

Abstract
A major architectural class in engineered binding proteins (“antibody mimics”) involves the presentation of recognition loops off a single-domain scaffold. This class of binding proteins, both natural and synthetic, has a strong tendency to bind a preformed cleft using a convex binding interface (paratope). To explore their capacity to produce high-affinity interfaces with diverse shape and topography, we examined the interface energetics and explored the affinity limit achievable with a flat paratope. We chose a minimalist paratope limited to two loops found in a natural camelid heavy-chain antibody (VHH) that binds to ribonuclease A. Ala scanning of the VHH revealed only three “hot-spot” side chains and additional four residues important for supporting backbone-mediated interactions. The small number of critical residues suggested that this is not an optimized paratope. Using selection from synthetic combinatorial libraries, we enhanced its affinity by >100 fold, resulting in variants with Kd as low as 180 pM with no detectable loss of binding specificity. High-resolution crystal structures revealed that the mutations induced only subtle structural changes but extended the network of interactions. This resulted in an expanded hot-spot region including four additional residues located at the periphery of the paratope with a concomitant loss of the so-called “O-ring” arrangement of energetically inert residues. These results suggest that this class of simple, single-domain scaffolds is capable of generating high-performance binding interfaces with diverse shape. More generally they suggest that highly functional interfaces can be designed without closely mimicking natural interfaces.
Keywords: antibody-antigen interaction, interface topography, binding hot spot, ligand efficiency, O-ring theory
Introduction
Protein-ligand interactions play fundamental roles in biological processes. Consequently, extensive research activities are underway to elucidate the molecular mechanisms underlying protein-ligand interactions and to exploit such knowledge for engineering proteins with novel interaction properties. Minimizing the complexity found in natural protein interaction interfaces is an important and challenging goal in this area of protein engineering. One aspect of such minimization is to reduce both the size of the interface and the size of the molecular scaffold (a protein supporting the interface),1; 2; 3 which complements minimization of chemical complexity in interfaces.4
Molecules of this type, also often referred to as antibody mimics, are designed based on the general architecture found in antibodies and other types of receptors where an interaction interface is constructed by multiple surface loops. In minimalist engineering, a single-domain scaffold, either natural or designed, is used for presenting functional residues, instead of larger, more complex natural scaffolds (e.g. the antigen-binding fragment (Fab) of the immunoglobulins). A common architecture of such scaffolds is a β-sandwich that present two or three recognition loops.1; 2; 3
Camelids produce a class of antibodies whose antigen recognition unit consists of a monomeric VH domain, VHH of about 120 amino acids.5 Thus, even though it is a natural antibody, a VHH domain may be viewed as a prototype of single-domain antibody mimics. Because VHH is monomeric, its binding surface includes only three CDRs.6 Nevertheless, the VHH domains commonly show a level of antigen binding affinity similar to that seen for the standard, heterodimeric antibodies that can utilize up to six CDRs. Numerous crystal structures of VHH in complex with an antigen provide a structural foundation to make VHH a particularly attractive platform for exploring the capacity of single-domain, β-sandwich scaffolds to generate diverse binding interfaces.
Recent structures of single-domain antibodies and antibody mimics, in complex with their respective targets, show that this class of binding proteins has a strong preference to interact with a preformed cleft in the target protein. A recent, comprehensive study by De Genst et al.7 showed that anti-hen eggwhite lysozyme (HEL) VHHs predominantly bind to the active site cleft. Their study found that one anti-HEL VHH that binds outside the cleft had a comparatively lower affinity (Kd = 70 nM) than the cleft-targeting VHHs (Kd as low as 77 pM). Likewise, an engineered fibronectin type III domain with a Tyr/Ser-binary interface was found to bind to the cleft of its target, maltose binding protein.8 This preference to bind to clefts was rationalized in terms of shape complementarity, because a small, single-domain scaffold presents recognition loops in a highly convex shape, which should fit well into a highly concave surface of its target.7
The observed bias in the mode of VHH-antigen interactions suggests that the single-domain antibody mimics might not be suitable for generating a high-affinity interface with a flat geometry. If this were correct, one might wish to use this class of scaffolds exclusively targeting to a concave surface and use/develop other types for targeting a flat surface. As antibody mimics are gaining acceptance as viable antibody alternatives, it is important to define the extent of the potential of this class of scaffold to bind to different topographical features. There are, however, examples of VHH that bind to a flat or convex epitope,9; 10 suggesting that single-domain antibodies and antibody mimics are capable of forming diverse interface shapes. Because there has not been detailed energetic characterization of VHH binding interfaces in general, it is not clear if such flat interfaces are extremely rare members of VHH paratope repertoire that were particularly successful in optimizing interactions in the paratope (following the convention in antibody-antigen interactions, a paratope denotes the interaction interface of an antibody).
