

Abstract
Severe low-renin hypertension has few known causes. Apparent mineralocorticoid excess (AME) is a genetic disorder that results in severe juvenile low-renin hypertension, hyporeninemia, hypoaldosteronemia, hypokalemic alkalosis, low birth weight, failure to thrive, poor growth, and in many cases nephrocalcinosis. In 1995, it was shown that mutations in the gene (HSD11B2) encoding the 11β-hydroxysteroid dehydrogenase type 2 enzyme (11β-HSD2) cause AME. Typical patients with AME have defective 11β-HSD2 activity, as evidenced by an abnormal ratio of cortisol to cortisone metabolites and by an exceedingly diminished ability to convert [11-3H]cortisol to cortisone. Recently, we have studied an unusual patient with mild low-renin hypertension and a homozygous mutation in the HSD11B2 gene. The patient came from an inbred Mennonite family, and though the mutation identified her as a patient with AME, she did not demonstrate the typical features of AME. Biochemical analysis in this patient revealed a moderately elevated cortisol to cortisone metabolite ratio. The conversion of cortisol to cortisone was 58% compared with 0–6% in typical patients with AME whereas the normal conversion is 90–95%. Molecular analysis of the HSD11B2 gene of this patient showed a homozygous C→T transition in the second nucleotide of codon 227, resulting in a substitution of proline with leucine (P227L). The parents and sibs were heterozygous for this mutation. In vitro expression studies showed an increase in the Km (300 nM) over normal (54 nM). Because ≈40% of patients with essential hypertension demonstrate low renin, we suggest that such patients should undergo genetic analysis of the HSD11B2 gene.
A form of severe low-renin hypertension called apparent mineralocorticoid excess (AME) first was described biochemically in 1977 (1). Similar patients subsequently were described, all of whom had prominent clinical signs and symptoms, including low birth weight, polyuria and polydipsia, failure to thrive, severe hypertension, hypokalemia, hypoaldosteronemia, nephrocalcinosis, and suppressed plasma renin activity (PRA), and it has been associated with sudden fatality. The deficiency of 11β-hydroxysteroid dehydrogenase type 2 enzyme has been demonstrated in patients with AME and explains the pathogenesis of the disease, which results from excess cortisol binding to the mineralocorticoid receptor (2). The cause of the disease was shown to be mutations in the HSD11B2 gene encoding the 11β-HSD2 enzyme (3). We report here on a form of low-renin hypertension in which a gene mutation produces a mild deficiency in the 11β-HSD2 enzyme. In contrast to previously described patients with AME, this new patient has low-renin hypertension and hypoaldosteronism but no other phenotypic features that would lead to the diagnosis of AME. Thus, the genetic mutation in the HSD11B2 gene, which results in a mild 11β-HSD2 enzyme deficiency, may be the cause of low-renin essential hypertension, the diagnostic basis of which is mostly unknown. Because 40% of patients with essential hypertension are associated with low renin, a number of these patients may have a mild form of AME. Further, as spironolactone causes ready remission, it is important to seek the diagnosis by genetic and clinical studies.
Two isoforms of 11β-HSD have been identified and characterized, both of which originally were suspected of involvement in AME. The type 1 isoform (11β-HSD1) was cloned and characterized by Tannin et al. (4); it is NADP-dependent and is expressed in several human tissues (4, 5). This type was ruled out as the cause of AME (6). Later, a defect in the type 2 isoform was shown to be the cause of AME (2). The type 2 isoform of the 11β-HSD enzyme is NAD-dependent (7). It normally protects humans from cortisol intoxication by converting cortisol to cortisone, which does not bind to the type 1 mineralocorticoid receptor. Because aldosterone is not metabolized by the 11β-HSD enzyme, it normally has unimpeded access to the mineralocorticoid receptor, which has equal affinity to aldosterone and cortisol (8, 9). In patients with AME, however, because cortisol is secreted in milligram amounts whereas aldosterone is secreted in microgram amounts, cortisol saturates the mineralocorticoid receptor in patients with deficient 11β-HSD2 enzyme activity. The excess cortisol binding to the mineralocorticoid receptor produces a hypermineralocorticoid state, which results in hypokalemia, sodium retention, and volume expansion, thus suppressing plasma renin and aldosterone secretion. Signs of mineralocorticoid excess caused by cortisol binding to the mineralocorticoid receptor in the absence of aldosterone is the hallmark of the disease, which has therefore been called “apparent mineralocorticoid excess.”
The cDNA for the enzyme 11β-HSD2 was cloned, and the gene (HSD11B2) encoding the enzyme was mapped to human chromosome 16q22 (10, 11). Immunohistological and activity studies localized the type 2 isoform to the distal nephron of the human kidney (12–16). Recently, researchers have identified mutations in the gene (HSD11B2) encoding 11β-hydroxysteroid dehydrogenase type 2. Thus far, 28 patients in 20 kindreds have had DNA analysis, revealing a total of 16 different mutations in the HSD11B2 gene (refs. 2, 3, 11, and 17–20; P. M. Stewart, personal communication) (Table 1). All of the patients had homozygous defects except three, who were compound heterozygotes (Table 1, Patients 3, 23, and 28). To date, there have been a total of ≈40 patients with AME and two 28-week fetuses with AME reported worldwide (1–3, 6, 11, 17, 21–36) (Table 1).
Table 1.
