargo interactions, additional experiments such as immunoelectron microscopy will be required to define the relative contributions of KLC and KHC to cargo attachment at high resolution, and to demonstrate the direct association of KHC with membranous organelles in Klc-dependent organelle jams.
Figure 5.

KHC and KLC accumulate in Klc-dependent organelle jams. Df(3L)8ex94/ Klc 1 larvae were incubated with antibodies to CSP (A, C) and antibodies to KHC (B) and KLC (D). KHC aggregates are observed in the segmental nerves of Klc mutant larvae. This observation would suggest that KHC can bind and transport intracellular cargoes in a KLC-independent manner; however, KLC protein accumulation is also observed in Klc mutant segmental nerves (D). Bar, 20 μM.
Discussion
Similar Axonal Transport Defects in Klc and Khc Mutants
The major conclusion of the experiments presented here is that the phenotype of Klc and Khc mutants are quite similar, especially with respect to disruption of fast axonal transport observed in both mutants (Saxton et al., 1991; Hurd and Saxton, 1996). Klc and Khc mutants exhibit paralysis that begins in the posterior end of the larva, which then proceeds anteriorly. This graded paralysis reflects the difference in the length of axons innervating the posterior segmental muscles in comparison to axons innervating more anterior segments (Hurd and Saxton, 1996). The cause of the locomotion defects and paralysis of Klc and Khc mutants may be formation of large aggregates of axonally transported organelles. These aggregations are similar to the clusters of anterograde and retrograde transports observed at the site of vertebrate nerve ligations (Smith, 1980; Tsukita and Ishikawa, 1980; Fahim et al., 1985; Hirokawa et al., 1990, Hirokawa et al., 1991). Immunostaining of organelle jams demonstrates that synaptic vesicle components, in addition to the neuronal motors KLP68D and cytoplasmic dynein, accumulate in Klc mutant axons. Electron microscopic analysis of Khc mutant axons demonstrate that many different types of membrane-bound organelles are present in organelle jams (Hurd and Saxton, 1996). Organelle jams may impair multiple anterograde and retrograde axonal transport and signaling pathways, resulting in a compromise of neuromuscular function similar to that observed in vertebrate neuropathies such as amyotrophic lateral sclerosis (Hurd and Saxton, 1996; for review see Bruijn and Cleveland, 1996).
Native kinesin is a heterotetramer composed of two heavy chains and two light chains (Bloom et al., 1988; Johnson et al., 1990; Kuznetsov et al., 1988). Although the high degree of light chain sequence conservation among higher eukaryotes (Cyr et al., 1991; Gauger and Goldstein, 1993; Wedaman et al., 1993; Beushausen et al., 1993; Cabeza et al., 1993; Fan and Amos, 1994; Chernajovsky et al., 1996) implies that the light chains are essential for kinesin function, their precise role is unclear. Existing suggestions for KLC function can be broken into two broad categories: positive and negative. If light chains are necessary for a positive function such as mediating kinesin–cargo interactions, then light chain mutations may have a phenotype similar to mutations in Khc that result in loss of motor activity. Alternatively, if the light chains have a negative function, for example, to inhibit kinesin activity by repressing motor activity or cargo binding, then Klc mutants may exhibit a phenotype different than the Khc loss-of-function phenotype. The results presented here demonstrate that the phenotypic consequences of loss of Klc and Khc function are very similar. Although it is difficult to predict the phenotypic consequences, if any, of kinesin hyperactivity in the absence of light chain function, we propose that the underlying defect in both Klc and Khc mutants is the inability of kinesin cargos to participate efficiently in fast axonal transport. Thus, two hypotheses consistent with the observed Klc mutant phenotype are (a) the inability of the kinesin motor domain to interact with some or all of its intracellular cargoes, or (b) the inability of the kinesin motor domain to be properly activated. However, further analysis will be required to determine unequivocally the in vivo role of KLC in kinesin-dependent axonal transport.
Both KLC and KHC May Be Important for Cargo Binding
Analysis of kinesin light chain function in other systems supports the hypothesis that the light chains are important for cargo binding. Adding light chain antibody to purified brain microsomes reduces the amount of microsome-bound kinesin by one-third relative to untreated control vesicles (Yu et al., 1992). Similar results are obtained when a different KLC antibody, specific to the conserved 42–amino acid TPR region of KLC, is incubated with microsomes (Stenoien and Brady, 1997). In contrast, an antibody specific for the coiled-coil region of KLC, which is likely to mediate KLC–KHC interactions (Gauger and Goldstein, 1993), does not inhibit kinesin–vesicle interactions (Stenoien and Brady, 1997). These results suggest that the region of KLC containing six conserved 42–amino acid repeats may be necessary for some kinesin–cargo interactions, perhaps by binding to membrane-bound receptors on the cargo surface. The loss of processive kinesin-dependent cargo transport observed in Klc mutants is consistent with inhibition of vesicle binding seen in anti-KLC inhibition experiments.