In this work, we performed extensive characterization of a VHH that has a small, flat interface (Figure 1A) and bind to its antigen, ribonuclease A (RNaseA) with a low nanomolar Kd originally isolated from an immunized animal.9 This VHH uses only two CDRs to form a particularly small interface among known VHHs, thus it can also be considered as a minimalist antibody-like interface in terms of the loop number (an interface consisting of a single recognition loop probably should be classified as “restrained peptides”). We established a yeast surface display system for VHH that was used to identify binding hotspots by scanning mutagenesis and to perform affinity maturation leading to variants with ∼100 fold higher affinity. A set of high-resolution x-ray crystal structures of the wild-type and affinity matured VHHs and scanning mutagenesis were used to rationalize the observed affinity enhancement. Together, our results indicate that a single-domain β-sandwich scaffold is capable of producing a high-performance recognition interface with a flat or concave geometry and revealed the synergistic contributions of the overall backbone geometry and of the side chains of a small number of residues. Implications in the minimal requirements for a highly functional protein-protein interaction interface and in protein engineering are discussed.
Figure 1. Yeast surface display characterization of the surface expression and antigen binding of cAb-RN05 VHH and Ala-scanning mutants.
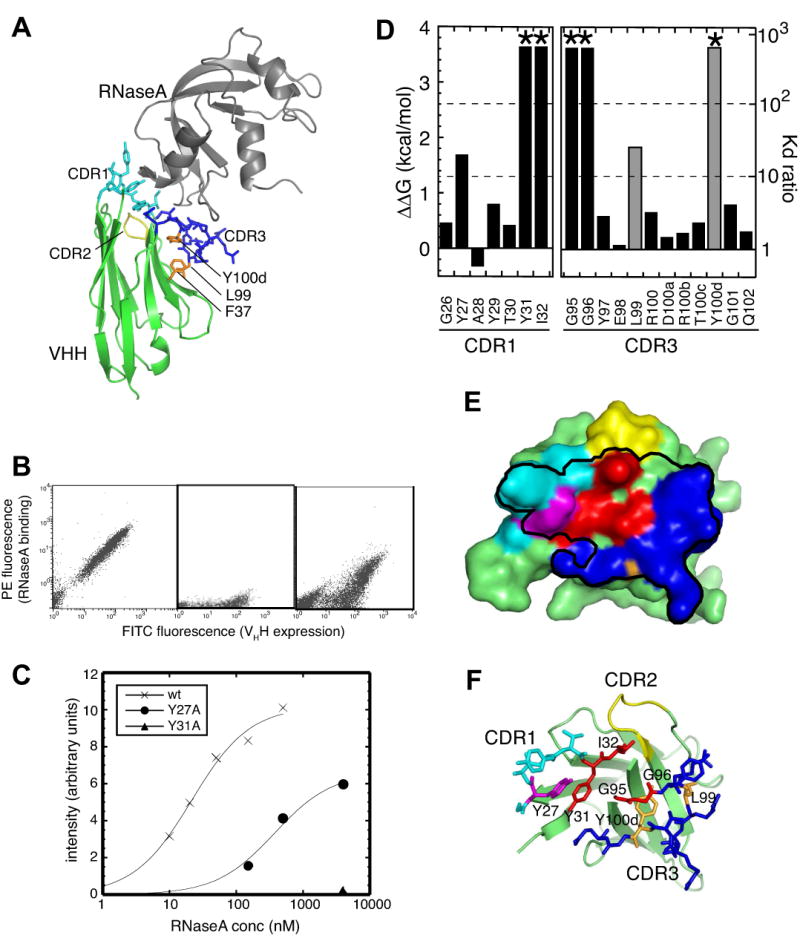
(A) Ribbon drawing of cAb-RN05 –VHH-RNaseA complex (PDB ID, 1BZQ). VHH is shown in green and RNaseA is in gray. CDR1, 2 and 3 of VHH are shown as stick models and in cyan, yellow and blue, respectively. The side chains of F37, L99 and Y100d, which form a mini core, are shown in orange and labeled.
(B) Flow cytometry analysis of yeast cells displaying cAb-RN05 VHH. The horizontal axis shows the intensity of FITC fluorescence emission that indicates the degree of surface expression of VHH. The vertical axis shows the intensity of PE fluorescence emission that indicates the degree of antigen (RNaseA) binding. Each dot represents data for a single cell. The left panels shows data taken with 1 μM RNaseA, the center panel shows the data without the antigen added to the reaction (negative control), and the right panel shows data with 1 μM RNaseA for the Y27A mutant that has reduced antigen binding affinity.
I Antigen titration of the wild-type and mutant VHHs as measured in the yeast surface display format. The sum of PE fluorescence intensity for 10,000 cells is plotted as a function of RNaseA concentration. The X's show data for wild type VHH, the circles for the Y27A mutant, and the triangle for the Y31A mutant. Y31A showed essentially no binding. The curves show the best fit of the 1:1 binding model.