Review of AME patients worldwide: signs and biochemical features at presentation and subsequent biochemical and genetic evaluation
| Patient | Kindred | Ethnicity | Age, years | Sex | Birth weight, kg | Blood pressure, mmHg | Blood pressure (90th centile for age) | Serum K+, mmol/ liter | THF + 5αTHF/THE | F secretion rate, mg/d | % Conversion, F → E | HSD11B2 mutation | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1* | 1 | American Indian | 1.0 | F | 1.8 | 180/140 | 105/69 | 2.2 | 32.1 | 0.08 | 0 | E356-1 Frameshift | 3 | |
| 2 | 2 | Zoroastrian | 9.0 | M | 2.0 | 250/180 | 110/71 | 3.5 | 9.1‡ | 0.47 | 0 | R337H, ΔY338 | 3 | |
| 3 | 3 | Italian-Moroccan | 4.0 | M | 2.3 | 160/110 | 104/67 | 3.1 | 33.0 | 0.72 | ND | L250R/D244N | 2 | |
| 4 | 4 | African American | 9.3 | F | 2.1 | 130/90 | 109/71 | 2.7 | 8.9 | 0.12 | 2 | R186C | 3 | |
| 5 | 4 | African American | 4.3 | F | 2.6 | 142/98 | 98/70 | 2.8 | 14.9 | 0.15 | 4.2 | R186C | 3 | |
| 6 | 5 | East Indian | 0.8 | M | 2.0 | 150/100 | 105/67 | 2.4 | 20.1 | 0.05 | 6 | R337H, ΔY338 | 3 | |
| 7 | 6 | East Indian | 2.5 | M | 2.3 | 150/100 | 91/68 | 1.79 | 12.5 | 0.83 | 2 | R337H, ΔY338 | 3 | |
| 8 | 7 | Middle Eastern | 10.9 | M | 2.1 | 170/110 | 115/73 | 1.7 | 27.9 | 0.36 | ND | R208C | 3 | |
| 9 | 7 | Middle Eastern | 9.3 | M | 2.4 | 160/118 | 110/71 | 2.9 | 27.3 | 0.24 | ND | R208C | 3 | |
| 10 | 8 | American Indian | 3.3 | M | 2.0 | 205/130 | 99/60 | 0.9 | 26.8 | 0.51 | 1.5 | L250P, L251S | 3 | |
| 11 | 9 | Persian | 14.0 | F | 2.2 | 220/160 | 116/79 | 2.8 | 8.91 | ND | ND | R337C | 17 | |
| 12 | 9 | Persian | 11.6 | M | 2.1 | 170/110 | 110/72 | 2 | 6.85 | ND | ND | R337C | 17 | |
| 13 | 9 | Persian | 4.0 | F | 2.4 | 160/100 | 98/70 | 3.1 | 6.7 | ND | ND | R337C | 17 | |
| 14 | 10 | Turkish | 0.1 | M | 2.5 | 155/115 | 99/60 | 3.0 | 13.8 | ND | 4.5 | N286-1 Frameshift | 2 | |
| 15 | 11 | Mennonite | 12.6 | F | 3.6 | 160/90 | 110/72 | 5.0 | 3.0 | 0.55 | 58.4 | P227L | This report | |
| 16 | 12 | American Indian | 9 | M | 170/100 | 110/71 | 14.4 | R208C | 11 | |||||
| 17 | 13 | Caucasian/South American Indian | 3 | F | 170/110 | 99/71 | 2.3 | 31.3 | R213C | 11, 23 | ||||
| 18 | 13 | Caucasian/South American Indian | 3.8 | F | 200/120 | 108/70 | 2 | 13.4 | R213C | 11, 23 | ||||
| 19* | 13 | Caucasian/South American Indian | 6 | M | – | ND | 23 | |||||||
| 20 | 14 | American Indian | 1 | F | 142/92 | 105/69 | – | 73.8 | L250P, L251S | 11 | ||||
| 21 | 15 | European-American Indian | 1.6 | M | 140/100 | 105/69 | 3.1 | 19.8 | L250P, L251S | 11, 35 | ||||
| 22 | 16 | Mexican American | 26 | F | 180/120 | 123/88 | – | 7.9 | Intron 3 (C to T) | 11 | ||||
| 23 | 17 | Irish American | 2.3 | M | 149/83 | 91/68 | – | 134 | Codon 232, Δ9nt/Codon 305, Δ11nt | 11 | ||||
| 24 | 18 | Asian-Pakistani | 3.5 | M | 141/117 | 100/70 | 2.1 | 20 | R374X | 18, 31 | ||||
| 25 | 18 | Asian-Pakistani | 1.4 | M | 144/91 | 105/69 | 3 | 50 | R374X | 18, 31 | ||||
| 26*† | 18 | Asian-Pakistani | – | M | – | – | – | – | – | – | – | R374X | 18, 31 | |
| 27*† | 18 | Asian-Pakistani | – | M | – | – | – | – | – | – | – | R374X | 18, 31 | |
| 28 | 19 | Japanese | 2 | M | 2.5 | 160/90 | 91/68 | 2.7 | 43.7 | R208H/R337H, ΔY338 | 19 | |||
| 29 | 20 | Brazilian | 7 | F | 160/120 | 110/73 | 1.8 | 29.8 | A328V | 20, 25 | ||||
| 30 | 21 | German | 3 | F | 175/110 | 99/71 | 2.8 | ND | 21 | |||||
| 31 | 22 | French | 4 | F | 2.1 | 140/60 | 98/70 | 2 | 0.61‡ | 1 | ND | 28 | ||
| 32* | 23 | American Indian | 2.7 | F | 180/120 | 101/71 | 2.7 | 9.78 | ND | 35 | ||||
| 33 | 24 | Caucasian | 2 | M | 110/65 | 91/68 | 2.2 | 15.87 | ND | 29 | ||||
| 34 | 25 | Northern European | 1.6 | M | 2.17 | 150/110 | 105/69 | 2.6 | 45 | ND | 33 | |||
| 35* | 26 | Caucasian | 0.4 | M | 2.36 | 200/100 | 105/69 | 1.8 | 68.8 | ND | 30 | |||
| 36 | 27 | Caucasian | 0.8 | F | 140/100 | 120/80 | 3.2 | 15‡ | 8 | R374X§ | 34 | |||
| 37 | 28 | Caucasian | 21 | M | 200/145 | 121/81 | 1.7 | 13.6 | 0 | ND | 24 | |||
| 38* | 18 | Asian-Pakistani | 3.5 | M | – | low | ND | ND | 18, 31 | |||||
| 39 | 29 | Turkish | 1.3 | M | low | 120/90 | 105/69 | low | 38.06 | ND | 32 | |||
| 40 | 30 | French | 2 | M | 3.1 | 140/80 | 91/68 | 2.5 | 22 | ND | 36 | |||
| 41 | 30 | French | 3.3 | M | 3.4 | 160/100 | 99/60 | 2.9 | 42 | ND | 36 | |||
| 42¶ | 31 | 3.3 | ND | 27 | ||||||||||
| 43¶ | 32 | 3.5 | ND | 27 | ||||||||||
| 44¶ | 33 | 9 | ND | 27 | ||||||||||
| Normal values | >2.5 | 3.2–5.2 | 1.0 | 11.5 | 90–95% | |||||||||
Died (Patient 19 died of stroke and may have been affected by AME. Patients 26 and 27 were still born. Patient 35 died after fever, persistently raised blood pressure, and hypokalemia. Patient 38, a twin of Patient 24, died of a diarrheal illness; AME is suspected).