A model proposing an exclusive role for KLC in mediating kinesin–cargo interactions is appealing, but other experimental evidence indicates that the COOH-terminal tail of KHC also participates in kinesin binding to intracellular cargoes. A bacterially expressed fragment of sea urchin KHC, including the stalk and tail domains, can bind to sea urchin egg microsomal membranes in a concentration-dependent and saturable manner, but the KHC stalk domain cannot (Skoufias et al., 1994), indicating that the COOH-terminal 200 amino acids of sea urchin kinesin has cargo-binding capacity. Injection of the sea urchin KHC stalk-tail domain into wounded sea urchin cells inhibits membrane resealing in a manner similar to that observed for a KHC motor domain–inhibitory antibody (Bi et al., 1997), providing in vivo evidence of a role for KHC in cargo binding. However, it is also possible that association of the injected KHC stalk-tail domain with an intracellular pool of KLC could result in inhibition of membrane resealing by a KLC-dependent mechanism.
The results of the experiments described above suggest that KLC is necessary for binding of some cargos, but not others. Individual cargos may contain distinct receptors, some of which interact with KLC, but others that bind KHC. Several lines of evidence support this model. First, purified KCl-washed vesicles bind kinesin in a saturable, concentration-dependent manner, and have the capacity to bind twice as much kinesin with light chains (10S native kinesin) as kinesin lacking light chains (7S kinesin; Skoufias et al., 1994). Second, purified vesicles bind twice as much nonphosphorylated kinesin as kinesin phosphorylated by A-kinase (Sato-Yoshitake et al., 1992), suggesting that some kinesin–cargo interactions, but not others, are blocked by phosphorylation. Third, washing synaptic vesicles with 1 M NaCl results in release of only 65% of membrane-bound kinesin (ibid.). Fourth and finally, incubation of purified microsomes with saturating amounts of anti-KLC inhibitory antibodies displaces only 30% of kinesin from the vesicle surface (Yu et al., 1992; Stenoien and Brady, 1997), demonstrating that, like incubation with NaCl, inhibition of KLC function does not result in quantitative release of kinesin from vesicles. These results all support a model in which kinesin can use multiple interaction pathways to bind intracellular cargoes. More speculatively, the cargo-binding domain of KLC (TPR motif) differs in proposed secondary structure from the putative KHC cargo-binding domain, which is predicted to be mostly alpha-helical coiled coil in structure (de Cuevas et al., 1992; Fan and Amos, 1994; our unpublished observations). Differences in the structure of the cargo-binding domains of KHC and KLC, as well as concomitant differences in the nature of their protein–protein interactions with intracellular cargos, may explain results suggesting multiple mechanisms of kinesin–cargo interactions.
Identification of kinesin homologs in the fungi Ustilago maydis (Lehmler et al., 1997), Neurospora crassa (Steinberg and Schliwa, 1995), and Syncephalum racemosum (Steinberg, 1997) further support this view of kinesin– cargo interactions. Like kinesins identified in higher organisms, fungal kinesins such as Neurospora Nkin and Ustilago Kin2 are necessary for intracellular transport events, including polarized secretion and hyphal extension (Lehmler et al., 1997; Seiler et al., 1997). However, biochemical purification of fungal kinesins demonstrate that they have no associated light chains. The absence of light chains from fungal kinesins may reflect less complex requirements for intracellular transport owing to simpler morphology, fewer cell types, and the less complex developmental profile of fungi relative to other organisms from which kinesin has been identified. Although the conservation of light chain structure throughout higher eukaryotes suggests that association of KLC with KHC is evolutionarily ancient, the light chains may have provided an additional level of complexity of the kinesin tail domain necessary during the evolution of increasingly complex pathways of intracellular transport. Interestingly, a KLC-like protein has recently been discovered in the cyanobacterium Plectonema boryanum, but a prokaryotic KHC was not identified (Celerin et al., 1997). Perhaps a protein similar to cyanobacterial KLC became associated with an ancestral KHC before the divergence of vertebrates and invertebrates. Higher vertebrates have added more layers of complexity by duplication and divergence of KHC and KLC (Aizawa et al., 1992; Niclas et al., 1994; Nakagawa et al., 1997; A. Rahman and L. Goldstein, unpublished results). Elaboration of kinesin tail structure and duplication of kinesin subunit-encoding genes facilitated an increase in the repertoire of intracellular cargoes with which kinesin can interact, and has enabled additional layers of regulatory complexity by cytosolic factors such as kinases and phosphatases (Sato-Yoshitake et al., 1992; Hollenbeck, 1993; Matthies et al., 1993; McIlvain et al., 1994; Lee and Hollenbeck, 1995; Okada et al., 1995; Lindesmith et al., 1997).