(D) Effects of Ala substitution on the affinity of VHH-antigen interaction, as expressed in ΔΔG (labeled on the left axis) and in the ratio of Kd (= Kdwild-type/Kdmutant; labeled on the right axis). *The Kd for Y31A, I32A, G95A, G96A, and Y100dA is estimated to be greater than 40 μM. The amino acid residue numbers are according to the Kabat convention.9 Data for residues that do not directly contact the antigen (L99 and Y100d) are shown in gray.
(E and F) The surface representation of wild-type cAb-RN05 VHH showing the paratope and binding hot spots. The black line encloses the surface of residues within 5Å from RNaseA. Residues where Ala substitution increased the Kd by >100 fold and by 10–100 fold are show in red and in magenta, respectively. L99 and Y100d, for which Ala substitution significantly reduced antigen binding but which are not in direct contact with the antigen, are shown in orange. The remaining residues in CDR1 and CDR3 are shown in light blue and dark blue, respectively. The CDR2 residues are in yellow. The VHH scaffold is shown as a cartoon model and CDR1 and CDR3 residues as stick models in (E).
Results
A high-resolution x-ray crystal structure of anti-RNaseA VHH
To obtain high-resolution x-ray crystal structures, we constructed an expression system using a synthetic gene for an anti-RNaseA VHH, cAb-RN05 VHH 9 (Supplementary Figure 1) that eliminated flexible residues and a His6-tag at the C-terminus that had been present in the original x-ray crystal structure (1BZQ). For brevity, we will refer to cAb-RN05 VHH simply as the wild-type VHH hereafter. Using this system, we determined the x-ray crystal structure of VHH-RNaseA complex at a 1.4 Å resolution, which revealed a much higher level of details than the original 2.8Å-resolution structure. As expected, the overall structure of the complex is essential identical to the previously determined structure. Additional descriptions of this structure are given near the end of the Results section along with the structure of an affinity-matured variant.
Yeast surface display of VHH
To facilitate quantitative binding characterization and library sorting, we established a yeast surface display system for the VHH (Figure 1 and Supplementary Figure 1). We first tested the surface expression of the VHH and found that the expression level reached the maximum after 18 hours of induction. Unlike yeast display of single-chain Fv (scFv), which commonly requires incubation at lower temperature (20 °C),11 VHH can be displayed robustly at 30°C. This observation suggests more efficient folding of the single-domain VHH molecule than the two-domain scFv, consistent with high-level secretion of a VHH domain from the yeast.12 The dissociation constant (Kd) for the antigen, RNaseA, of the VHH as measured from titration using yeast surface display was 23 ± 3 nM (Figure 1C), consistent with the Kd value of 35nM as previously determined for a purified sample of the same VHH using surface plasmon resonance by Decanniere et al.9 These results indicate that the VHH displayed on the yeast surface is fully functional.
Hot spot identification by alanine-scanning mutagenesis
The thermodynamic contribution of the side chain of each of VHH residues in the VHH-RNaseA interface were determined using alanine-scanning mutagenesis.13 Residues 26-32 in CDR1 and 95-102 in CDR3 were mutated (all numbers are according to the Kabat numbering scheme; Figure 1D). These include all residues that are within 4 Å of RNaseA atoms in the complex and also all the solvent-exposed residues in CDR1 and CDR3. We did not examine CDR2 residues (residues 51-55) because none of them make direct contacts with the antigen. A28 was changed to a Gly, and four Gly residues within the CDRs were respectively replaced with an alanine. All these mutant proteins were expressed on the yeast surface at similar levels. It has been shown that the surface expression level is well correlated with the integrity and stability of a displayed protein.11 Therefore, these results suggest that the mutations in the CDRs did not disrupt the global fold of the VHH.
Effects of alanine substitution on antigen binding varied greatly among the mutants (Figure 1D). Critical residues revealed by alanine scanning in CDR1 are Y27, Y31, and I32, all of which make direct contact with RNaseA. G26 has a positive φ angle, but the Ala substitution a minimal effect, suggesting that this position can accommodate a bulkier side chain and possibly changes in backbone conformation.
Among the 12 residues in CDR3, only 4 positions, G95, G96, L99 and Y100d, caused significant increase in Kd (> 10 fold) upon alanine substitution. Among these residues (Figure 1D), only G95 and G96 make a direct contact with the antigen. The Gly-to-Ala mutation increases the size of the side chain and tends to be more disruptive than the other Ala substitutions. In addition, G95 has a positive φ angle, and Ala substitution of this residue may have caused a significant change in the backbone conformation. While it is difficult to compare quantitatively the effects of these Gly-to-Ala mutations with the rest of the mutations tested here, the structure and the mutation results clearly show that the backbone mediated interactions between these Gly residues and RNaseA are important.
The side chains of Y97, R100b and T100c make contacts with RNaseA, but Ala-substitution of these residues resulted in marginal changes. These results are consistent with the structure of the VHH-RNaseA complex9 in which backbone atoms of CDR3 residues are involved in antigen interactions (Supplementary Table 1). Although Ala scanning cannot directly probe the energetic contributions of backbone atoms, these backbone-mediated interactions are insensitive to perturbations caused by side chain replacement.