Placental DNA.
THF/THE.
Personal communication from P. M. Stewart.
Unreported potential AME Patients.
ND, not done; F, cortisol; B, cortisone.
In a recent report of our extensive personal experience, genotype, biochemical features, and phenotype of 14 patients with severe low-renin hypertension caused by AME (Patients 1–14, Table 1) were examined (2). All of the patients described had characteristic signs of a severe 11β-HSD2 defect. Birth weights were significantly lower than their unaffected sibs, and the patients were short, underweight, and hypertensive for age. Variable damage of one or more organs (kidneys, retina, heart, and central nervous system) was found in all of the patients except one. The follow-up studies of end organ damage after 2 to 13 years of treatment in six patients demonstrated significant improvement in all patients.
The urinary metabolites of cortisol demonstrated an abnormal ratio with predominance of cortisol metabolites (2); that is, [tetrahydrocortisol (THF) + 5αTHF]/tetrahydrocortisone (THE) was 6.7 to 33 whereas the normal ratio is 1.0. Infusion of [11-3H]cortisol was used in patients to examine their ability to convert cortisol to cortisone by measuring the release of tritiated water. All of the previously reported patients with AME who were examined in this way exhibited little release of tritiated water compared with normal, indicating their failure to convert cortisol to cortisone.
Because of the small number of patients with identical mutations, it was difficult to correlate genotype with phenotype. For example, Patient 1 had one of the most severe mutations, resulting in the truncation of the enzyme 11β-HSD2, and died at the age of 16 years while under treatment whereas Patient 14, with a similarly severe mutation, is thriving at age 3. Three patients (Patients 2, 6, and 7) with identical homozygous mutations from different families had varying degrees of severity in clinical and biochemical features.
Herein we report studies of a patient with a mild form of low-renin hypertension. When the patient was referred with the diagnosis of AME but without hypokalemia and other classic signs and symptoms of the disease, 11β-HSD2 deficiency appeared unlikely. However, the diagnosis of AME proved correct on further examination. Though she demonstrated only mild hypertension and hypoaldosteronism and was otherwise asymptomatic, the patient clearly had AME based on biochemical evidence and later was proven to be homozygous for the P227L mutation of the HSD11B2 gene.
CASE REPORT
This patient is a member of a highly inbred Mennonite population (Fig. 1). She was born by vaginal delivery with a normal birth weight (3.6 kg), similar to her unaffected her sibs (Table 1). She grew normally without typical symptoms of other patients with AME, such as polyuria and polydipsia, failure to thrive, or developmental delay. After the detection of asymptomatic mild low-renin hypertension during a routine sports physical at age 12.5, she was referred to a Kansas medical center for further evaluation. An extensive evaluation by her local Mennonite physician included normal serum electrolytes, urine analysis, urine culture, renal sonogram, renal scan, intravenous pyelogram, renal arteriogram, and urinary catecholamines. The patient was considered to be a potential AME patient by T.W., the local physician and co-author, and was referred to The New York Hospital–Cornell Medical Center Children’s Clinical Research Center for further evaluation. On examination at The New York Hospital–Cornell Medical Center at 13 years of age, her height was 146.4 cm (10th centile), her weight was 46.4 kg (50th centile), her body mass index was 21.8, and she was Tanner stage II for pubic hair and breast development. Her blood pressure was 148/92 (90th centile for age and sex, 124/78), she was normokalemic (serum K+ concentration 4.1 mmol/liter), and did not have alkalosis (CO2 = 26 mmol/liter) (Table 2). Her endocrine evaluation revealed that her PRA was low (0.7 ng/ml/hr) and that her hormone levels were unremarkable, with the exception of her serum aldosterone and urinary pH 1 aldosterone, which were both undetectable (see Tables 2, 3, and 4).
Figure 1.

Pedigree of Mennonite family showing consanguinity (50).
Table 2.
Clinical and steroid hormone data
| Age, years | Height, cm (centile) | Weight, kg (centile) | Blood pressure mmHg | Blood pressure, mmHg (90th centile for age) | K+, mmol/ liter | CO2, mmol/ liter | Serum F., μg/dl | Serum Aldo, ng/dl | Urine pH1 Aldo, μg/24 h | PRA ng/ml⋅h | F secretion rate, mg/m2/d | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12.6 | 147.5 (10) | 45 (40) | 160/90 | 123/79 | 5.0 | ND | ND | <1.2 | <1 | UD | At presentation in Kansas | |
| 13 | 149 (20) | 47.4(50) | 148/92 | 124/78 | 4.1 | 26 | 9.3 | UD | UD | 0.7 | ND | Admission to New York Hospital |
| 13.3 | 149 (20) | 45.8 (50)* | 141/81 | 124/78 | 4.5 | 28 | 12.9 | 14 | ND | 6.6 | 0.9 | Spironolactone (25 mg/day) |
| Chlorothiazide (125 mg/day) | ||||||||||||
| Normal | 158 (50) | 47 (50)† | 110/72 | 3.2–5.2 | 22–32 | 5–15 | 4–18 | 5–20 | 0.3–5.0 | 12.5 |
*Body mass index = 20.8.
