

Abstract
The small GTPase racE is essential for cytokinesis in Dictyostelium. We found that this requirement is restricted to cells grown in suspension. When attached to a substrate, racE null cells form an actomyosin contractile ring and complete cytokinesis normally. Nonetheless, racE null cells fail completely in cytokinesis when in suspension. To understand this conditional requirement for racE, we developed a method to observe cytokinesis in suspension. Using this approach, we found that racE null cells attempt cytokinesis in suspension by forming a contractile ring and cleavage furrow. However, the cells form multiple blebs and fail in cytokinesis by regression of the cleavage furrow. We believe this phenotype is caused by the extremely low level of cortical tension found in racE null cells compared to wild-type cells. The reduced cortical tension of racE null cells is not caused by a decrease in their content of F-actin. Instead, mitotic racE null cells contain abnormal F-actin aggregates. These results suggest that racE is essential for the organization of the cortical cytoskeleton to maintain proper cortical integrity. This function of racE is independent of attachment to a substrate, but can be bypassed by other signaling pathways induced by adhesion to a substrate.
The rho family of small GTPases participates in a broad range of cellular processes including cytoskeletal organization, endocytosis, and regulation of cell growth (Tapon and Hall, 1997). Individual rho proteins can modulate the organization of distinct actin structures in response to different stimuli. In particular, rhoA organizes actin into stress fibers, whereas rac1 controls the formation of lamellipodia and cdc42 controls filopodia formation (Hall, 1994). Rho proteins are also thought to have an important role in cytokinesis. Inhibition of rhoA activity leads to a failure in cytokinesis in sand dollar eggs, and in Xenopus and Drosophila embryos (Kishi et al., 1993; Mabuchi et al., 1993; Drechsel et al., 1996). Similarly, inhibition of cdc42 activity in Xenopus embryos and mammalian cells results in a cytokinesis defect (Drechsel et al., 1996; Dutartre et al., 1996). However, the role of these proteins is not unique to cytokinesis, as inhibition of their function affects a variety of cellular processes. In contrast, a genetic screen designed to isolate cytokinesis mutants in Dictyostelium identified a novel rho protein, called racE, which plays a specific role during cytokinesis. Dictyostelium racE null mutants have a strong defect in cytokinesis: they fail to divide in suspension and become large and multinucleate (Larochelle et al., 1996). Importantly, many other motile processes remain unaffected in these mutant cells.
Further genetic experiments in Dictyostelium have also highlighted the role of rho proteins in cytokinesis. Mutations in two related Dictyostelium proteins, gapA and Dgap1, also cause a failure in cytokinesis (Faix and Dittrich, 1996; Adachi et al., 1997; Lee et al., 1997). These two proteins are interesting because they are related to the mammalian protein IQGAP-1, which is thought to be an effector of rac and cdc42 (Kuroda et al., 1996; McCallum et al., 1996). It is not yet known whether these proteins act through the same pathway as racE or that of another rho protein.
Whereas members of the rho family of small GTPases clearly play vital roles in cytokinesis, the precise stages they regulate are not known. Cytokinesis occurs in multiple steps that require exquisite regulation. First, the plane of division must be determined at the equatorial cortical region of the cell. Next, the actomyosin contractile ring must be assembled and induced to contract to effect the ingression of the cleavage furrow. Finally, the juxtaposed membranes must be resolved to produce two independent cells. Which of these stages is affected when rho function is disturbed? The microinjection studies in Xenopus and Drosophila embryos suggest that rhoA and cdc42 function is required either for the assembly of the contractile ring or for the ingression of the cleavage furrow (Drechsel et al., 1996). In contrast, the Dictyostelium GAP1 mutants can form a contractile ring and cleavage furrow, but frequently fail to separate the two daughter cells (Adachi et al., 1997). Thus, different rho proteins may have distinct functions at specific stages of cytokinesis. The challenge is now to sort out those individual functions and to identify their mechanisms of action.
Recent localization studies of racE demonstrated that this protein is distributed at the cortex of the cell throughout all stages of the cell cycle (Larochelle et al., 1997). Furthermore, a constitutively active mutant of racE is completely functional during cytokinesis. These results demonstrated that the activation of racE by GTP is essential for proper cytokinesis, and that racE exerts its function at the cortex of the cell. However, the stage of cytokinesis that is perturbed by the loss of racE remains to be determined.
To answer this question and to delineate the role of racE in cell division, we monitored the progress of cytokinesis in cells lacking racE. To do so, we developed a novel method to study live cells in suspension. We found that racE null cells initially form a contractile ring that constricts forming a cleavage furrow. However, ingression of the cleavage furrow never completes, and cytokinesis fails. Instead, as the cells attempt to divide, they form large blebs and actin aggregates. Furthermore, we found that the loss of racE causes a dramatic reduction in the level of cortical tension in Dictyostelium cells. These results suggest that racE plays a critical role in the organization of the cortical cytoskeleton, which is required for cortical tension and for contraction of the cleavage furrow during cytokinesis.
Materials and Methods
Cell Culture Conditions for the Enrichment of Mitotic Cells
Previous work has shown that washing Dictyostelium cells from a saturated culture into fresh medium produces a semisynchronous culture of dividing cells (Krefft and Weijer, 1989). We found that DH1 cells (the parent strain in our genetic studies) did not recover well from saturated cultures. Fortunately, we found that it is not necessary to force the cells to reach saturation to obtain good semisynchronous cultures. Wild-type DH1 and racE mutant cells were cultured in 100-mm Petri dishes in HL5 media, and passaged before reaching full confluence. To enrich for mitotic cells, log phase cells at about 8.0 × 105 cells/ml were gently removed from the substrate by pipetting, and pelleted in a Beckman GS-6R centrifuge (Fullerton, CA) at 228 g for 5 min at 10°C. The cells were washed twice in 6–10 ml of fresh HL5 medium. The cells were pelleted a third time, and resuspended in HL5 medium at a density of about 2.0 × 105 cells/ml. For the study of adherent cells, the cells were allowed to settle onto glass coverslips. A high number of mitotic racE null cells were observed between 5–7 h after the washing procedure. Wild-type DH1 cells divided between 9–11 h after washing.
A New Method to Observe Dividing Cells in Suspension
A common technique to study cells that are not attached to a coverslip or Petri dish is to place the cells in a hanging drop. Cells settle at the liquid/ air interface and are easily observed in a single focal plane. However, we observed that under these conditions, Dictyostelium cells remained unattached for only a short time. After a few min the cells spread out and translocated like cells attached to a solid substrate. It has been reported that a layer of denatured protein at the air/liquid interface can serve as a good substrate for cell attachment (Giaever and Keese, 1983); this protein layer may serve as a substrate for Dictyostelium cells. We also tested the ability of Dictyostelium cells to remain unattached at the interface of culture medium and fluorocarbon liquids. Again, similarly to what has been reported for mammalian cells (Keese and Giaever, 1983, 1991) we found that Dictyostelium cells were able to attach at the interface within a few minutes of incubation.
We then turned to explore the use of additives to the culture medium that would maintain Dictyostelium cells in suspension. We found that the addition of 40% percoll to the nutrient medium successfully kept the cells from settling down. However, it was very hard to maintain cells in the focal plane due to thermal convection currents within our observation chamber. In contrast, the addition of 0.03% low-melting agarose to the culture medium maintained the cells in suspension, while greatly reducing the amount of convection currents. Furthermore, myosin II or racE null cells cultured in this agarose suspension were not able to divide and became large multinucleate cells (data not shown). Thus, although this concentration of agarose is sufficient to prevent cells from settling, it does not have enough tensile strength to provide a substrate for attachment.