Organelle Jams Result from a Failure of Processive Kinesin-dependent Transport
An interesting property of Klc and Khc mutants is the stochastic distribution of organelle jams in larval segmental nerve axons. If kinesin cargos were unable to enter the axon when active kinesin levels are greatly reduced, then cargos would be concentrated in the cell body and proximal parts of the axon. Instead, organelle jams appear to be randomly distributed along the length of the axon (Hurd and Saxton, 1996; this analysis). In vitro experiments studying the movement of kinesin-coated latex beads along microtubules, or microtubules along a kinesin-coated glass coverslip, have demonstrated that the processivity of kinesin-dependent movement is directly proportional to the number of kinesin molecules on the latex bead or glass coverslip (Howard et al., 1989; Block et al., 1990). Similarly, the in vivo processive movement of kinesin cargos may also be dependent on the intracellular concentration of kinesin. In Klc and Khc mutants, the number of kinesin molecules on its intracellular cargoes may decrease as the concentration of maternally supplied kinesin is exhausted. Therefore, kinesin-dependent transport becomes less processive, resulting in nucleation of an axonal traffic jam (Hurd and Saxton, 1996). Comparative analysis of the amount of kinesin bound to wild-type and kinesin mutant organelles, as well as assays designed to study the processivity of kinesin-dependent transport in kinesin mutants will help us better understand the underlying mechanism of the organelle jam phenotype.
An unresolved issue is the nature of the cargos transported in the axon by kinesin, as well as how kinesin binds these cargoes. Presumably, Khc mutants result in failure to transport all kinesin cargos as the mechanochemical head domain is encoded by KHC. However, the loss of Klc function may inhibit the transport of only a subset of kinesin cargoes, given that the tail domain of KHC may be able to bind to vesicles in a KLC-independent manner. If this hypothesis is correct, why are the Khc and Klc phenotypes so similar? The underlying cause of the organelle jam phenotype may be the failure to transport processively a small percentage of kinesin cargoes. Perhaps the less processive kinesin cargos serve as nucleation points for organelle jams, and other axonal cargos such as those transported by KLP68D and cytoplasmic dynein become trapped in these kinesin-dependent organelle jams. We propose that the nature of the cargo precipitating the organelle jam itself may be irrelevant, and that many different kinds of membrane-bound organelles can potentially serve as nucleation points for organelle jams. Preliminary data suggest that mutations in several different complementation groups can result in formation of organelle jams (M. A. Martin, A. Gassman, and W. M. Saxton, personal communication; M. McGrail, A. Bowman, and L. Goldstein, unpublished results). Therefore, the presence or absence of organelle jams in the larval segmental nerve may provide a sensitive assay for identifying parallel pathways of fast axonal transport, as well as additional components necessary for kinesin-dependent transport.
Acknowledgments
We would like to thank Kathy Matthews and the Indiana Stock Center for providing fly stocks, Maria Dolph and Jenny Xu for technical assistance; Bill Saxton, Mary Ann Martin, and Daryl Hurd for valuable advice and communicating results before publication; Tom Hays, J. Troy Littleton, Bill Saxton, and Konrad Zinsmaier for generously providing antibodies; Bill Harris and Bill Hagar for providing equipment for video microscopy and imaging; and members of the Goldstein lab, especially Nelson Barton and Roman Sakowicz, for advice and invigorating discussions.
Abbreviations used in this paper
- CSP
cysteine string protein
- DHC
dynein heavy chain
- KHC
kinesin heavy chain
- KLC
kinesin light chain
- TPR
tetratricopeptide repeat
- KLH
keyhole limpet hemocyanin
- SYT
synaptotagmin
- TPR
tetratrico peptide repeat
Footnotes
This research was supported by National Institutes of Health grants to L.S.B. Goldstein and K. Zinn. L.SB. Goldstein is an investigator of the Howard Hughes Medical Institute. J.G. Gindhart was supported by a National Institutes of Health postdoctoral fellowship. C.J. Desai was supported by an American Cancer Society postdoctoral fellowship.
Address all correspondence to Lawrence S.B. Goldstein, Howard Hughes Medical Institute, Division of Cellular and Molecular Medicine, Department of Pharmacology, University of California, San Diego, La Jolla, CA 92093-0683. Phone: 619-534-9702; FAX: 619-534-9701. E-mail: lgoldstein@ucsd.edu
The current address of Joseph G. Gindhart is Department of Biology, University of Massachusetts Boston, 100 Morrissey Blvd., Boston, MA 02125. The current address of Chand J. Desai is Center for Molecular Neuroscience, 406 MBRI, Vanderbilt University Medical Center, 1211 22nd Ave. S., Nashville, TN 37232.