The other two CDR3 residues that showed significant increase in Kd upon mutation, L99 and Y100d, are not directly involved in antigen binding. Rather, their side chains point away from RNaseA to the “side” of the VHH scaffold, and, together with a highly conserved F37, they form a mini-core that sequesters the hydrophobic surface corresponding to the VL-binding interface of the VH domain in the conventional, heterodimeric antibody (Figure 1A). Thus, the large effects by the mutations of these residues can be attributed to their roles as “scaffolding” residues that maintain the proper conformation of CDR3 and hence the paratope. The position equivalent to Y100d was found to be important for maintaining the stability of another VHH.14
Ala scanning revealed that only three side chains in direct contact with the antigen are critical and the identity of an additional four positions are important for backbone-mediated interactions. In this work, we define a hot spot as a position that makes direct contact with the antigen and exhibits >10 fold increase in Kd upon Ala substitution. The hot spot residues revealed by Ala-scanning mutagenesis are localized near the center of the paratope (Figure 1E and F), which are surrounded by less important residues. This architecture is reminiscent of the “O-ring” structure seen in other high-affinity protein-protein interaction interfaces.15; 16
Affinity maturation leads to picomolar Kd
To determine whether the affinity of this small, flat paratope of VHH can be significantly increased, we performed affinity maturation experiments. Experiments were done in two stages as outlined in Figure 2A. First, we constructed separate phage display libraries for the CDR1 residues and the CDR3 residues, respectively. In order to sample a large fraction of possible sequences, we designed libraries so that their theoretical sizes are similar to the size attainable by phage display methods (∼1010). Two libraries were made for CDR1. In the first library, termed CDR1-NNT, all positions in CDR1 were randomized using the NNT codon (N is an equal mixture of A, T, G and C) that encodes 15 amino acids (Gln, Glu, Lys, Met and Trp are excluded). The theoretical size of this library is 2.7×108. In the second CDR1 library, termed CDR1-NNK, all 20 amino acid types were allowed at each of the “cold spots” (G26, A28, Y29 and T30) while keeping the hot-spot residues unchanged (its theoretical size is 106). For CDR3, we kept the hot-spot residues (G95 and G96) and the scaffolding residues (L99 and Y100d) unchanged and diversified the remaining nine residues using the NNT codon as described above. The theoretical library size (4.2×109) was comparable to the actual library size. We sorted these libraries separately to enrich functional CDR1 and CDR3 sequences, which were then combined into a single yeast display library. This yeast display library was sorted based on the off rate to enrich high-affinity clones (Figure 2A).11
Figure 2. Affinity maturation of cAb-RN05 VHH and characterization of a selected clone.
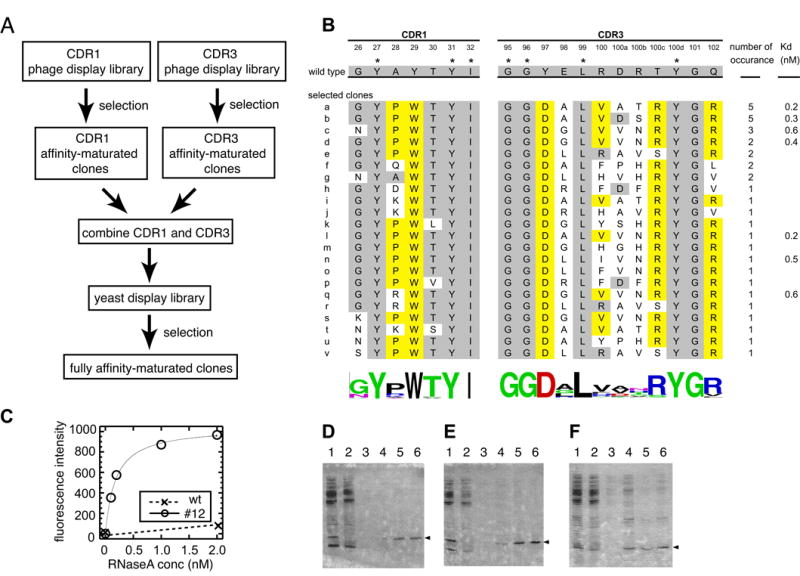
(A) A flow chart showing the overall strategy of affinity maturation. (B) Amino acid sequences of the wild-type VHH and selected mutants. Kd values for a subset of clones are also shown. Residues that were held constant in the combinatorial libraries are marked with an asterisk. Residues that are identical to the wild type are shaded in gray and those represent a new concensus are in yellow. Degrees of sequence diversity among the selected clones are shown using the WebLOGO respresentation.41; 42 (C) Antigen titration curves for the wild type (crosses) and an affinity-matured clone a measured using yeast surface display. Average florescence intensity is plotted as a function of antigen concentration. (D-F) Specificity test using immobilized VHH and E. coli lysate. Data for the wild-type VHH (D), clone a (E) and human SUMO4 (F) are shown. Lane 1 shows the sample (a mixture of a protein with a His6-tag and E. coli lysate) applied to the Ni-affinity column, lane 2 shows the flow-through fraction, lane 3 shows buffer wash fraction, and lanes 4-6 show elution fractions. Co-elution of the protein with a His6-tag (marked with the triangles) and E. coli proteins, as seen in lane 4 of panel F indicate interactions of the protein with E. coli proteins.