Body mass index = 18.8.
ND, Not done; UD, undetectable; Aldo, aldosterone; F, cortisol.
Table 3.
Urinary metabolites on admission to The New York Hospital–Cornell Medical Center
| Urinary metabolites | Patient with mild AME | Typical AME | Normal controls |
|---|---|---|---|
| THF (μg/d) | 340 | 0.1–479.5 | 1469 ± 585 |
| THE (μg/d) | 320 | UD–502.3 | 2589 ± 1292 |
| 5αTHF (μg/d) | 624 | UD–647.1 | 1373 ± 701 |
| THF/THE | 1.1 | 2.1–2.2 | <1 |
| THF + 5α THF/THE | 3.0 | 6.7–33 | 1 |
UD, undetectable.
Table 4.
ACTH stimulation test at admission to The New York Hospital–Cornell Medical Center
| Time | 17-OHP, ng/dl | Δ5, ng/dl | Aldo, ng/dl | DOC, ng/dl | B, μg/dl | F, μg/dl | Δ4, ng/dl | T, ng/dl | DS, ng/dl | DHEA, ng/dl | E2, ng/dl | DHT, ng/dl |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 19 | 40 | B1 | 19 | 0.51 | 18.6 | 80 | 22 | 65 | 135 | 4.9 | 7 |
| 60 | 55 | 221 | B1 | 53 | 2.05 | 35.5 | 91 | 26 | 86 | 240 | 3.7 | 7 |
17-OHP, 17-hydroxyprogesterone; Δ5, 17-hydroxypregnenolone; Aldo, aldosterone; DOC, deoxycorticosterone; B, corticosterone; F, cortisol; Δ4, Δ4-androstenedione; T, testosterone; DS, dehydroepiandrosterone sulfate; DHEA, dehydroepiandrosterone; Es, estradiol; DHT, dihydrotestosterone.
MATERIALS AND METHODS
This patient was studied under an institutionally approved protocol on the Children’s Clinical Research Center of The New York Hospital–Cornell Medical Center. Blood pressures were measured every 2 hr with a mercury sphygmomanometer after the patient had been supine for at least 10 min throughout her hospitalization. The patient was given a diet calculated for calories, sodium, and potassium by the Children’s Clinical Research Center kitchen. Her 24-hr urines were collected and checked for urinary steroids, sodium, potassium, calcium, and creatinine. Blood samples for adrenal steroids, PRA, and electrolytes were collected daily at 8 a.m.
Hormone Analysis.
Hormone studies were performed after all antihypertensive medications had been discontinued for 10 days. Serum cortisol, aldosterone, deoxycortisone, corticosterone, testosterone, dehydroepiandrosterone, Δ4-androstenedione, and estradiol were measured according to reported methods (37–41). PRA was measured by the method of Sealey et al. (42). Urinary steroid metabolites were measured by assays as described by Shackleton et al. (43). Corticotropin (ACTH) stimulation testing was performed by injecting 0.25 mg of Cortrosyn i.v. Serum hormones were assayed at 0 and 60 min after injection.
Metabolic Studies.
Cortisol secretion rate and cortisol half-life were determined as described (1, 2, 22, 44). To establish the dysfunction of the 11β-HSD2 enzyme, the metabolism of cortisol to cortisone was determined by measuring the release of tritiated water after [11-3H]cortisol infusion, as described by Hellman et al. (45). The tritiated water was recovered from the plasma by lyophilization.
Molecular Analysis.
In this patient, exons 2 through 5 of the HSD11B2 gene were sequenced, with the exception of the first 110 base pairs of exon 5. DNA sequencing was done after two rounds of PCR. In the first PCR, genomic DNA (100–500 ng obtained from peripheral blood leukocytes) as described in ref. 46 was denatured for 10 min at 98°C and was amplified using primers 54 GTGACTCTGGTTTTGGCAAGGA and 58 AAGTACAGTACATGCTTCCCTGTGG. The following reagents were added to the denatured DNA: 50 mM KCl, 10 mM Tris⋅HCl (pH 8.3), 1.5 mM MgCl2, 0.01% gelatin, 0.75 units Taq polymerase (Life Technologies, Grand Island, NY), 200 μM dNTP, and 0.3 μM of each primer. The samples were subjected to denaturation at 94°C for 2 min. Five cycles consisting of 94°C for 1 min, 60°C for 1.5 min, and 72°C for 10 min were performed, followed by 30 cycles consisting of 94°C for 1 min, 60°C for 1.5 min, and 72°C for 4 min. A final cycle consisted of 94°C for 1 min, 60°C for 1.5 min, and 72°C for 10 min.
The second PCR was performed in a 50-μl volume containing 2 μl from the first PCR with 15 pmols of forward primer and 5 pmols of reverse biotin primer, as described in ref. 2. The remaining reagents were identical to that of the first PCR. The samples were denatured for 1 min at 95°C and then for 4 cycles of 1 min at 95°C, 30 sec at 58°C, 10 min at 72°C, 30 cycles of 30 sec at 95°C, 30 sec at 58°C, 1 min at 72°C, 1 cycle of 1 min at 95°C, 1.5 min at 58°C, and 10 min at 72°C. The 5′ end of exon 2 was PCR-amplified directly without the first round of PCR by using 100–500 ng of genomic DNA, following the identical conditions of the second PCR by using primers F1 and B1 (2).
DNA Sequencing.