References
- Aizawa H, Sekine Y, Takemura R, Zhang Z, Nangaku M, Hirokawa N. Kinesin family in murine central nervous system. J Cell Biol. 1992;119:1287–296. doi: 10.1083/jcb.119.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NR, Pereira AJ, Goldstein LSB. Motor activity and mitotic spindle localization of the Drosophilakinesin-like protein KLP61F. Mol Biol Cell. 1995;6:1563–1574. doi: 10.1091/mbc.6.11.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beushausen S, Kladakis A, Jaffe H. Kinesin light chains: identification and characterization of a family of proteins from the optic lobe of the squid Loligo pealii. . DNA Cell Biol. 1993;12:901–909. doi: 10.1089/dna.1993.12.901. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Morris RL, Liao G, Alderton JM, Scholey JM, Steinhardt RA. Kinesin- and myosin-driven steps of vesicle recruitment for Ca2+-regulated exocytosis. J Cell Biol. 1997;138:999–1008. doi: 10.1083/jcb.138.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier E, Vaessin H, Shepherd S, Lee K, McCall K, Barbel S, Ackerman L, Carretto R, Uemura T, Grell E, et al. Searching for pattern and mutation in the Drosophilagenome with a P-lacZ vector. Genes Dev. 1989;3:1273–1287. doi: 10.1101/gad.3.9.1273. [DOI] [PubMed] [Google Scholar]
- Block SM, Goldstein LSB, Schnapp BJ. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- Bloom GS, Endow SA. Motor proteins 1: kinesins. Protein Profile. 1995;2:1105–1171. [PubMed] [Google Scholar]
- Bloom GS, Wagner MC, Pfister KK, Brady ST. Native structure and physical properties of bovine brain kinesin and identification of the ATP-binding subunit polypeptide. Biochemistry. 1988;27:3409–3416. doi: 10.1021/bi00409a043. [DOI] [PubMed] [Google Scholar]
- Brady ST. A novel brain ATPase with properties expected for the fast axonal transport motor. Nature. 1985;317:73–75. doi: 10.1038/317073a0. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Cleveland DW. Mechanisms of selective motor neuron death in ALS: insights from transgenic mouse models of motor neuron disease. Neuropathol Appl Neurobiol. 1996;22:373–387. doi: 10.1111/j.1365-2990.1996.tb00907.x. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, McIlvain JMJ, Sheetz MP, Argon Y. Lytic granules from cytotoxic T cells exhibit kinesin-dependent motility on microtubules in vitro. J Cell Sci. 1993;104:151–162. doi: 10.1242/jcs.104.1.151. [DOI] [PubMed] [Google Scholar]
- Cabeza AY, Shih LC, Hardman N, Asselbergs F, Bilbe G, Schmitz A, White B, Siciliano MJ, Lachman LB. Cloning and genetic characterization of the human kinesin light-chain (KLC) gene. DNA Cell Biol. 1993;12:881–892. doi: 10.1089/dna.1993.12.881. [DOI] [PubMed] [Google Scholar]
- Celerin M, Gilpin AA, Dossantos G, Laudenbach DE, Clarke MW, Beushausen S. Kinesin light chain in a eubacterium. DNA Cell Biol. 1997;16:787–795. doi: 10.1089/dna.1997.16.787. [DOI] [PubMed] [Google Scholar]
- Chernajovsky Y, Brown A, Clark J. Human kinesin light (beta) chain gene: DNA sequence and functional characterization of its promoter and first exon. DNA Cell Biol. 1996;15:965–974. doi: 10.1089/dna.1996.15.965. [DOI] [PubMed] [Google Scholar]
- Cooley L, Kelley R, Spradling A. Insertional mutagenesis of the Drosophila genome with single Pelements. Science. 1988;239:1121–1128. doi: 10.1126/science.2830671. [DOI] [PubMed] [Google Scholar]
- Cyr JL, Pfister KK, Bloom GS, Slaughter CA, Brady ST. Molecular genetics of kinesin light chains: generation of isoforms by alternative splicing. Proc Natl Acad Sci USA. 1991;88:10114–10118. doi: 10.1073/pnas.88.22.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlstrom AB, Pfister KK, Brady ST. The axonal transport motor ‘kinesin' is bound to anterogradely transported organelles: quantitative cytofluorimetric studies of fast axonal transport in the rat. Acta Physiol Scand. 1991;141:469–476. doi: 10.1111/j.1748-1716.1991.tb09107.x. [DOI] [PubMed] [Google Scholar]