Video Light Microscopy of Attached and Suspended Cells
Cells were viewed in a custom microscopy chamber kindly loaned from D. Kiehart (Duke University, Durham, NC). A complete diagram of this chamber and a detailed description of the microscope setup was described recently (Kiehart et al., 1994). Briefly, the chamber consists of a stainless steel ring fixture with a glass coverslip placed over the top for use on an upright microscope. A clear Teflon sheet that allows gas exchange is fixed to the bottom of the chamber with a metal ring.
For the study of adherent cells, the chamber was filled with fresh HL5, and coverslips containing attached cells were placed face down over the top. For the study of cells in suspension, cells were prevented from settling by the addition of 0.03% low gelling temperature agarose to the HL5 medium. A 0.1% stock solution of low gelling temperature agarose (sea plaque; FMC Bioproducts, Rockland, ME) in HL5 medium was made fresh each time. The stock solution was chilled to 19°C and diluted 1:3 with the cells in suspension. The mixture was immediately loaded into the viewing chamber and covered with a clean coverslip.
The chamber was viewed on an upright Zeiss (Oberkochen, Germany) axioplan microscope. A 610-nm red filter was placed at the field aperture to reduce light toxicity. A 40× phase dry objective with 0.6 NA was used in conjunction with a 2× optivar setting to maintain the maximum range of focus throughout the chamber. Some examples of attached cells were viewed with a 100× 1.4 NA oil objective with a 1× optivar setting. Cells were recorded in real time to videotape using a CCD camera (Dage-MTI, Inc., Michigan City, IN) and a Sony S-VHS VCR (model SVO-9500MD; Park Ridge, NJ).
Video Image Processing
Videotaped images were digitized and processed for time-lapse sequence with the Perception video hardware (Digital Processing Systems Inc., Florence, KY), for the windows NT platform. For montage sequences and Quicktime movies, video images were also digitized in National Institutes of Health (NIH) image 1.61 (available at http://rsb.info.nih.gov) on a Macintosh Power PC equipped with an LG3 frame grabber card (Scion Corp. Frederick, MD). Contrast levels were adjusted using Adobe Photoshop 3.0 (San Jose, CA). Quicktime movies created from NIH image files were compressed with the Movie Cleaner Lite 1.2 software (Terran Interactive, Inc., San Jose, CA).
Fluorescence Microscopy
For the study of cells on a substrate, cells were prepared and attached to coverslips as described above and placed on parafilm inside a humid chamber. At the desired time points, the HL5 media was removed by blotting and replaced by 0.2 ml of fix solution (3.7% formaldehyde in PBS) for 15–30 min. The fix solution was removed by blotting and replaced by 0.2 ml of 0.05% Triton X-100 for 2 min to permeabilize the cells. The detergent solution was blotted and the coverslips were immediately washed three times for 5 min in PBS. To visualize nuclei, the coverslips were then stained with 0.1 μg/ml 4,6-diamidino-2-phenylindole (DAPI),1 50 mM ammonium chloride in PBS for 15 min, followed by three 5-min washes in PBS. This fixation was sufficient for preserving the green fluorescent protein (GFP)-myosin II signal. For F-actin localization, coverslips were also incubated with 0.2 ml 66 nM rhodamine–phalloidin (Molecular Probes, Inc., Eugene, OR) in PBS for 30 min in a covered humid chamber at room temperature. The coverslips were washed three times for 5 min in PBS, and rinsed briefly in dH2O before being mounted onto glass slides with 0.01 ml mounting media (50% glycerol, 100 mg/ml 1,4-diazabicyclo- [2.2.2]octane in PBS). The slides were then sealed with clear nail polish and stored at 4°C.
For the study of cells in suspension conditions, cells were removed from their culture flask at the desired time points and were fixed in suspension. The cells were mixed with an equal volume of 2× fix solution (7.4% formaldehyde in PBS), mixed gently by inversion for 15–30 min, and then centrifuged for 5 min in a clinical centrifuge (International Equipment Co., Needham Heigths, MA) at top speed. The supernatant was aspirated, reserving 0.4–0.6 ml covering the pelleted cells. The fixed cells were carefully resuspended in the reserved volume and were allowed to attach to poly-DL-lysine–coated coverslips for 15–30 min in a moist chamber. From this point, samples were treated identically to the fixed attached cells described above.
Images from the fixed cells were taken on an upright Zeiss axiophot microscope with a 1.4-NA 100× oil objective, and an optivar setting of 1.0× or 1.6×. Pictures were taken using IPlab software (Signal Analytics Corp., Vienna, VA) with a Star I cooled CCD camera (Photometrics Ltd., Tucson, AZ), converted to PICT format and transferred to Adobe Photoshop for contrast adjustment. The Photoshop sharpen filter was used on differential interference contrast (DIC) images.
Measurements of nuclear distance and equatorial diameter in pixels were taken from PICT files in NIH image. The data was plotted with regression curves in Sigma Plot 4.16 (Jandel Scientific, Corte Madera, CA).
Cortical Tension Measurements
To measure the cortical tension of Dictyostelium cells, we used the method of micropipet aspiration (Evans and Yeung, 1989). Briefly, individual cells in suspension were aspirated by a glass micropipet of ∼6–7 μm in diameter. The cell was deformed by aspirating part of the cell into the pipet and the critical pressure (ΔP) required to form a hemispherical projection into the pipet was measured. To obtain ΔP, an aspiration pressure was set to a small value and increased slowly until the cell was aspirated into a hemispherical projection that remained stationary in the pipet. The cortical tension (Tc) was calculated from the following equation: ΔP = 2 Tc (1/Rp − 1/Rc) where Rp and Rc are the radii of the micropipet and the cell, respectively.
This equation is based on the “cortical shell-liquid core” model (Yeung and Evans, 1989). This model includes the assumption that the inside of the cell will not produce resistance to the deformation and that the cell possesses a constant surface tension.
Quantification of F-actin
The relative ratios of F-actin in wild-type and racE null cells were measured as previously described (Insall et al., 1996) with the following modifications. For the measurement of F-actin levels in adherent culture, cells were grown to confluence in 100-mm Petri dishes. Cells were suspended in the culture media and seeded in triplicate onto 60-mm plates (1 ml of cells with 4 ml fresh HL5), and allowed to attach overnight. On the following day, the media and unattached cells were aspirated and gently replaced with 0.5-ml fresh HL5. An equal volume of 2× fix/stain mixture was added (7.4% formaldehyde, 0.2% Triton X-100, 0.2 μM rhodamine-phalloidin in 2× GB buffer: 20 mM KPO4, 25 mM Pipes, pH 6.8, 5 mM EGTA, 2 mM MgSO4), and the cells were allowed to stand in the dark for 1 h. By fixing cells directly in the Petri dishes, we find that the vast majority become tightly bound to the bottom of the dish. The supernatant was carefully removed and reserved for protein quantification using the Bio-Rad DC protein assay (Bio-Rad Laboratories, Hercules, CA). Cells fixed in the dishes were allowed to stand in the dark for 1 h in 1 ml 0.1% saponin in 2× GB. The supernatant was removed and discarded. Methanol (1 ml) was added, and the plates were sealed with parafilm and shaken in the dark for 4 h at 55 rpm. The methanol containing the extracted rhodamine– phalloidin was removed and saved. Particulate debris was spun down in an Eppendorf centrifuge at 14,000 rpm for 1 min. The supernatant was removed and added to 2 ml of fresh methanol and stored covered at −20°C. Rhodamine–phalloidin fluorescence was measured in a FluoroMax spectrofluorometer (Spex Industries, Inc., Edison, NJ) at an excitation wavelength of 550 nm and an emission of 580 nm.