Sequencing of the HSD11B2 gene was performed using solid phase single-strand sequencing with the Sequenase Dye Primer Kit (Applied Biosystems) containing M13 primers. Single-stranded DNA from the PCR fragments was purified with streptavidin-bound magnetic beads (Dynal, Oslo), following the procedure in bulletin 21 from Applied Biosystems. After denaturation to remove the nonbiotinylated DNA strand, the bound DNA strand then was sequenced. Sequencing was done following the procedure described in the sequencing manual supplied with the sequencing kit with the following modifications: For the A and C reactions, 2 μl instead of 1 μl of the respective dye primers were used; and, instead 1 μl of the Sequenase enzyme, 2 μl were used. For the G and T reactions, 4 μl instead of 2 μl of the respective dye primers were used; and, instead of 2 μl of the Sequenase enzyme, 4 μl were used. The reactions were incubated at 37°C for 15 min, and then the four reactions were stopped by pooling them into 200 μl of TT buffer (10 mM Tris⋅HCl, pH 8.0/0.1% Tween 20). The beads were concentrated with a magnet and were washed with an additional 200 μl of TT buffer. The sequencing products were electrophoresed and analyzed with an Applied Biosystems Model 373A sequencer.
In Vitro Expression Studies.
Expression studies were performed as described (47, 48). The pALTER vector containing the insert from pHSD2 (10) was mutagenized with the following oligonucleotide: GACATGCCATATCTGTGCTTGGGGGCC. The mutated insert was subcloned in the mammalian expression vector pcDNAI (Invitrogen) to give the mutant plasmid pP227L.
Chinese hamster ovary modified cell line (CHOP) cells were transfected, and homogenates were prepared and analyzed as described (10), with the exception of the presence or absence of 20% glycerol. The addition of glycerol had little or no effect on the percent conversion of cortisol to cortisone. The Km was determined in homogenates over the range of 25 nM to 400 nM cortisol. The Km in whole cells was determined over a range of 25nM to 800 nM cortisol. For Western blot analysis, 50 mg of transfected CHOP-cell homogenate protein was loaded per lane. Antibody HUH21 (12) was used at 1 mg/ml.
RESULTS
Metabolic Studies.
To study the patient’s cortisol secretion rate, she was infused with [1,2-3H]cortisol. Her secretion rate was 0.55 mg/day, which is extremely low (see Table 1). To study the patient’s ability to convert cortisol to cortisone, the patient was infused with [11-3H]cortisol. The release of tritiated water was used to measure the conversion of cortisol to cortisone. The patient converted 58.4% of [11-3H]cortisol to cortisone. Normal conversion is 90% to 95% whereas previously studied patients with AME converted only 0% to 6% (2, 3, 22, 26, 49). The patient’s 24-hr urinary cortisol metabolites are shown in Table 3. Her (THF + 5αTHF)/THE ratio was 3.0, whereas patients with AME previously studied by us had (THF + 5αTHF)/THE ratios of 6.7 to 33 (2), and it has been reported to be as high as 134 (7).
Adrenal Hormone Studies.
The patient’s baseline and 60-min ACTH-stimulated adrenal hormones were all within the normal limits or close to normal, with the exception of aldosterone (see Table 4). As with most patients with AME, after ACTH stimulation, her aldosterone level remained undetectable.
Gene Mutational Analysis.
DNA analysis of the patient’s HSD11B2 gene revealed a homozygous C to T transition in the second nucleotide of codon 227 (CCG to CTG), resulting in a substitution of a proline for a leucine (P227L) (Fig. 2). The patient’s parents and her two siblings are heterozygous for this mutation.
Figure 2.

Mutations in the gene for 11β-hydroxysteroid dehydrogenase type 2 in patients with AME. These patients were investigated by our group, and the phenotypes are well described. The HSD11B2 gene has five exons, is 6.2 kb long, and has been mapped to chromosome 16q22. The mutation in Patient 15 is reported here (identified by an arrow). All mutations found in affected patients are homozygous except for Patient 3, who is a compound heterozygote.
In Vitro Expression Studies.
In vitro expression studies were used to determine the kinetics and activity of the 11β-HSD2 enzyme with a P227L mutation. Wild-type pHSD2 or mutant pP227L plasmids were transfected into CHOP cells. Kinetic analyses using cell homogenates revealed a Km of 54 nM (data not shown) from cells transfected with pHSD2 and a Km of 350 nM from cells transfected with pP227L (Fig. 3). Kinetic analyses also were done in whole cells and revealed similar Km [62 nM for cells transfected with the wild-type pHSD2 plasmid and 300 nM for cells transfected with the pP227L plasmid (data not shown)].
Figure 3.

Determination of Km value (Michaelis–Menten constant) for the metabolism of cortisol by 11β-HSD2 enzyme in CHOP cell homogenates transfected with pP227L in the presence of 20% glycerol (transfection with pHSD2 resulted in a Km of 54 nM).
Whole cell expression studies resulted in similar conversion activities. The conversion of cortisol to cortisone using the wild-type pHSD2 plasmid was 15%, and the mutant pP227L plasmid resulted in 16% conversion (data not shown). However, expression studies using cell homogenates resulted in only 2% conversion of cortisol to cortisone with pP227L whereas pHSD2 resulted in 90% conversion (data not shown). The presence or absence of 20% glycerol made no difference in the percent conversion. Western analysis showed that comparable amounts of mutant and wild-type 11β-HSD2 were expressed (Fig. 4).
Figure 4.