- Dalby B, Pereira AJ, Goldstein LSB. An inverse PCR screen for the detection of P element insertions in cloned genomic intervals in Drosophilamelanogaster. Genetics. 1995;139:757–766. doi: 10.1093/genetics/139.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cuevas M, Tao T, Goldstein LS. Evidence that the stalk of Drosophilakinesin heavy chain is an alpha-helical coiled coil. J Cell Biol. 1992;116:957–965. doi: 10.1083/jcb.116.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai CJ, Gindhart JG, Goldstein LSB, Zinn K. Receptor tyrosine phosphatases are required for motor axon guidance in the Drosophila embryo. Cell. 1996;84:599–609. doi: 10.1016/s0092-8674(00)81035-1. [DOI] [PubMed] [Google Scholar]
- Desai CJ, Krueger NX, Saito H, Zinn K. Competition and cooperation among receptor tyrosine phosphatases control motoneuron growth cone guidance in Drosophila. . Development. 1997;124:1941–1952. doi: 10.1242/dev.124.10.1941. [DOI] [PubMed] [Google Scholar]
- Dillman JF, III, Dabney LP, Pfister KK. Cytoplasmic dynein is associated with slow axonal transport. Proc Natl Acad Sci USA. 1996;93:141–144. doi: 10.1073/pnas.93.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahim MA, Lasek RJ, Brady ST, Hodge AJ. AVEC-DIC and electron microscopic analyses of axonally transported particles in cold-blocked squid giant axons. J Neurocytol. 1985;14:689–704. doi: 10.1007/BF01170822. [DOI] [PubMed] [Google Scholar]
- Fan J, Amos LA. Kinesin light chain isoforms in Caenorhabditis elegans. . J Mol Biol. 1994;240:507–512. doi: 10.1006/jmbi.1994.1465. [DOI] [PubMed] [Google Scholar]
- Futterer A, Kruppa G, Kramer B, Lemke H, Kronke M. Molecular cloning and characterization of human kinectin. Mol Biol Cell. 1995;6:161–170. doi: 10.1091/mbc.6.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauger AK, Goldstein LS B. The Drosophila kinesin light chain. Primary structure and interaction with kinesin heavy chain. J Biol Chem. 1993;268:13657–13666. [PubMed] [Google Scholar]
- Gho M, McDonald K, Ganetzky B, Saxton WM. Effects of kinesin mutations on neuronal functions. Science. 1992;258:313–316. doi: 10.1126/science.1384131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons BH, Asai DJ, Tang WJ, Hays TS, Gibbons IR. Phylogeny and expression of axonemal and cytoplasmic dynein genes in sea urchins. Mol Biol Cell. 1994;5:57–70. doi: 10.1091/mbc.5.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindhart JG, Goldstein LS B. Tetratrico peptide repeats are present in the kinesin light chain. Trends Biochem Sci. 1996;21:52–53. [PubMed] [Google Scholar]
- Goldstein LS. With apologies to Scheherazade: tails of 1001 kinesin motors. Annu Rev Genet. 1993;27:319–351. doi: 10.1146/annurev.ge.27.120193.001535. [DOI] [PubMed] [Google Scholar]
- Hackney DD, Levitt JD, Suhan J. Kinesin undergoes a 9 S to 6 S conformational transition. J Biol Chem. 1992;267:8696–8701. [PubMed] [Google Scholar]
- Hackney DD, Levitt JD, Wagner DD. Characterization of alpha-2 beta-2 and alpha-2 forms of kinesin. Biochem Biophys Res Commun. 1991;174:810–815. doi: 10.1016/0006-291x(91)91490-4. [DOI] [PubMed] [Google Scholar]
- Hartenstein V, Jan YN. Studying Drosophila embryogenesis with P-lacZ enhancer trap lines. Roux's Arch Dev Biol. 1992;201:194–220. doi: 10.1007/BF00188752. [DOI] [PubMed] [Google Scholar]
- Heck MM, Pereira A, Pesavento P, Yannoni Y, Spradling AC, Goldstein LSB. The kinesin-like protein KLP61F is essential for mitosis in Drosophila. J Cell Biol. 1993;123:665–679. doi: 10.1083/jcb.123.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Pfister KK, Yorifuji H, Wagner MC, Brady ST, Bloom GS. Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell. 1989;56:867–78. doi: 10.1016/0092-8674(89)90691-0. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Kobayashi N, Pfister KK, Bloom GS, Brady ST. Kinesin associates with anterogradely transported membranous organelles in vivo. J Cell Biol. 1991;114:295–302. doi: 10.1083/jcb.114.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Yoshida T, Kawashima T. Brain dynein (MAP1C) localizes on both anterogradely and retrogradely transported membranous organelles in vivo. J Cell Biol. 1990;111:1027–1037. doi: 10.1083/jcb.111.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. The distribution, abundance and subcellular localization of kinesin. J Cell Biol. 1989;108:2335–2342. doi: 10.1083/jcb.108.6.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. Phosphorylation of neuronal kinesin heavy and light chains in vivo. J Neurochem. 1993;60:2265–2275. doi: 10.1111/j.1471-4159.1993.tb03513.x. [DOI] [PubMed] [Google Scholar]