After library sorting, we identified 24 unique clones from 36 randomly chosen clones (Figure 2B). All of the high-affinity clones originated from the CDR1-NNK library. Many of clones that exhibited a level of binding similar to the wild type originated from the CDR1-NNT library, and the hot-spot residues in the wild type (Y27, Y31 and I32) were completely conserved among them (data not shown), consistent with the Ala scanning results. These results indicate that the Trp residue at position 29 plays an important role in enhancing the affinity, which was encoded in the CDR1-NNK library but not in the CDR1-NNT library).
In CDR3, the amino acid sequences exhibit a high level of convergence at positions that were not part of the hot spots in the wild type (97, 100c 101 and 102), suggesting their importance among the affinity matured clones. In contrast, residues 98, 100, 100a and 100b retained high levels of sequence diversity, suggesting that their side chains are not involved in specific interactions.
We determined Kd values of seven clones using yeast surface display, all of which were in the sub-nM range (Figure 2B). The Kd values of the best clone (clone a) was 180 (± 18) pM (Figure 2C), corresponding to an improvement of >100 fold relative to the wild type whose Kd determined using yeast display was 23 nM. Using the yeast surface display format, we estimated the koff of the mutant to be greater than 10 hours and that of the wild type on the order of ∼20 minutes (data not shown). These values are consistent with the notion that the reduction of Kd was achieved primarily by a reduction of koff, as expected for an outcome from off-rate based selection.
We produced a soluble protein sample of clone a that contains all the consensus residues (Figure 2B). The protein was monomeric as tested with size exclusion chromatography (data not shown). We analyzed its binding kinetics using surface plasmon resonance. Its extremely slow koff made it impossible to precisely determine the Kd, but the data were consistent with the mutant's much lower Kd than the wild type (data not shown).
Clone a maintained a high level of binding specificity. We immobilized the wild-type and affinity-matured VHH samples to agarose beads, and tested interactions between E. coli lysate and the immobilized VHH. We found no significant binding of E. coli proteins either to the wild type or the affinity-matured VHH (Figure 2 D and E), while a “sticky” control (human SUMO4) showed interactions with many types of E. coli proteins (Figure 2F). Note that this is a highly stringent test for binding specificity, because only weak affinity with a Kd in the sub μM to low μM ranges, i.e. ∼10,000 times higher than the Kd of the mutant VHH for RNaseA, is required for this type of interaction. Thus, despite the ∼100 fold increase in affinity, binding specificity was not compromised in the affinity maturation process.
High-resolution x-ray crystal structures of the wild type and affinity-matured VHHs complexed with RNaseA
To attempt to elucidate the structural basis of the large magnitude of affinity improvement, we determined the x-ray crystal structure of the affinity matured clone a at a 1.9 Å resolution. The overall structure of the affinity-matured complex is nearly identical to the wild-type structure, with the RMSD for the Cα atoms for the VHH and RNaseA between the two structures of 0.38 and 0.49 Å, respectively. There was, however, a small change in the relative orientation between VHH and RNaseA (Figure 3A).
Figure 3. High-resolution x-ray crystal structures of wild-type and affinity-matured VHHs in complex with RNaseA.
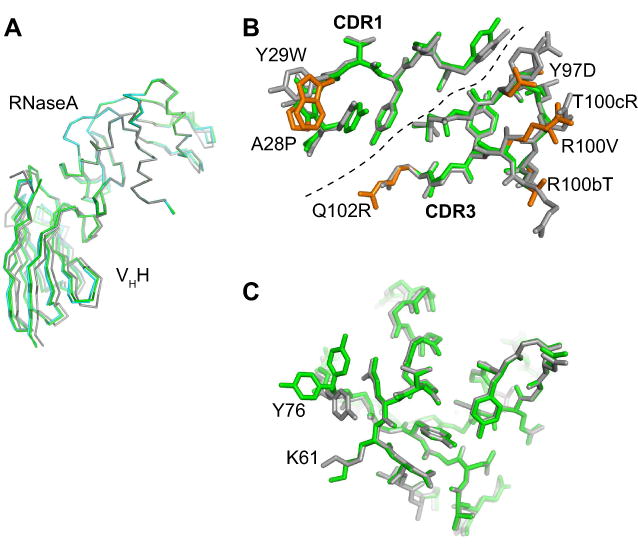
(A) A comparison of the two structures after superposition of the RNaseA portion. The wild type is shown in gray and the mutant in green. (B) CDR1 and CDR3 residues of the wild-type (gray) and mutant (green) VHHs. The dashed line divides CDR1 and CDR3. Mutations in the affinity-matured VHH are shown in orange and labeled. (C) The epitope on RNaseA in the wild-type (gray) and mutant (green) complexes. Y76 has two conformers in the mutant structure.