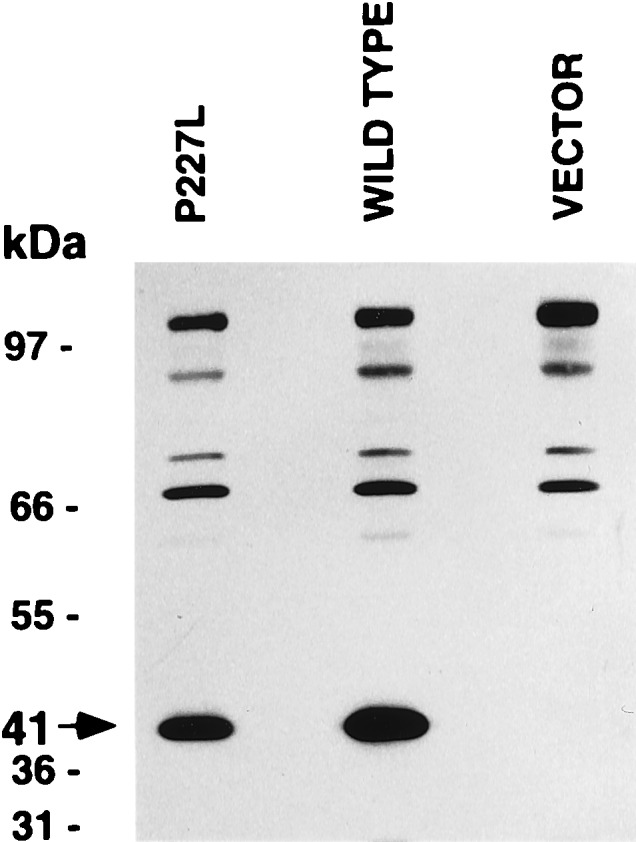
Western blot analysis of homogenates from CHOP cells transfected with pP227L, wild type (pHSD2) or pcDNA1 vector.
DISCUSSION
Essential hypertension has been estimated to occur in 15 million residents in the United States, and ≈40% are associated with low renin. Because mild low-renin hypertension can affect a young patient, it is imperative that it is detected as early as possible for prophylactic therapy and careful monitoring of the patient.
Apparent mineralocorticoid excess is a severe form of low-renin hypertension that begins in childhood. Thus far, it has been rarely reported, with ≈40 patients identified worldwide. DNA analysis has now been performed in 29 patients with AME (Table 1; Fig. 2). We have examined the phenotype of 15 of these patients at The New York Hospital–Cornell Medical Center, including the mild case (Table 1, Patients 1–15). Before this report, all of the patients have displayed symptoms typical of AME, such as low birth weight, failure to thrive, poor growth, severe hypertension, hypokalemia, hyporeninemia, and hypoaldosteronemia. The patient reported in this article did not report symptoms, though mild hypertension relative to her age and weight, low PRA, and low aldosterone were found on examination. She also had moderately abnormal findings on her electroencephalogram: Rare left front-to-central sharp waves occurred spontaneously accompanied by no observed behavioral change, which is suggestive of a seizure disorder of left front-to-central origin but is not diagnostic.
From the data presented in Table 1, it is evident that the genetic defect in HSD11B2 is mild in this patient. She lacks (i) low birth weight, (ii) severe hypertension, (iii) failure to thrive, (iv) polyuria and polydipsia, (v) hypokalemia, and (vi) nephrocalcinosis and other significant end organ damage. Indeed, her major manifestations of AME are hyporeninemia, hypoaldosteronemia, and mild hypertension. In addition, the studies of conversion of cortisol to cortisone indicate that this patient has 11β-HSD2 activity intermediate between severely affected patients with AME and normals. The P227L construct gave a Km for cortisol of 350 nM as compared with a Km of 62 nM for the wild-type construct in whole cells. This is in contrast to the Km in an expression study of a severely affected patient with AME, which was totally devoid of activity. The Km of severely affected patients can be as high as 1,010 nM, as compared with the normal control of 110 nM (47). These studies indicate that the mutation P227L is less severe than the mutations reported in other patients with AME.
Most reported patients with AME have homozygous mutations. Homozygosity for a rare mutation classically is explained by consanguinity, endogamy, or a founder effect. Historical evidence among reported patients with AME suggests that many are members of consanguineous populations (2, 3). The patient described here is from a consanguineous Mennonite family (of the Alexanderwohl Church) (see Pedigree, Fig. 1). It is possible that the mild form of AME seen in this patient may be prevalent in this inbred Mennonite population. A study is in place in which the Mennonite population will be analyzed to determine whether there are other cases of this mild form of AME. The gene frequency, heterozygote frequency, and disease frequency for AME in this population of 2,000 inbred Mennonites will be calculated according to the Hardy–Weinberg Equation (Fig. 5). If 2,000 members of the Mennonite population are screened for the gene mutation observed in our patient, the degree of error in the estimated prevalence will be at most ±2%.
Figure 5.

Calculation of the frequency of a mild form of AME among an inbred Mennonite population of 2,000 members. This calculation would be a minimum frequency if no additional patients with AME are found.
40% of patients with essential hypertension are associated with low renin, a number of these patients may have a mild form of AME. As spironolactone causes ready remission, it is important to seek the diagnosis in this population by clinical studies and genetic analysis of the HSD11B2 gene.
Acknowledgments
We express our appreciation to Laurie Vandermolen for her editorial assistance in the preparation of this manuscript. Significant sections of the work on which the data are reported herein were supported by U.S. Public Health Service Grant HD00072 and General Clinical Research Center Grant RR 06020. Transportation for the patient and her parents was made possible by the Mary T. Harriman Fund.