- Howard J, Hudspeth AJ, Vale RD. Movement of microtubules by single kinesin molecules. Nature. 1989;342:154–158. doi: 10.1038/342154a0. [DOI] [PubMed] [Google Scholar]
- Hurd DD, Saxton WM. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics. 1996;144:1075–1085. doi: 10.1093/genetics/144.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd DD, Stern M, Saxton WM. Mutation of the axonal transport motor kinesin enhances paralytic and suppresses Shakerin Drosophila. Genetics. 1996;142:195–204. doi: 10.1093/genetics/142.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang MY, Sheetz MP. Cargo-activated ATPase activity of kinesin. Biophys J. 1995;68:283s–285s. [PMC free article] [PubMed] [Google Scholar]
- Johnson CS, Buster D, Scholey JM. Light chains of sea urchin kinesin identified by immunoadsorption. Cell Motil Cytoskeleton. 1990;16:204–213. doi: 10.1002/cm.970160307. [DOI] [PubMed] [Google Scholar]
- Kumar J, Yu H, Sheetz MP. Kinectin, an essential anchor for kinesin-driven vesicle motility. Science. 1995;267:1834–1837. doi: 10.1126/science.7892610. [DOI] [PubMed] [Google Scholar]
- Kuznetsov SA, Vaisberg EA, Shanina NA, Magretova NN, Chernyak VY, Gelfand VI. The quaternary structure of bovine brain kinesin. EMBO (Eur Mol Biol Organ) J. 1988;7:353–356. doi: 10.1002/j.1460-2075.1988.tb02820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov SA, Vaisberg YA, Rothwell SW, Murphy DB, Gelfand VI. Isolation of a 45-kDa fragment from the kinesin heavy chain with enhanced ATPase and microtubule-binding activities. J Biol Chem. 1989;264:589–595. [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lamb JR, Tugendreich S, Heiter P. Tetratrico peptide repeat interactions: to TPR or not to TPR? . Trends Biochem Sci. 1995;20:257–259. doi: 10.1016/s0968-0004(00)89037-4. [DOI] [PubMed] [Google Scholar]
- Lee H, Simon JA, Lis JT. Structure and expression of ubiquitin genes of Drosophila melanogaster. . Mol Cell Biol. 1988;8:4727–4735. doi: 10.1128/mcb.8.11.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KD, Hollenbeck PJ. Phosphorylation of kinesin in vivo correlates with organelle association and neurite outgrowth. J Biol Chem. 1995;270:5600–5605. doi: 10.1074/jbc.270.10.5600. [DOI] [PubMed] [Google Scholar]
- Lehmler C, Steinberg G, Snetselaar KM, Schliwa M, Kahmann R, Bolker M. Identification of a motor protein required for filamentous growth in Ustilago maydis. EMBO (Eur Mol Biol Organ) J. 1997;16:3464–3473. doi: 10.1093/emboj/16.12.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold PL, McDowall AW, Pfister KK, Bloom GS, Brady ST. Association of kinesin with characterized membrane-bounded organelles. Cell Motil Cytoskeleton. 1992;23:19–33. doi: 10.1002/cm.970230104. [DOI] [PubMed] [Google Scholar]
- Lindesmith L, McIlvain JJ, Argon Y, Sheetz MP. Phosphotransferases associated with the regulation of kinesin motor activity. J Biol Chem. 1997;272:22929–22933. doi: 10.1074/jbc.272.36.22929. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Bellen HJ, Perin MS. Expression of synaptotagmin in Drosophilareveals transport and localization of synaptic vesicles to the synapse. Development. 1993;118:1077–1088. doi: 10.1242/dev.118.4.1077. [DOI] [PubMed] [Google Scholar]
- Matthies HJ, Miller RJ, Palfrey HC. Calmodulin binding to and cAMP-dependent phosphorylation of kinesin light chains modulate kinesin ATPase activity. J Biol Chem. 1993;268:11176–11187. [PubMed] [Google Scholar]
- McGrail M, Hays TS. The microtubule motor cytoplasmic dynein is required for spindle orientation during germline cell divisions and oocyte differentiation in Drosophila. . Development. 1997;124:2409–2419. doi: 10.1242/dev.124.12.2409. [DOI] [PubMed] [Google Scholar]
- McIlvain JJ, Burkhardt JK, Hamm AS, Argon Y, Sheetz MP. Regulation of kinesin activity by phosphorylation of kinesin-associated proteins. J Biol Chem. 1994;269:19176–19182. [PubMed] [Google Scholar]
- Mooseker MS, Cheney RE. Unconventional myosins. Annu Rev Cell Dev Biol. 1995;11:633–675. doi: 10.1146/annurev.cb.11.110195.003221. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Tanaka Y, Matsuoka E, Kondo S, Okada Y, Noda Y, Kanai Y, Hirokawa N. Identification and classification of 16 new kinesin superfamily (KIF) proteins in mouse genome. Proc Natl Acad Sci USA. 1997;94:9654–96599. doi: 10.1073/pnas.94.18.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclas J, Navone F, Hom BN, Vale RD. Cloning and localization of a conventional kinesin motor expressed exclusively in neurons. Neuron. 1994;12:1059–1072. doi: 10.1016/0896-6273(94)90314-x. [DOI] [PubMed] [Google Scholar]