For the measurement of F-actin levels in suspension, cells were washed from confluent 100-mm Petri dishes. Volumes were adjusted so that both wild-type and mutant concentrations were ∼1 × 106 cells/ml. One flask of each cell type was shaken for 4 h at 220 rpm, and 0.5-ml samples were taken in triplicate. An equal volume of 2× fix/stain was added to the cells in Eppendorf tubes, and the suspension was mixed on a Nutator (Clay Adams, Parsippany, NJ) in the dark for 1 h. The cells were pelleted in an Eppendorf centrifuge at 14,000 rpm for 1 min, and the supernatant was reserved for protein quantification. The cells were resuspended in 0.1% saponin wash and mixed in the dark for 1 h. The cells were pelleted and the supernatant was discarded. The cells were resuspended in 1 ml methanol and mixed in the dark for 4 h. As with the attached cells, the particulate debris was spun down and discarded. The supernatant was added to 2 ml methanol and stored at −20°C until measured in the fluorometer.
To normalize the relative amount of F-actin, the fluorescence intensity values were divided by the protein concentration obtained for each cell sample.
Results
RacE Null Cells Form a Successful Cleavage Furrow on a Substrate
We have shown previously that Dictyostelium racE is essential for cytokinesis (Larochelle et al., 1996). When in suspension, racE null mutant cells fail to divide and become large and multinucleate. However, when attached to a substrate, racE null cells grow at normal rates and are small and uninucleate. To gain insight into the function of racE in Dictyostelium, we used first video light microscopy to determine how racE null cells divide while attached to a substrate.
Similarly to wild-type cells (Fig. 1 A), racE null cells formed a cleavage furrow that divided the cells 3–5 min after furrow formation (Fig. 1 B). After division was completed, wild-type cells rapidly crawled away from one another by the extension of large pseudopods (Fig. 1 A, white arrowheads). In contrast, racE null cells did not form these distinct cell processes and, instead, moved using a single broad lamellipodium at the leading edge (Fig. 1 B, bracket). Multiple filopodia were also prominent at the front of these cells (Fig. 1 B, black arrowheads). Thus, whereas racE null cells are able to form a normal cleavage furrow on a substrate, the motile behavior of dividing cells is clearly affected by the loss of racE function.
Figure 1.
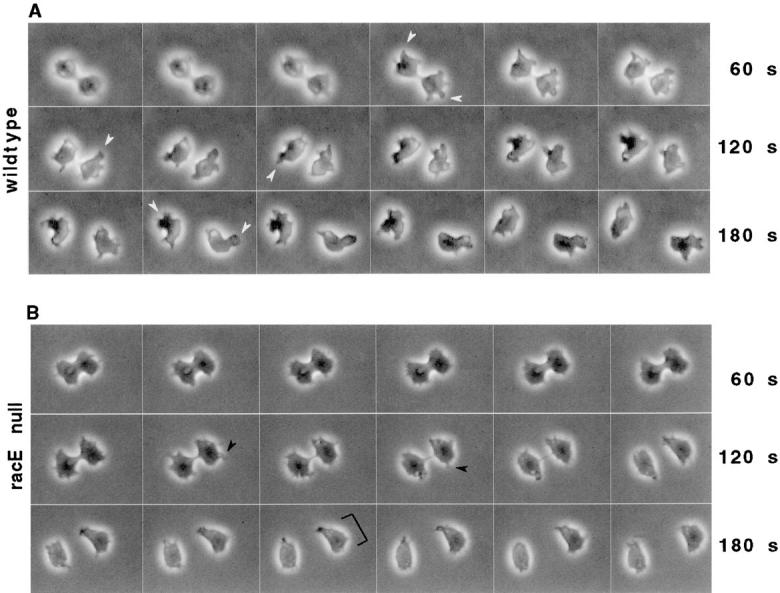
Dictyostelium wild-type and racE null cells complete cytokinesis normally when attached to a substrate. Dictyostelium wild-type (strain DH1) and racE null (strain 24EH6) cells were allowed to attach to coverslips and were visualized by video light microscopy. (A) Wild-type cells form a cleavage furrow that divides the cells 3–5 min after furrow formation. As the daughter cells separate, they form large pseudopods (white arrowheads) to migrate away from each other. (B) RacE null cells also form a cleavage furrow that successfully divides the cell. However, the daughter cells do not form large pseudopods. Instead, they have multiple filopodia (white arrowheads) and a broad leading edge (bracket). The time between each frame is 10 s. QuickTime movies of the images in this manuscript can be accessed at http://note.cellbio. duke.edu/Faculty/∼http://DeLozanne/Gerald1998a.html
Because Dictyostelium cells can use traction forces to produce cleavage furrows on a substrate independently of an actomyosin contractile ring (Neujahr et al., 1997), we investigated the distribution of myosin II in dividing racE null cells. To this end we introduced into wild-type and racE null cells an expression vector for the production of a fusion protein between the GFP and the myosin II heavy chain (MHC; Moores et al., 1996). This fusion protein has been shown to function normally in Dictyostelium cells, and localizes to the contractile ring to the same extent as myosin II (Moores et al., 1996). We fixed the expressing cells and stained them with DAPI to stage phases of mitosis by the morphology of the cell's nuclei (Kitanishi-Yumura and Fukui, 1987). Both wild-type and racE mutant cells concentrated GFP-myosin II into the furrowing region of dividing cells (Fig. 2). These results revealed that racE null cells attached to a substrate can form a contractile ring at the appropriate place and time to successfully divide by cytokinesis. Why then do these cells fail to divide in suspension cultures?
Figure 2.

Wild-type and racE null cells form a contractile ring rich in myosin II when attached to a substrate. Both cell types were transfected with an expression vector for the production of GFP-myosin II heavy chain (Moores et al., 1996). Adherent cells were fixed and stained with DAPI to visualize their nuclei. (A–C) Dividing wild-type cells have a characteristic dumbbell shape and compact nuclei. In these cells, GFP-myosin II is highly concentrated in the contractile ring. (D–I) When racE null cells divide on a substrate, they also have a normal dumbbell shape, with compact nuclei and a myosin II contractile ring.
RacE Null Cells Initiate Furrowing but Then Fail to Divide in Suspension
To address how cytokinesis fails in racE null cells grown in suspension, we developed a method to observe cells that are not attached to a substrate (see Materials and Methods). We used this method to determine the behavior of wild-type and racE null cells undergoing cell division. Wild-type cells divided in suspension similarly to cells attached to a substrate. A cleavage furrow formed at the equatorial plane and the cells separated within 5 min of furrow formation (Fig. 3 A). Because the daughter cells were not able to attach to a substrate, they did not crawl away from each other after separation. Instead, they tumbled around one another and extended large pseudopods (Fig. 3 A, white arrowheads).
Figure 3.