The affinity maturation process did not significantly change the backbone conformations of CDR1 and CDR3 (Figure 3B). The RMSD for the Cα atoms for all the CDR1 and CDR3 residues between the two structures was 0.34 Å. The side chains of the conserved residues also showed little conformation changes upon affinity maturation (Figure 3B). Likewise, the epitope residues of RNaseA had very similar conformations between the two complexes (Figure 3C). Only the Y76 side chain had clearly different conformations between the two structures. In the affinity-matured complex, it had two conformers, both of which were distinct from its conformation in the wild-type complex. This movement of Y76 exposed K61 of RNaseA with which the indole side chain of VHH Y29W interacts (We denote a residue mutated in the affinity complex in the format of (original amino acid)-(position)-(new amino acid) such as Y29W).
The affinity-matured interface buries a slightly smaller amount of surface areas than that of the wild-type complex (Table 1), but it has a slightly larger number of atoms that are in close contact (≤ 4 Å) with the antigen than the wild type. In contrast to the VHH side of the interface, 37% more antigen atoms are in close contact with the VHH in the affinity-matured complex, suggesting a more efficient paratope. A small increase of the shape complementarity (SC) value (0.78 vs. 0.76; Table 1) is consistent with this view.
Table 1.
Interface characteristics of wild-type and affinity-matured cAb-RN05 and related proteins.
| Epitope | Paratope | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ligand | Kd (nM) | ΔG (kcal/mol) | # of atoms1 | Ligand Efficiency | Area (Å2) | # of atoms1 | Efficiency | Area (Å2) | SC2 | Planarity |
| cAb-RN05 | ||||||||||
| WT | 23 | 10.3 | 46 | 0.22 | 589 | 47 | 0.22 | 613 | 0.76 | 2.13 |
| matured | 0.15 | 13.2 | 63 | 0.21 | 562 | 51 | 0.26 | 592 | 0.78 | 2.02 |
| Other VHH | ||||||||||
| D3-L11 (1ZVY) | 0.14 | 13.3 | 43 | 0.31 | 729 | 58 | 0.23 | 649 | 0.77 | 2.43 |
| D2-L29 (1ZV5) | 10 | 10.8 | 45 | 0.24 | 759 | 60 | 0.18 | 634 | 0.75 | 2.88 |
| cAb-Lys3 (1JTT) | 7.5 | 10.9 | 68 | 0.16 | 690 | 54 | 0.20 | 793 | 0.75 | 3.3 |
| cAb-Lys2 (1RI8) | 0.077 | 13.6 | 52 | 0.26 | 795 | 67 | 0.20 | 771 | 0.71 | 3.08 |
| D2-L19 (1RJC) | 3 | 11.5 | 59 | 0.19 | 741 | 60 | 0.19 | 644 | 0.79 | 2.88 |
| D2-L24 (1ZVH) | 80 | 9.5 | 63 | 0.15 | 528 | 51 | 0.19 | 593 | 0.82 | 1.65 |
| cAb-Hu6 (1OP9) | 0.7 | 12.3 | 45 | 0.27 | 556 | 43 | 0.29 | 577 | 0.76 | 1.61 |
| Small molecule | ||||||||||
| SP4206-IL2R (1PY2) | 68.8 | 9.6 | 44 | 0.22 | 422 | 30 | 0.32 | 572 | 0.77 | 2.01 |
Atoms within 4Å of the binding partner.
Surface complementarity.21
The H-bonds in the interface were highly conserved. In the high-resolution wild-type complex there are nine direct H-bonds at the VHH–RNaseA interface with six main chain atoms participating in them (Supplementary Table 1). Of these six, five are formed by CDR3 main chain carbonyl groups (G95, G96, R100b and T100c), and one by the NH group of I32 in CDR1. All of these H-bonds are preserved in the affinity matured VHH complex (Supplementary Figure 2 and Supplementary Table 2). The wild-type VHH side chains are minimally involved in direct H-bonding interactions with RNaseA. Only Y27 in CDR1 forms direct H-bonds with RNaseA via its side chain, which are also preserved in the affinity-matured complex. There are no salt bridges across the interfaces in either complex.
In the wild-type complex we found four first shell water molecules that coordinate to the CDR main chain and mediate the VHH-RNaseA interactions (Supplementary Table 2). The equivalent water molecules are found in the affinity-matured complex, but three of them formed additional H-bonds to the main chain groups of VHH (Supplementary Figure 2 and Supplementary Table 2). Together, the crystal structures revealed that the affinity maturation process did not cause significant structural perturbations to the VHH structure or to the VHH-RNaseA interactions that existed in the wild-type complex.