- O'Hare K, Rubin GM. Structures of Ptransposable elements and their sites of insertion and excision in the Drosophila melanogaster genome. Cell. 1983;34:25–35. doi: 10.1016/0092-8674(83)90133-2. [DOI] [PubMed] [Google Scholar]
- Okada Y, Sato-Yoshitake R, Hirokawa N. The activation of protein kinase A pathway selectively inhibits anterograde axonal transport of vesicles but not mitochondria transport or retrograde transport in vivo. J Neurosci. 1995;15:3053–3064. doi: 10.1523/JNEUROSCI.15-04-03053.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted JB. Affinity purification of antibodies from diazotized paper blots of heterogeneous protein samples. J Biol Chem. 1981;256:11955–11957. [PubMed] [Google Scholar]
- Parfitt K, Reist N, Li J, Burgess R, Deitcher D, DiAntonio A, Schwarz TL. Drosophilagenetics and the functions of synaptic proteins. Cold Spring Harbor Symp Quant Biol. 1995;60:371–377. doi: 10.1101/sqb.1995.060.01.041. [DOI] [PubMed] [Google Scholar]
- Penningroth SM, Rose PM, Peterson DD. Evidence that the 116 kDa component of kinesin binds and hydrolyzes ATP. FEBS Lett. 1987;222:204–210. doi: 10.1016/0014-5793(87)80220-x. [DOI] [PubMed] [Google Scholar]
- Pesavento PA, Stewart RJ, Goldstein LSB. Characterization of the KLP68D kinesin-like protein in Drosophila: possible roles in axonal transport. J Cell Biol. 1994;127:1041–1048. doi: 10.1083/jcb.127.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister KK, Wagner MC, Stenoien DL, Brady ST, Bloom GS. Monoclonal antibodies to kinesin heavy and light chains stain vesicle-like structures, but not microtubules, in cultured cells. J Cell Biol. 1989;108:1453–1463. doi: 10.1083/jcb.108.4.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrotta V. Vectors for P-mediated transformation in Drosophila. . Biotechnology. 1988;10:437–456. doi: 10.1016/b978-0-409-90042-2.50028-3. [DOI] [PubMed] [Google Scholar]
- Robertson HM, Preston CR, Phillis RW, Johnson-Schlitz DM, Benz WK, Engels WR. A stable genomic source of Pelement transposase in Drosophila melanogaster. Genetics. 1988;118:461–470. doi: 10.1093/genetics/118.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GM, Spradling AC. Genetic transformation of Drosophilawith transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- Sato-Yoshitake R, Yorifuji H, Inagaki M, Hirokawa N. The phosphorylation of kinesin regulates its binding to synaptic vesicles. J Biol Chem. 1992;267:23930–23936. [PubMed] [Google Scholar]
- Saxton WM, Hicks J, Goldstein LSB, Raff EC. Kinesin heavy chain is essential for viability and neuromuscular functions in Drosophila, but mutants show no defects in mitosis. Cell. 1991;64:1093–1102. doi: 10.1016/0092-8674(91)90264-y. [DOI] [PubMed] [Google Scholar]
- Saxton WM, Porter ME, Cohn SA, Scholey JM, Raff EC, McIntosh JR. Drosophila kinesin: characterization of microtubule motility and ATPase. Proc Natl Acad Sci USA. 1988;85:1109–1113. doi: 10.1073/pnas.85.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholey JM. Kinesin-II, a membrane traffic motor in axons, axonemes, and spindles. J Cell Biol. 1996;133:1–4. doi: 10.1083/jcb.133.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholey JM, Heuser J, Yang JT, Goldstein LSB. Identification of globular mechanochemical heads of kinesin. Nature. 1989;338:355–357. doi: 10.1038/338355a0. [DOI] [PubMed] [Google Scholar]
- Searles LL, Greenleaf AL, Kemp WE, Voelker RA. Sites of P element insertion and structures of Pelement deletions in the 5′ region of Drosophila melanogaster RpII215. Mol Cell Biol. 1986;6:3312–3319. doi: 10.1128/mcb.6.10.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler S, Nargang FE, Steinberg G, Schliwa M. Kinesin is essential for cell morphogenesis and polarized secretion in Neurospora crassa. EMBO (Eur Mol Biol Organ) J. 1997;16:3025–3034. doi: 10.1093/emboj/16.11.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Boguski MS, Goebl M, Heiter P. A repeating amino acid motif in CDC23 defines a family of proteins and a new relationship among genes required for mitosis and RNA synthesis. Cell. 1990;60:307–317. doi: 10.1016/0092-8674(90)90745-z. [DOI] [PubMed] [Google Scholar]