ABBREVIATIONS
- AME
apparent mineralocorticoid excess
- PRA
plasma renin activity
- THF
tetrahydrocortisol
- THE
tetrahydrocortisone, CHOP, modified Chinese hamster ovary cell line
References
- 1.New M I, Levine L S, Biglieri E G, Pareira J, Ulick S. J Clin Endocrinol Metab. 1977;44:924–933. doi: 10.1210/jcem-44-5-924. [DOI] [PubMed] [Google Scholar]
- 2.Dave-Sharma S, Wilson R C, Harbison M D, Newfield R, Razzaghy-Azar M, Krozowski Z, Funder J W, Shackleton C H L, Bradlow H L, Wei J, et al. J Clin Endocrinol Metab. 1998;83:2244–2254. doi: 10.1210/jcem.83.7.4986. [DOI] [PubMed] [Google Scholar]
- 3.Wilson R C, Harbison M D, Krozowski Z S, Funder J W, Shackleton C H L, Hanauske-Able H M, Wei J Q, Hertecant J, Moran A, Neiberger R E, Balfe J W, et al. J Clin Endocrinol Metab. 1995;80:3145–3150. doi: 10.1210/jcem.80.11.7593417. [DOI] [PubMed] [Google Scholar]
- 4.Tannin G M, Agarwal A K, Monder C, New M I, White P C. J Biol Chem. 1991;266:16653–16658. [PubMed] [Google Scholar]
- 5.Whorwood C B, Mason J I, Rickets M L, Howie A J, Stewart P M. Mol Cell Endocrinol. 1995;110:R7–R11. doi: 10.1016/0303-7207(95)03546-j. [DOI] [PubMed] [Google Scholar]
- 6.Nikkila H, Tannin G M, New M I, Taylor N F, Kalaitzoglou G, Monder C, White P C. J Clin Endocrinol Metab. 1993;77:687–691. doi: 10.1210/jcem.77.3.8370690. [DOI] [PubMed] [Google Scholar]
- 7.Naray-Fejes-Toth A, Fejes-Toth G. Steroids. 1994;59:105–110. doi: 10.1016/0039-128x(94)90085-x. [DOI] [PubMed] [Google Scholar]
- 8.Lan, N. C., Matulich, D. T., Stockigt, J. R., Biglieri, E. G., New, M. I., Winter, J. S., McKenzie, J. K. & Baxter, J. D. (1980) Circ. Res. 46 (Suppl. I) 94–100. [PubMed]
- 9.Lombes M, Kenouch S, Souque A, Farman N, Rafestin-Oblin M E. Endocrinology. 1994;135:834–840. doi: 10.1210/endo.135.3.8070376. [DOI] [PubMed] [Google Scholar]
- 10.Albiston A L, Obeyesekere V R, Smith R E, Krozowski Z S. Mol Cell Endocrinol. 1994;105:R11–R17. doi: 10.1016/0303-7207(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 11.Mune T, Rogerson F M, Nikkila H, Agarwal A K, White P C. Nat Genet. 1995;10:394–399. doi: 10.1038/ng0895-394. [DOI] [PubMed] [Google Scholar]
- 12.Krozowski Z, MaGuire J A, Stein-Oakley A N, Dowling J, Smith R E, Andrews R K. J Clin Endocrinol Metab. 1995;80:2203–2209. doi: 10.1210/jcem.80.7.7608280. [DOI] [PubMed] [Google Scholar]
- 13.Krozowski Z, Albiston A L, Obeyesekere V R, Andrews R K, Smith R E. J Steroid Biochem Mol Biol. 1995;55:457–464. doi: 10.1016/0960-0760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 14.Zhetcho K, Walker P D, Reeves W B. Kidney Int. 1996;49:271–281. doi: 10.1038/ki.1996.39. [DOI] [PubMed] [Google Scholar]
- 15.Naray-Fejes-Toth A, Watlington C O, Fejes-Toth G. Endocrinology. 1991;129:17–21. doi: 10.1210/endo-129-1-17. [DOI] [PubMed] [Google Scholar]
- 16.Naray-Fejes-Toth A, Rusvai E, Fejes-Toth G. Am J Physiol. 1994;266:F76–F80. doi: 10.1152/ajprenal.1994.266.1.F76. [DOI] [PubMed] [Google Scholar]
- 17.Wilson R C, Krozowski Z S, Li K, Obeyesekere V R, Razzaghy-Azar M, Harbison M D, Wei J Q, Shackleton C H L, Funder J W, New M I. J Clin Endocrinol Metab. 1995;80:2263–2266. doi: 10.1210/jcem.80.7.7608290. [DOI] [PubMed] [Google Scholar]
- 18.Stewart P M, Krozowski Z S, Gupta A, Milford D V, Howie J A, Sheppard M C. Lancet. 1996;347:88–91. doi: 10.1016/s0140-6736(96)90211-1. [DOI] [PubMed] [Google Scholar]
- 19.Kitanaka S, Katsumata N, Tanae A, Hibi I, Takeyama K, Fuse H, Kato S, Tanaka T. J Clin Endocrinol Metab. 1997;82:4054–4058. doi: 10.1210/jcem.82.12.4455. [DOI] [PubMed] [Google Scholar]
- 20.Li A, Li K X Z, Marui S, Krozowski Z S, Batista M C, Whorwood C B, Arnhold I J P, Shackleton C H L, Mendonca B B, Stewart P M. J Hypertens. 1997;15:1397–1402. doi: 10.1097/00004872-199715120-00005. [DOI] [PubMed] [Google Scholar]
- 21.Werder E A, Zachmann M, Vollmin J A, Veyrat R, Prader A. Res Steroids. 1974;6:385–389. [Google Scholar]
- 22.Ulick S, Levine L S, Gunczler P, Zanconato G, Ramirez L C, Rauh W, Rosler A, Bradlow H L, New M I. J Clin Endocrinol Metab. 1979;49:757–764. doi: 10.1210/jcem-49-5-757. [DOI] [PubMed] [Google Scholar]
- 23.Shackleton C H, Rodriguez J, Arteaga E, Lopez J M, Winter J S. Clin Endocrinol. 1985;22:701–712. doi: 10.1111/j.1365-2265.1985.tb00160.x. [DOI] [PubMed] [Google Scholar]