Expanded binding hot spots in the affinity matured VHH
The strong convergence of amino acid sequences in the affinity-matured clones (Figure 2B) suggests that many of the paratope residues contribute significantly to binding. To quantify these contributions, we performed Ala-scanning analysis of clone a, whose x-ray crystal structure had been determined. We analyzed 12 residues that showed a high level of sequence convergence in the selection and excluded three residues that were not diversified in the CDR3 library (G95, G96, and Y100d) and four residues that did not show sequence convergence (positions 98, 100, 100a and 100b; Figure 2B).
In CDR1, the side chain contributions of positions 27 and 29 increased (Figure 5). Although the A28P mutation was common and T30 was highly conserved among the selected clones, their side chains did not significantly contribute to binding, as tested using Ala replacement. Thus, the abundance of the A28P mutation and the conservation of T30 are likely to be due to incomplete sampling of sequences in the library. In CDR3, three residues (Y97D, T100cR and Q102R) significantly contributed to binding (Figure 4), while their counterparts in the wild-type VHH only marginally contributed (Figure 1). G101 was fully conserved, but, similar to A28P and T30, Ala substitution had little effect. Therefore, the affinity maturation process converted four “cold spot” residues into hot spots. The hot spot residues account for 76 % of the paratope surface of the affinity-matured VHH. By comparison, only 35 % of the wild-type paratope surface is from the hot spot residues. Interestingly, the expansion of the hot spots in the affinity-matured VHH abolished the “O-ring” architecture of the wild-type protein where hot spots are surrounded by neutral residues (compare Figure 1D and Figure 4B).
Figure 5. Molecular interactions involving mutated residues in the affinity-matured VHH.
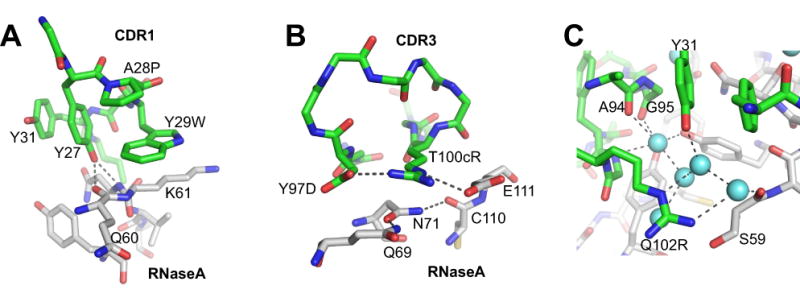
The carbon atoms of VHH are in green and those of RNaseA are in white. The nitrogen and oxygen atoms are in blue and red, respectively. (A) Interactions involving CDR1 residues. H-bonds are depicted with dashed lines. (B) Interactions involving triads of polar residues. (C) Interactions involving Q102R and interface water molecules (spheres in light blue). See the main text for more details.
Figure 4. Energetic contributions of the paratope residues of the affinity-matured VHH probed with Ala-scanning mutagenesis.
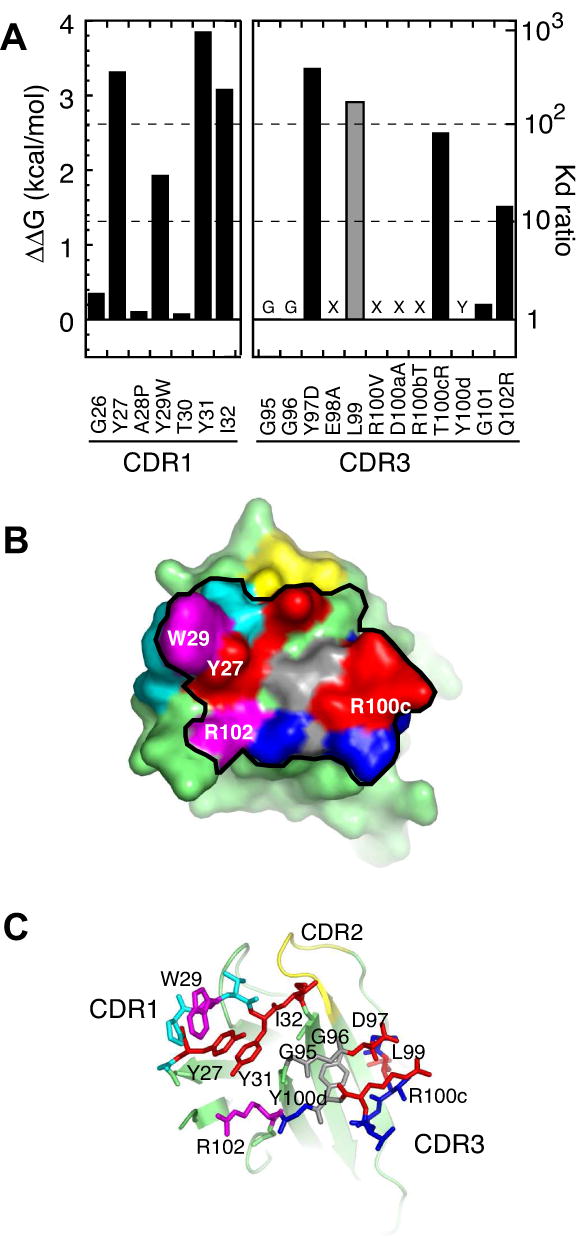
(A) Effects of Ala substitution on antigen binding expressed in terms of ΔΔG (the left axis) and the ratio of Kd (= Kdwild-type/Kdmutant; the right axis). Positions 95, 96, 98, 100-100b and 100d were not tested because (i) Gly residue is critical (labeled with “G”), (ii) no sequence convergence was found in library screening (labeled with “X”) or (iii) the Tyr is involved in scaffolding (labeled with “Y”). The value for L99, a noncontacting residue, is shown in gray. (B) The surface representation of the affinity-matured VHH. The black line encloses residues within 5 Å of antigen atoms. Red and cyan surfaces are for residues where Ala substitution causes >100 fold and 10-100 fold increase in Kd, respectively. Gray surfaces indicate critical residues that were not tested with Ala scanning. The remaining CDR1 and CDR3 residues are shown in light blue and dark blue, respectively. The four residues that expand the hot spot (W29, D97, R100c and R102) are labeled. (C) shows CDR1 and CDR3 residues as sticks and the VHH scaffold as a cartoon model.