- Skoufias DA, Cole DG, Wedaman KP, Scholey JM. The carboxyl-terminal domain of kinesin heavy chain is important for membrane binding. J Biol Chem. 1994;269:1477–1485. [PubMed] [Google Scholar]
- Smith RS. The short term accumulation of axonally transported organelles in the region of localized lesions of single myelinated axons. J Neurocytol. 1980;9:39–65. doi: 10.1007/BF01205226. [DOI] [PubMed] [Google Scholar]
- Steinberg G. A kinesin-like mechanoenzyme from the zygomycete Syncephalastrum racemosum shares biochemical similarities with conventional kinesin from Neurospora crassa. Eur J Cell Biol. 1997;73:124–131. [PubMed] [Google Scholar]
- Steinberg G, Schliwa M. The Neurospora organelle motor: a distant relative of conventional kinesin with unconventional properties. Mol Biol Cell. 1995;6:1605–1618. doi: 10.1091/mbc.6.11.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien DL, Brady ST. Immunochemical analysis of kinesin light chain function. Mol Biol Cell. 1997;8:675–689. doi: 10.1091/mbc.8.4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower J, Karpen GH, Craig N, Spradling AC. Preferential transposition of Drosophila Pelements to nearby chromosomal sites. Genetics. 1993;133:347–359. doi: 10.1093/genetics/133.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima I, Yu H, Steuer ER, Sheetz MP. Kinectin, a major kinesin-binding protein on ER. J Cell Biol. 1992;118:1121–1131. doi: 10.1083/jcb.118.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubota S, Schedl P. Hybrid dysgenesis-induced revertants of insertions at the 5′ end of the rudimentary gene in Drosophila melanogaster: transposon- induced control mutations. Genetics. 1986;114:165–182. doi: 10.1093/genetics/114.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Ishikawa H. The movement of membranous organelles in axons. Electron microscopic identification of anterogradely and retrogradely transported organelles. J Cell Biol. 1980;84:513–530. doi: 10.1083/jcb.84.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Fletterick R. The design plan of kinesin motors. Annu Rev Cell Dev Biol. 1997;13:745–777. doi: 10.1146/annurev.cellbio.13.1.745. [DOI] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP. Identification of a novel force- generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee RB, Sheetz MP. Targeting of motor proteins. Science. 1996;271:1539–1544. doi: 10.1126/science.271.5255.1539. [DOI] [PubMed] [Google Scholar]
- Wedaman KP, Knight AE, Kendrick JJ, Scholey JM. Sequences of sea urchin kinesin light chain isoforms. J Mol Biol. 1993;231:155–158. doi: 10.1006/jmbi.1993.1267. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Nakata T, Okada Y, Hirokawa N. KIF3A/B: a heterodimeric kinesin superfamily protein that works as a microtubule plus end-directed motor for membrane organelle transport. J Cell Biol. 1995;130:1387–1399. doi: 10.1083/jcb.130.6.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JT, Laymon RA, Goldstein LS. A three-domain structure of kinesin heavy chain revealed by DNA sequence and microtubule binding analyses. Cell. 1989;56:879–889. doi: 10.1016/0092-8674(89)90692-2. [DOI] [PubMed] [Google Scholar]
- Yang JT, Saxton WM, Stewart RJ, Raff EC, Goldstein LSB. Evidence that the head of kinesin is sufficient for force generation and motility in vitro. Science. 1990;249:42–47. doi: 10.1126/science.2142332. [DOI] [PubMed] [Google Scholar]
- Yu H, Nicchitta CV, Kumar J, Becker M, Toyoshima I, Sheetz MP. Characterization of kinectin, a kinesin-binding protein: primary sequence and N-terminal topogenic signal analysis. Mol Biol Cell. 1995;6:171–183. doi: 10.1091/mbc.6.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Toyoshima I, Steuer ER, Sheetz MP. Kinesin and cytoplasmic dynein binding to brain microsomes. J Biol Chem. 1992;267:20457–20464. [PubMed] [Google Scholar]
- Zhang P, Spradling AC. Efficient and dispersed local Pelement transposition from Drosophila females. Genetics. 1993;133:361–373. doi: 10.1093/genetics/133.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]