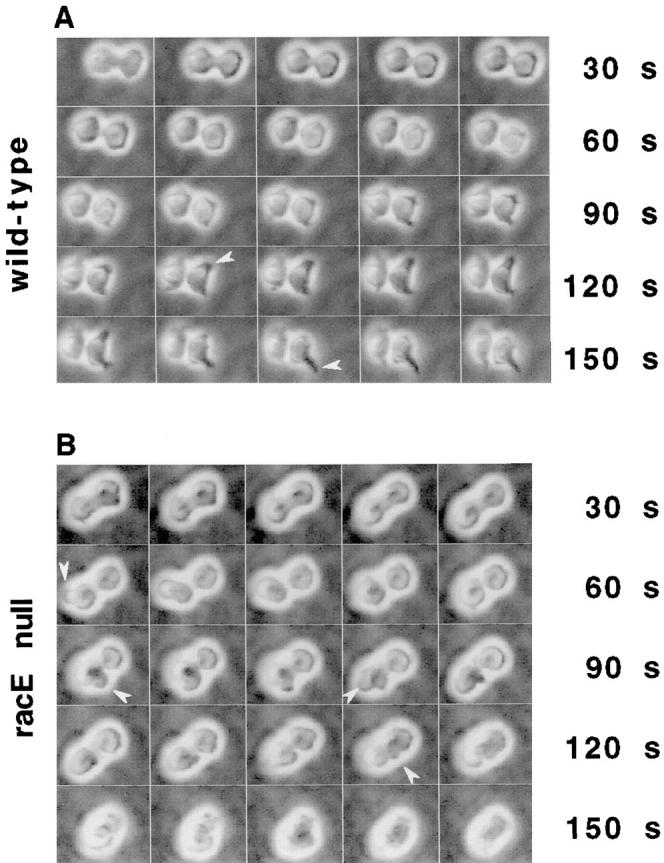
RacE null cells form blebs and fail to complete cytokinesis when grown in suspension culture. Wild-type and racE null cells were placed in a 0.03% agarose solution to maintain them in suspension conditions and were observed by video microscopy. (A) Wild-type cells form a cleavage furrow that rapidly divides the cells. After cleavage is complete, the daughter cells attempt to migrate away from each other by the extension of pseudopods (arrowheads). (B) RacE null cells also form a cleavage furrow but do not divide successfully. As the cells attempt to divide, they form blebs (arrowheads) and their furrows regress.
Interestingly, racE null cells also initiated the formation of a cleavage furrow in suspension. However, the furrow never completed constriction and the cells eventually rounded up and failed to divide (Fig. 3 B). In 6 of 21 cells observed, the cleavage furrow began constriction, halted, and then slowly reverted to form a nonpolarized spherical cell. In the other 15 cases, however, the cells displayed violent shape changes. Some cells extended large blebs (Fig. 3 B, arrowheads), localized frequently near the polar regions of the dumbbell-shaped cell. In other cells, the extension of blebs was accompanied by strong cortical contractions in different regions of the cell. Ultimately, the fate of division was uniform: the cleavage furrow became disrupted and the cell reverted to a spherical shape.
The behavior of racE null cells is very different in attached or suspended conditions. We have not observed blebbing in attached racE null cells at any time during cytokinesis (data not shown). Also, we have observed blebbing behavior in only a small percentage of attached interphase racE null cells. These results suggest that attachment of a substrate can organize the cortical cytoskeleton of racE null cells for normal function. This hypothesis was supported by one remarkable observation. Fortuitously, we observed a racE null cell that initiated cell division in suspension while slowly settling onto the substrate. The cell formed a cleavage furrow (Fig. 4, white arrowhead) but failed in cytokinesis, as furrowing regressed before the cell attached to the substrate (Fig. 4, black arrowheads). Once attached, however, new furrowing was initiated and successfully completed division within 3 min (Fig. 4, white arrow). Thus, attachment to a substrate appeared to be sufficient to allow racE null cells to complete cytokinesis.
Figure 4.

Attachment to a substrate allows a racE null cell to divide. A racE null cell suspended in medium containing less that 0.03% agarose slowly settled onto the substrate. Notice how other attached cells come into the plane of focus. The racE null cell attempts to divide in suspension by forming a cleavage furrow (white arrowhead), but it fails and its furrow begins to regress (black arrowheads). The cell attaches to the coverslip and becomes “phase-dark.” Remarkably, the cell quickly forms a new cleavage furrow (white arrow), which constricts to completion. Each frame = 12 s.
Our observations suggested that racE null cells cannot constrict their cleavage furrows in suspension to the same extent as wild-type cells. To explore this phenotype in more detail, we determined the equatorial diameter of cells in suspension as a function of the distance between the two daughter nuclei (which is a measure of the stage of mitosis of a cell). We fixed wild-type and racE null cells in suspension and stained them with DAPI to easily observe their nuclei. We found that wild-type cells had a tight inverse relationship between their equatorial diameter and internuclear distance (R value = 0.74; Fig. 5 A). As wild-type cells progressed through mitosis, their cleavage furrows constricted. In contrast, racE null cells displayed only a loose relationship between their equatorial diameter and internuclear distance (R value = 0.31; Fig. 5 B). RacE null cells progressed through mitosis, but full constriction of their cleavage furrows did not follow.
Figure 5.
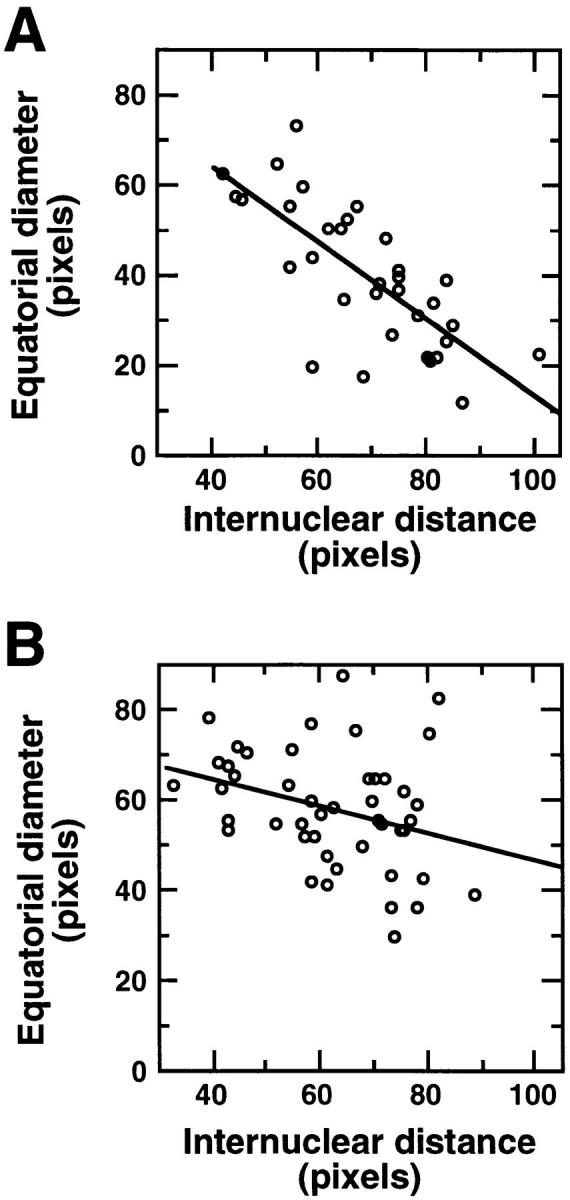
The cleavage furrow of racE null cells does not constrict to the same extent as that of wild-type cells. Wild-type (A) and racE null (B) cells were fixed in suspension culture and stained with DAPI to visualize their nuclei. Cells in anaphase (which display two condensed nuclei with no nucleoli) were photographed and measured. The internuclear distance increases as the cells progress through anaphase and telophase. In wild-type cells, an increase in the internuclear distance is correlated with a constriction of the cleavage furrow (equatorial diameter). In racE null cells the cleavage furrow fails to constrict to the same extent as in wild-type cells. The R values for these linear regressions are: wild-type = 0.74; mutant = 0.31.