- 24.Stewart P M, Corrie J E, Shackleton C H, Edwards C R. J Clin Invest. 1988;82:340–349. doi: 10.1172/JCI113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batista M C, Mendonca B B, Kater C E, Arnhold I J, Rocha A, Nicolau W, Bloise W. J Pediatr. 1986;109:989–993. doi: 10.1016/s0022-3476(86)80282-7. [DOI] [PubMed] [Google Scholar]
- 26.Monder C, Shackleton C H, Bradlow H L, New M I, Stoner E, Iohan F, Lakshmi V. J Clin Endocrinol Metab. 1986;63:550–557. doi: 10.1210/jcem-63-3-550. [DOI] [PubMed] [Google Scholar]
- 27.Kitanaka S, Tanae A, Hibi I. Clin Endocrinol. 1996;44:353–359. doi: 10.1046/j.1365-2265.1996.677500.x. [DOI] [PubMed] [Google Scholar]
- 28.Sann L, Revol A, Zachmann M, Legrand J C, Bethenod M. J Clin Endocrinol Metab. 1976;43:265–271. doi: 10.1210/jcem-43-2-265. [DOI] [PubMed] [Google Scholar]
- 29.Shackleton C H, Honour J W, Dillon M J, Chantler C, Jones R W. J Clin Endocrinol Metab. 1980;50:786–802. doi: 10.1210/jcem-50-4-786. [DOI] [PubMed] [Google Scholar]
- 30.Honour J W, Dillon M J, Levin M, Shah V. Arch Dis Child. 1983;58:1018–1020. doi: 10.1136/adc.58.12.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milford D V, Shackleton C H L, Stewart P M. Clin Endocrinol. 1995;43:241–246. doi: 10.1111/j.1365-2265.1995.tb01923.x. [DOI] [PubMed] [Google Scholar]
- 32.Muller-Berghaus J, Homoki J, Michalk D V, Querfeld U. Acta Paediatr. 1996;85:111–113. doi: 10.1111/j.1651-2227.1996.tb13903.x. [DOI] [PubMed] [Google Scholar]
- 33.Fiselier T J, Otten B J, Monnens L A, Honour J W, van Munster P J. Horm Res. 1982;16:107–114. doi: 10.1159/000179490. [DOI] [PubMed] [Google Scholar]
- 34.Harinck H I, van Brummelen P, Van Seters A P, Moolenaar A J. Clin Endocrinol. 1984;21:505–514. doi: 10.1111/j.1365-2265.1984.tb01388.x. [DOI] [PubMed] [Google Scholar]
- 35.Winter J S D, McKenzie J K. In: Juvenile Hypertension. New M I, Levine S, editors. New York: Raven; 1977. pp. 123–132. [Google Scholar]
- 36.Gourmelen M, Saint-Jacques I, Morineau G, Soliman H, Julien R, Fiet J. Eur J Endocrinol. 1996;135:238–244. doi: 10.1530/eje.0.1350238. [DOI] [PubMed] [Google Scholar]
- 37.Abraham G E, Corrales P C, Teller R C. Anal Lett. 1972;5:915. [Google Scholar]
- 38.Abraham G E, Manlimos F S, Solis M, Wickman A C. Clin Biochem. 1975;8:374–378. doi: 10.1016/s0009-9120(75)93886-2. [DOI] [PubMed] [Google Scholar]
- 39.Korth-Schutz S, Levine L S, New M I. J Clin Endocrinol Metab. 1976;42:117–124. doi: 10.1210/jcem-42-1-117. [DOI] [PubMed] [Google Scholar]
- 40.Rauh W, Levine L S, Gottesdiener K, Chow D, Oberfield S E, Gunczler P, Pareira J, New M I. J Clin Endocrinol Metab. 1979;49:52–57. doi: 10.1210/jcem-49-1-52. [DOI] [PubMed] [Google Scholar]
- 41.Sonino N, Chow D, Levine L S, New M I. Clin Endocrinol. 1981;14:31–39. doi: 10.1111/j.1365-2265.1981.tb00362.x. [DOI] [PubMed] [Google Scholar]
- 42.Sealey J E, Campbell G, Preibisz J J. Renin, Aldosterone, Peripheral Vein, Renal Vein, and Urinary Assays. New York: Raven; 1990. [Google Scholar]
- 43.Shackleton C H L. J Steroid Biochem Mol Biol. 1993;45:127–140. doi: 10.1016/0960-0760(93)90132-g. [DOI] [PubMed] [Google Scholar]
- 44.DiMartino-Nardi J, Stoner E, Martin K, Balfe J W, Jose P A, New M I. Clin Endocrinol. 1987;27:49–62. doi: 10.1111/j.1365-2265.1987.tb00838.x. [DOI] [PubMed] [Google Scholar]
- 45.Hellman L, Nakada F, Zumoff B, Fukushima D, Bradlow H L, Gallagher T F. J Clin Endocrinol Metab. 1971;33:52–62. doi: 10.1210/jcem-33-1-52. [DOI] [PubMed] [Google Scholar]
- 46.Wilson R C, Wei J Q, Cheng K C, Mercado A B, New M I. J Clin Endocrinol Metab. 1995;80:1635–1640. doi: 10.1210/jcem.80.5.7745011. [DOI] [PubMed] [Google Scholar]
- 47.Obeyesekere V R, Ferrari P, Andrews R K, Wilson R C, New M I, Funder J W, Krozowski Z S. J Clin Endocrinol Metab. 1995;80:3381–3383. doi: 10.1210/jcem.80.11.7593456. [DOI] [PubMed] [Google Scholar]
- 48.Ferrari P, Obeyesekere V R, Li K, Wilson R C, New M I, Funder J W, Krozowski Z S. Mol Cell Endocrinol. 1996;119:21–24. doi: 10.1016/0303-7207(96)03787-2. [DOI] [PubMed] [Google Scholar]
- 49.Ulick S, Ramirez L C, New M I. J Clin Endocrinol Metab. 1977;44:799–802. doi: 10.1210/jcem-44-4-799. [DOI] [PubMed] [Google Scholar]
- 50.Wedel D C. The Story of Alexanderwohl. Newton, KA: Mennonite Press; 1974. [Google Scholar]