RacE Null Cells in Suspension Localize Myosin II to the Presumptive Cleavage Furrow, but Fail to Constrict Late in Cytokinesis
Because the racE null cells formed a cleavage furrow in suspension conditions, it seemed likely that they were able to assemble a contractile ring at the appropriate location. To test this possibility, we determined the localization of GFP-myosin II in wild-type and racE null cells fixed in suspension. RacE null cells fixed in suspension frequently displayed small aggregates of GFP-myosin II, but we believe these are not caused by the loss of racE; we found similar aggregates in wild-type cells as has been reported previously (Yumura and Uyeda, 1997). Contrary to what has been reported recently (Neujahr et al., 1997), we found that wild-type cells displayed a clearly defined equatorial localization of GFP-myosin II. Early in anaphase, GFP-myosin II formed a broad equatorial ring that constricted into a narrow ring in telophase (Fig. 6, A–C). Similarly, racE null cells also formed a broad equatorial ring of GFP-myosin II early in anaphase. However, we never observed constricted rings in telophase (Fig. 6, D–F). These results confirmed that racE null cells in suspension localized GFP-myosin II to contractile rings, yet failed to constrict through late cytokinesis.
Figure 6.

RacE null cells localize myosin II to a contractile ring in suspension conditions. Wild-type (A–C) and racE null (D–F) cells expressing GFP-myosin II were fixed in suspension cultures and stained with DAPI to visualize their nuclei. Under these conditions, wild-type cells concentrate myosin II in the cleavage furrow. These results differ from recently published observations that suggest myosin II does not localize to the furrow region of suspended cells (Neujahr et al., 1997). RacE null cells also localize myosin II in a contractile ring but their furrows never constrict to completion.
RacE Is Required for Cortical Tension and Actin Organization
The blebbing behavior of dividing racE null cells in suspension suggested that the cortex of these cells may be particularly weak. To explore this possibility, we measured the cortical tension of suspended wild-type and racE null cells using the micropipet aspiration method (Evans and Yeung, 1989). Remarkably, whereas wild-type cells displayed about 1.55 dyne/cm, the cortical tension of racE null cells was only 0.28 dyne/cm (Fig. 7). This drop in cortical tension was specifically due to the loss of racE; ectopic expression of constitutively active racE in the racE null cells increased the cortical tension to wild-type levels (Fig. 7). In contrast, expression of constitutively inactive racE did not restore the cortical tension of racE null cells (Fig. 7). Therefore the presence of activated racE at the cortex of Dictyostelium cells is essential for normal cortical tension.
Figure 7.

RacE is essential for maintaining cortical tension in Dictyostelium. The cortical tension of interphase cells in suspension culture was measured by the micropipet aspiration method (Evans and Yeung, 1989). RacE null cells have a dramatic reduction in their cortical tension compared to wild-type cells. Ectopic expression of constitutively active racE (V20-racE; Larochelle et al., 1996) restores the tension of racE null cells to wild-type levels. A constitutively inactive racE protein (N25-racE; Larochelle et al., 1996) is ineffective in restoring cortical tension in racE null cells. Bars indicate mean ± standard deviation of 20 measurements.
One potential role for racE may be to stimulate the polymerization of F-actin at the cortex. However, when we measured the total content of F-actin in racE null cells, we found that it was very similar to that of wild-type cells (Fig. 8). Interestingly, both wild-type and racE null cells have a decreased level of F-actin in suspension compared to attached conditions (Fig. 8). Similarly, F-actin levels also decrease when mammalian cells are placed in suspension (Cunningham, 1995).
Figure 8.

RacE null cells have a normal content of F-actin in attached and suspension conditions. Cells were fixed, attached, or in suspension conditions and their F-actin content was determined. RacE null cells (open bars) have F-actin levels comparable to those of wild-type cells (hatched bars) under both conditions. Interestingly, the level of F-actin is much lower in cells grown in suspension culture. The ordinate represents the units of rhodamine–phalloidin fluorescence (×10−7) divided by the protein concentration of each sample (mg/ml). Bars indicate the mean ± standard deviation of three measurements.
Because racE does not seem to play a role in actin polymerization, we explored the possibility that racE is involved in the organization of cortical actin. We determined the distribution of F-actin in wild-type and racE null cells in suspension conditions. Wild-type and racE null cells were fixed in suspension and stained with DAPI and rhodamine phalloidin. Under these conditions, rhodamine phalloidin staining was observed throughout the cortex of the cells and was most concentrated in actin-rich structures, such as crowns and filopodia. Interestingly, we found that F-actin underwent a dramatic redistribution during mitosis. In interphase cells, F-actin was highly concentrated in large crowns and filopodia (Fig. 9, A and B, and I–J). On the other hand, mitotic cells lacked crowns and had a large number of filopodia on their entire surface (Fig. 9, C–H, K–P). This redistribution was similar in wild-type and racE null cells.
Figure 9.

F-Actin is reorganized in mitotic Dictyostelium cells but is not concentrated in the cleavage furrow. Cells were fixed in suspension cultures and stained with DAPI (A, C, F, I, K, and L) and rhodamine–phalloidin (B, D, E, G, H, J, L, M, O, and P). During interphase (recognized by the large, decondensed nuclei) F-actin is concentrated in filopodia, crowns, and ruffles of both wild-type and racE null cells (A and B; I and J). During mitosis, both cell types loose their crowns and ruffles and are covered by many filopodia (C–H and K–P). F-actin is not particularly concentrated at the cleavage furrow of dividing cells. Two focal planes are shown for mitotic cells stained with rhodamine phalloidin. (A–H) Wild-type cells; (I and J) racE null cells.
In contrast to what has been observed in attached cells by the agar overlay method (Kitanishi-Yumura and Fukui, 1989), F-actin did not seem to be more enriched in the cleavage furrow region of dividing wild-type cells in suspension (Fig. 9, C–H). This might suggest that the extensive attachment of cells under agar enhances the concentration of F-actin in the contractile ring to a higher level than is necessary for cytokinesis in suspension (Gerisch et al., 1993). Similar to wild-type cells, racE null cells fixed in suspension also contained F-actin in their cortices. Interestingly, ∼40% (n = 42) of mitotic racE null cells also displayed abnormal aggregates of cortical F-actin (Fig. 9 M and Fig. 10, A–I). These aggregates were often localized immediately beneath the plasma membrane and could sometimes be observed by DIC microscopy (data not shown). The disruption of the cortical cytoskeleton of racE null cells is especially prevalent in cells in suspension conditions. We have not detected abnormal actin aggregates in racE null cells when attached to a substrate or in wild-type cells under any condition (data not shown).
Figure 10.
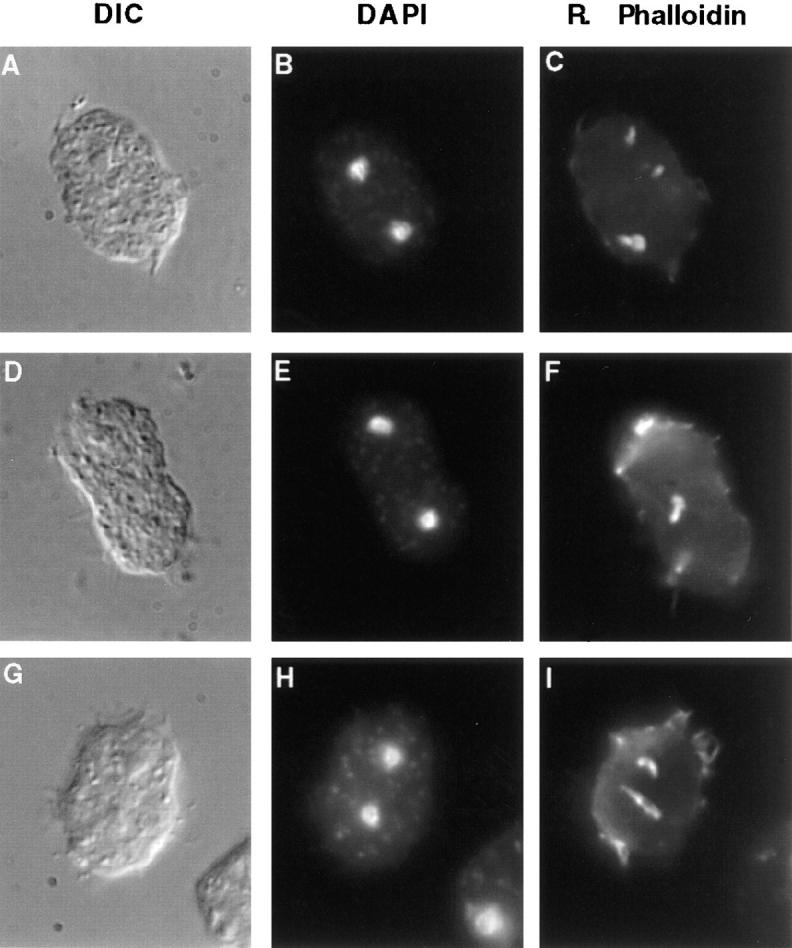
RacE null cells have abnormal F-actin inclusions. Cells were fixed in suspension and stained as in Fig. 9. These three cells are examples of about 40% (n = 42) of mitotic racE null cells that display abnormal F-actin aggregates. These inclusions were not observed in racE null cells during interphase or in any wild-type or myosin II null cells at any stage of the cell cycle. The inclusions appear to form only as racE null cells attempt to divide in suspension.
Discussion
Our phenotypic characterization of racE null cells shows that racE is not involved in the spatial or temporal control of contractile ring formation. RacE null cells can localize myosin II to the cleavage furrow at the correct placement and timing of the cell cycle. Furthermore, we have shown here that the requirement for racE in cytokinesis is specific to cells grown in suspension. When allowed to attach to a substrate, a racE null cell forms a competent contractile ring, rich in actin and myosin II, which constricts and cleaves the cell into two. Therefore, unlike myosin II mutants (Fukui et al., 1990; Neujahr et al., 1997; Zang et al., 1997), which do not form contractile rings and divide by traction-mediated processes, racE null cells divide on a substrate by normal cytokinesis.
Despite their normal cytokinesis when attached, racE null cells are absolutely unable to divide in suspension conditions. This led us to develop methods to directly observe cells undergoing mitosis in suspension. Using these methods we found that, when racE null cells attempt to divide in suspension, they still localize myosin II to the equatorial region and form a contractile ring. RacE null cells begin furrow ingression, but then are unable to complete constriction of the furrow. Ultimately, cytokinesis fails.
The most striking aspect of the cytokinesis failure in racE null cells is that they often suffer violent blebbing. Blebbing occurs only after the cells have initiated furrow ingression. The cells are usually able to recover from the initial blebs while furrow ingression proceeds. However, the furrow is eventually disrupted by a bleb or it stops advancing and regresses. It should be remarked that blebbing is not always associated with the failure in cytokinesis of racE null cells. Several cells have been observed that do not display any obvious blebs, yet their furrows also initiate ingression, stop, and regress. This phenotype is reminiscent of the behavior of HeLa tissue culture cells when treated with dihydrocytochalasin B (Martineau et al., 1995). At concentrations below those required to completely disrupt the actin cytoskeleton, cytochalasin-treated cells can form cleavage furrows that ingress almost to completion. However, like racE null cells, these treated HeLa cells also form blebs and their furrows revert. This similar phenotype suggests that, like cytochalasin-treated cells, racE null cells contain a disrupted actin cortex that causes bleb formation and regression of the cleavage furrow.
In many instances bleb formation indicates a disruption of cortical integrity caused by stress. Blebs are known in diverse cell types during anoxia, ATP depletion, and apoptosis (Cunningham, 1995; Schindl et al., 1995; Laster and Mackenzie, 1996; Chen et al., 1997). However, blebs are not always the symptoms of physical or chemical stress, but can be legitimate protrusive structures in many cell types. Blebs are often observed during lobopodia and pseudopodia formation (Keller and Bebie, 1996). Interestingly, blebs can also appear normally in dividing cells as internal pressures change (Erickson and Trinkaus, 1976; Laster and Mackenzie, 1996; Burton and Taylor, 1997).
In general, bleb formation can occur at any point where the internal hydrostatic pressure exceeds the tension of the actin-associated cortical gel. This can happen in a variety of ways: a rise in internal pressure, a decrease in F-actin cross-linking, a decrease in F-actin content, a lower affinity between the cortex and the plasma membrane (Cunningham, 1995), or any combination of the above.
Direct evidence for a disrupted actin-cortex defect in racE null cells was revealed by the cortical tension measurements in suspension. Because these measurements were made on interphase cells, we can now assert that this defect is not restricted to cytokinesis. We showed that a constitutively active racE construct, which restores growth in suspension, also restores cortical tension. Accordingly, a constitutively inactive racE construct, does not restore either phenotype. The reduced cortical tension and the inability to complete cytokinesis in suspension both rely on the activity of racE. Thus, it is possible that racE null cells fail to divide in suspension because their cortical tensions are not strong enough to withstand the intracellular pressure produced during cytokinesis. Another possible scenario is that the cortical defect, which is responsible for reduced cortical tension, also acts directly on the furrow. For example, if racE helps to tightly bind the cortex to the membrane, then we could expect both the appearance of blebbing at the poles and “slippage” of the contractile ring in the furrow.
A potential role for racE, based on the role of other members of the rho family of GTPases, is to induce polymerization of F-actin at the cortex of the cell. However, we have shown that racE null cells have levels of F-actin similar to those of wild-type cells. An alternative possibility is that racE contributes to the organization of the cortical cytoskeleton. More specific evidence for problems with cortical organization comes from the actin aggregates, which we found in racE null cells in suspension. These aggregates often extended from the membrane into the cell interior, and sometimes were visible by DIC microscopy (data not shown). Actin aggregates are not observed in cells in adherent culture nor in mononucleate racE null cells in suspension (Larochelle et al., 1996), but only appear in mitotic or multinucleate cells in suspension (Fig. 10). Notably, actin aggregates are not seen in myosin II null multinucleate cells fixed in suspension (data not shown). Thus, the actin aggregates may result specifically from the failure in cytokinesis of racE null cells. Analysis of these structures at the EM level, and observations in live cells using GFP probes will help elucidate the precise nature of the cytokinesis defect in racE null cells.
We showed previously that racE is found throughout the cortex of the cell (Larochelle et al., 1996), a distribution consistent with a general role in organizing the cortical actin cytoskeleton. In this light, it is remarkable that racE null cells can still carry out many other cortical functions. For example, racE null cells can contract their cortices in response to azide treatment and cap membrane receptors when challenged with concanavalin A (Larochelle et al., 1996). These cells can also phagocytose and pinocytose at normal levels even in suspension conditions. Furthermore, as we have shown here, racE null cells form normal membrane ruffles, crowns, and filopodia either attached or in suspension conditions. Therefore, the cortical defect of racE null cells affects specifically the integrity of the cortex and the function of the contractile ring.
The question still remains, however, as to how adherent racE null cells complete cytokinesis. Why is the defect of these cells suppressed when they are adhered to a substrate? We have never observed a racE null cell that completes cleavage in suspension, yet, a cell that failed to divide in suspension divided almost immediately once it attached and spread on a substrate (Fig. 4). It appears that all that racE null cells require for successful cytokinesis is the physical state of attachment. Motility undoubtedly provides some assistance for these cells, as they begin to crawl before cytokinesis has completed. However, this is unlikely to be their only advantage on a substrate, as blebbing and actin aggregation are also suppressed in dividing racE null cells when adhered to a substrate.
The screen used to identify racE mutants, although designed to find strains with phenotypes similar to myosin II null cells, was also a screen for mutants exhibiting differences between adherent and nonadherent conditions. It is becoming apparent that differences between these two conditions can turn out to be quite profound states in themselves. Perhaps the most striking example of this is the myosin II null cell line itself, which has been recently shown to form furrows on a substrate by a process termed attachment-assisted mitotic cleavage (Neujahr et al., 1997). As another example, two different myosin I double mutants exhibit conditional pinocytosis defects when grown in suspension, but are indistinguishable from wild-type when grown on a substrate (Novak et al., 1995). The different behavior of attached and suspended cells might result from either the stabilization of the cortical cytoskeleton by attachment to the substrate, from the activation of an attachment-mediated signaling pathway that controls the organization of the cytoskeleton, or from the increased F-actin content of attached cells.
In a very simplistic view, an attached cell is most easily distinguished from a suspended cell by its cytoskeletal organization (Fig. 11). An adherent cell has a top and a bottom, with dramatically different cytoskeletal structures (Novak et al., 1995; Fishkind et al., 1996). The bottom of the cell usually has an abundantly organized cytoskeletal base. Comparatively, the top of the cell has less cytoskeletal material, and is sparsely organized except for surface protrusions. In this scheme, a cell in suspension can be viewed as having its entire surface sparsely organized as “top,” with no stabilizing base.
Figure 11.

The role of racE in cortical tension and cytokinesis. We postulate that racE plays a role in organizing the cortical-actin cytoskeleton by an attachment independent process. (A) Cells on a substrate receive a signal through attachment that stimulates the polymerization of F-actin. The increase in F-actin may be sufficient to suppress any cytoskeletal organization defects in racE null cells. Alternatively, the attachment signal may also organize the cytoskeleton in a racE-independent manner. (B) In suspension conditions, cells are not stimulated by attachment and have less F-actin. RacE is required under these conditions to organize the cytoskeleton and preserve cortical integrity (left). In the absence of racE, the cortical cytoskeleton is less organized, leading to breaches in the cortex and formation of blebs (right). Similarly, the contractile ring forms but fails to constrict to completion.
It is conceivable that the simple adhesion of a cell to a substrate may enhance the stability of its cortex. In fact, it has been postulated that cells may have a mechanical feedback system that allows them to respond to different environments of resistance with appropriate force and organization of the cortical cytoskeleton (Fishkind et al., 1996). A similar argument has been used to explain the concentration of cytoskeletal components in the cortex of Dictyostelium cells covered by agar overlay (Gerisch et al., 1993). In this view, the weak cortex of racE null cells may be stabilized enough by substrate adhesion to allow them to complete cytokinesis and avoid blebbing.
We have shown that Dictyostelium cells have less overall F-actin in suspension compared to adherent conditions. Similar results have been obtained with mammalian cells (Cunningham, 1995). Thus, an attachment-mediated signaling pathway promotes the polymerization of actin in both systems. Therefore, any mutation that causes a destabilization of the cortex (as in racE null cells) may be expressed differently in attached or suspended conditions. When the cells are adhered to a substrate, the increased level of F-actin may be sufficient to stabilize the cortex and suppress any phenotype (Fig. 11 A). In suspended conditions, the reduced level of F-actin may enhance the defect in cortical organization and result in an observable phenotype (Fig. 11 B).
To delineate the exact role of racE, it will be necessary to determine the defect in cortical organization in racE null cells. It will also be important to identify the molecular partners that interact with racE in vivo and that help exert its function.
Acknowledgments
We would like to thank Dan Kiehart and members of his laboratory (Duke University, Durham, NC) for their help in using their equipment. Dan Felsenfeld (Duke University) was instrumental in helping us process video images. Taro Uyeda (National Institute for Advanced Interdisciplinary Research, Tsukuba, Japan) offered helpful suggestions on the use of agarose for suspension culture observations. We also thank Hugh Crenshaw for microscopy instruction and access to equipment in the Howard Hughes Microscopy Laboratory for Undergraduate Instruction, Departments of Botany and Zoology (Duke University). We thank the members of the O'Halloran, Titus, and De Lozanne laboratories for their help and support throughout this work. Denis Larochelle (Duke University) provided us with the racE null cells expressing active and inactive forms of racE. Sheri Moores and James Spudich (Stanford University, Stanford, CA) provided us with the GFP–MHC expression vector.
This work was supported by an R29 grant and a minority supplement award to A. De Lozanne from the National Institutes of Health (NIH) (GM48745). H. Ping Ting-Beall was supported by a grant from NIH (R01-HL23728).
Abbreviations used in this paper
- DAPI
4,6-diamindino-2-phenylindole
- DIC
differential interference contrast
- GFP
green fluorescent protein
- MHC
myosin II heavy chain
Footnotes
Address all correspondence to Arturo De Lozanne, Department of Cell Biology, Box 3709, Duke University Medical Center, Durham, NC 27710. Tel.: (919) 681-6851. Fax: (919) 681-7978. E-mail: a.delozanne@cellbio.duke.edu
References
- Adachi H, Takahashi Y, Hasebe T, Shirouzu M, Yokoyama S, Sutoh K. DictyosteliumIQGAP-related protein specifically involved in the completion of cytokinesis. J Cell Biol. 1997;137:891–898. doi: 10.1083/jcb.137.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton K, Taylor DL. Traction forces of cytokinesis measured with optically modified elastic substrata. Nature. 1997;385:450–454. doi: 10.1038/385450a0. [DOI] [PubMed] [Google Scholar]
- Chen J, Dai J, Grant RL, Doctor RB, Sheetz MP, Mandel LJ. Loss of cytoskeletal support is not sufficient for anoxic plasma membrane disruption in renal cells. Am J Physiol. 1997;272:C1319–C1328. doi: 10.1152/ajpcell.1997.272.4.C1319. [DOI] [PubMed] [Google Scholar]
- Cunningham CC. Actin polymerization and intracellular solvent flow in cell surface blebbing. J Cell Biol. 1995;129:1589–1599. doi: 10.1083/jcb.129.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Hall A, Glotzer M. A requirement for Rho and Cdc42 during cytokinesis in Xenopusembryos. Curr Biol. 1996;7:12–23. doi: 10.1016/s0960-9822(06)00023-6. [DOI] [PubMed] [Google Scholar]
- Dutartre H, Davoust J, Gorvel JP, Chavrier P. Cytokinesis arrest and redistribution of actin-cytoskeleton regulatory components in cells expressing the Rho GTPase CDC42Hs. J Cell Sci. 1996;109:367–377. doi: 10.1242/jcs.109.2.367. [DOI] [PubMed] [Google Scholar]
- Erickson CA, Trinkaus JP. Microvilli and blebs as sources of reserve surface membrane during cell spreading. Exp Cell Res. 1976;99:375–384. doi: 10.1016/0014-4827(76)90595-4. [DOI] [PubMed] [Google Scholar]
- Evans E, Yeung A. Apparent viscosity and cortical tension of blood granulocytes determined by micropipet aspiration. Biophys J. 1989;56:151–160. doi: 10.1016/S0006-3495(89)82660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faix J, Dittrich W. DGAP1, a homologue of rasGTPase activating proteins that controls growth, cytokinesis, and development in Dictyostelium discoideum. FEBS Lett. 1996;394:251–257. doi: 10.1016/0014-5793(96)00963-5. [DOI] [PubMed] [Google Scholar]
- Fishkind DJ, Silverman JD, Wang Y-L. Function of spindle microtubules in directing cortical movement and actin filament organization in dividing cultured cells. J Cell Sci. 1996;109:2041–2051. doi: 10.1242/jcs.109.8.2041. [DOI] [PubMed] [Google Scholar]
- Fukui Y, De Lozanne A, Spudich J A. Structure and function of the cytoskeleton of a Dictyosteliummyosin-defective mutant. J Cell Biol. 1990;110:367–378. doi: 10.1083/jcb.110.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerisch G, Albrecht R, De Hostos E, Wallraff E, Heizer C, Kreitmeier M, Muller-Taubenberger A. Actin-associated proteins in motility and chemotaxis of Dictyosteliumcells. Symp Soc Exp Biol. 1993;47:297–315. [PubMed] [Google Scholar]
- Giaever I, Keese CR. Behavior of cells at fluid interfaces. Proc Natl Acad Sci USA. 1983;80:219–222. doi: 10.1073/pnas.80.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu Rev Cell Biol. 1994;10:31–54. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- Insall RH, Borleis J, Devreotes PN. The aimless RasGEF is required for processing of chemotactic signals through G-protein-coupled receptors in Dictyostelium. Curr Biol. 1996;6:719–729. doi: 10.1016/s0960-9822(09)00453-9. [DOI] [PubMed] [Google Scholar]
- Keese CR, Giaever I. Cell growth on liquid interfaces: role of surface active compounds. Proc Natl Acad Sci USA. 1983;80:5622–5626. doi: 10.1073/pnas.80.18.5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keese CR, Giaever I. Substrate mechanics and cell spreading. Exp Cell Res. 1991;195:528–532. doi: 10.1016/0014-4827(91)90406-k. [DOI] [PubMed] [Google Scholar]
- Keller HU, Bebie H. Protrusive activity quantitatively determines the rate and direction of cell locomotion. Cell Motil Cytoskeleton. 1996;33:241–251. doi: 10.1002/(SICI)1097-0169(1996)33:4<241::AID-CM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Kiehart DP, Montague RA, Rickoll WL, Foard D, Thomas GH. High-resolution microscopic methods for the analysis of cellular movements in Drosophilaembryos. Methods Cell Biol. 1994;44:507–532. doi: 10.1016/s0091-679x(08)60929-2. [DOI] [PubMed] [Google Scholar]
- Kishi K, Sasaki T, Kuroda S, Itoh T, Takai Y. Regulation of cytoplasmic division of Xenopus embryo by rho p21 and its inhibitory GDP/GTP exchange protein (rhoGDI) J Cell Biol. 1993;120:1187–1195. doi: 10.1083/jcb.120.5.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitanishi-Yumura T, Fukui Y. Reorganization of microtubules during mitosis in Dictyostelium: dissociation from MTOC and selective assembly/disassembly in situ. Cell Motil Cytoskeleton. 1987;8:106–117. [Google Scholar]
- Kitanishi-Yumura T, Fukui Y. Actomyosin organization during cytokinesis: reversible translocation and differential redistribution in Dictyostelium. . Cell Motil Cytoskeleton. 1989;12:78–89. doi: 10.1002/cm.970120203. [DOI] [PubMed] [Google Scholar]
- Krefft M, Weijer CJ. Expression of a cell surface antigen in Dictyostelium discoideumin relation to the cell cycle. J Cell Sci. 1989;93:199–204. doi: 10.1242/jcs.93.1.199. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Fukata M, Kobayashi K, Nakafuku M, Nomura N, Iwamatsu A, Kaibuchi K. Identification of IQGAP as a putative target for the small GTPases, cdc42 and rac1. J Biol Chem. 1996;271:23363–23367. doi: 10.1074/jbc.271.38.23363. [DOI] [PubMed] [Google Scholar]
- Larochelle D A, Vithalani K, De Lozanne A. A novel member of the rho family of small GTP-binding proteins is specifically required for cytokinesis. J Cell Biol. 1996;133:1321–1330. doi: 10.1083/jcb.133.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle DA, Vithalani KK, De Lozanne A. The role of DictyosteliumracE in cytokinesis: Mutational analysis and localization studies by use of green fluorescent protein. Mol Biol Cell. 1997;8:935–944. doi: 10.1091/mbc.8.5.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laster SM, Mackenzie JM. Bleb formation and F-actin distribution during mitosis and tumor necrosis factor-induced apoptosis. Microsc Res Tech. 1996;34:272–280. doi: 10.1002/(SICI)1097-0029(19960615)34:3<272::AID-JEMT10>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Lee S, Escalante R, Firtel RA. A ras GAP is essential for cytokinesis and spatial patterning in Dictyostelium. . Development. 1997;124:983–996. doi: 10.1242/dev.124.5.983. [DOI] [PubMed] [Google Scholar]
- Mabuchi I, Hamaguchi Y, Fujimoto H, Morii N, Mishima M, Narumiya S. A rho-like protein is involved in the organisation of the contractile ring in dividing sand dollar eggs. Zygote. 1993;1:325–331. doi: 10.1017/s0967199400001659. [DOI] [PubMed] [Google Scholar]
- Martineau SN, Andreassen PR, Margolis RL. Delay of HeLa cell cleavage into interphase using dihydrocytochalasin B: retention of a postmitotic spindle and telophase disc correlates with synchronous cleavage recovery. J Cell Biol. 1995;131:191–205. doi: 10.1083/jcb.131.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum SJ, Wu WJ, Cerione RA. Identification of a putative effector of cdc42Hs with high sequence similarity to the rasGAP-related protein IQGAP1 and a cdc42Hs binding partner with similarity to IQGAP2. J Biol Chem. 1996;271:21732–21737. doi: 10.1074/jbc.271.36.21732. [DOI] [PubMed] [Google Scholar]
- Moores SL, Sabry JH, Spudich JA. Myosin dynamics in live Dictyostelium cells. Proc Natl Acad Sci USA. 1996;93:443–446. doi: 10.1073/pnas.93.1.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neujahr R, Heizer C, Gerisch G. Myosin II-independent processes in mitotic cells of Dictyostelium discoideum: redistribution of the nuclei, re-arrangement of the actin system, and formation of the cleavage furrow. J Cell Sci. 1997a;110:123–137. doi: 10.1242/jcs.110.2.123. [DOI] [PubMed] [Google Scholar]
- Neujahr R, Heizer C, Albrecht R, Ecke M, Schwartz J-M, Weber I, Gerisch G. Three-dimensional patterns and redistribution of myosin II and actin in mitotic Dictyosteliumcells. J Cell Biol. 1997b;139:1793–1804. doi: 10.1083/jcb.139.7.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak KD, Peterson MD, Reedy MC, Titus MA. Dictyosteliummyosin I double mutants exhibit conditional defects in pinocytosis. J Cell Biol. 1995;131:1205–1221. doi: 10.1083/jcb.131.5.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindl M, Wallraff E, Deubzer B, Witke W, Gerisch G, Sackmann E. Cell-substrate interactions and locomotion of Dictyosteliumwild-type and mutants defective in three cytoskeletal proteins: a study using quantitative reflection interference contrast microscopy. Biophys J. 1995;68:1177–1190. doi: 10.1016/S0006-3495(95)80294-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapon N, Hall A. Rho, rac and cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- Yeung A, Evans E. Cortical shell-liquid core model for passive flow of liquid-like spherical cells into micropipets. Biophys J. 1989;56:139–149. doi: 10.1016/S0006-3495(89)82659-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumura S, Uyeda TQP. Transport of myosin II to the equatorial region without its own motor activity in mitotic Dictyosteliumcells. Mol Biol Cell. 1997;8:2089–2099. doi: 10.1091/mbc.8.10.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang J-H, Cavet G, Sabry JH, Wagner P, Moores SL, Spudich JA. On the role of myosin II in cytokinesis: division of Dictyosteliumcells under adhesive and nonadhesive conditions. Mol Biol Cell. 1997;8:2617–2629. doi: 10.1091/mbc.8.12.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]