Structural basis for the contribution of new hot spot residues to antigen binding
As described above, the Ala-scanning analysis identified five residues, Y27, Y29W, Y97D, T100cR and Q102R, that show significantly enhanced contributions to binding in the affinity-matured complex. While rationalizing affinity changes based on interactions observed in the time-averaged structure of the VHH-RNaseA complexes has limitations, there are several specific, well-defined features that probably play a significant role in the enhanced binding.
The indole ring of Y29W in CDR1 introduces extensive interactions with the side chain of RNaseA K61 (Figure 5A). In the wild-type interface, Y76 of RNaseA folds over K61 and thus masks this hydrophobic patch (Figure 3C).
In both complexes, Y27 hydroxyl forms H-bonds with the backbone NH group of RNaseA N62 and with the backbone carbonyl group of RNaseA Q60. The distance of the latter H-bond is significantly shorter in the affinity-matured complex (2.62 Å) than in the wild type (3.45 Å; Supplementary Table 1). Furthermore, Y27 forms a face-to-edge interaction with the indole ring of Y29W on one side and a face-to-face interaction with Y31 on the other (Figure 5A). These enhanced interactions rationalize the greater impact of the Y27A mutation in the affinity-matured complex (3.3 kcal/mol in the affinity matured complex versus 1.7 kcal/mol in the wild type).
The guanidinium group of T100cR in CDR3 interacts with the carboxyl group of RNaseA E111 and also forms a direct H-bond with the carboxyl group of Y97D in CDR3, another hot spot residue (Figure 5B). This H-bonded Glu-Arg-Asp triad sits on the H-bonded triad of Q69, N71 and C110 of RNaseA, strongly suggesting that this network of polar residues cooperatively forms high-affinity interface. By contrast, the wild-type complex does not contain such a network.
The Q102R side chain atoms do not form direct interactions with other residues, and its importance seems to lie in binding water molecules in the interface. Its guanidinium group forms H-bonds with two water molecules that bridge VHH and RNaseA. These water molecules are a part of a larger network of H-bonded water molecules (Figure 5C). Taken together, the mutations introduced a number of cooperative networks that seem to synergistically contribute to affinity enhancement without increasing the interface size.
Discussion
Energetics of the VHH-antigen interfaces
This work represents the first set of Ala-scanning analysis of VHH-antigen interfaces to characterize their binding determinants. As expected from the extensive involvement of the VHH backbone atoms in antigen binding, Ala-scanning of the wild-type VHH revealed that a relatively few side chains (Y27, Y31 and I32) make critical direct interactions with the antigen (Figure 1). Among the residues involving in backbone-mediated contacts, only G95 and G96 did not tolerate Ala substitution. By comparison, eight to ten side chains are found to be hot spots (>10 fold increase in Kd upon Ala-substitution) in the paratopes of conventional, heterodimeric antibodies17; 18; 19; 20. Similarly, nine residues are hot spots in the growth hormone receptor that consists of loops from two domains.15 Thus, the wild-type VHH paratope achieves high-affinity interaction using much fewer hot spot residues than these examples of larger interfaces.
The high efficiency of the small VHH interface is manifested in its shape complementarity (Table 1). Its shape compelmentarity (SC) value (0.76) is much higher than 0.64-0.68 found for conventional antibodies and at the high end of the range (0.70-0.76) for oligomeric proteins and protease-inhibitor complexes.21 Interestingly, we found that other VHH interfaces also have very high SC values regardless of the paratope shape (Table 1), suggesting that the camelid immune system is capable of generating small and high-efficiency interfaces in VHH in general. A higher shape complementarity may be a general requirement for achieving high affinity using a smaller paratope. Alternatively, because of the single-domain architecture with fewer CDR loops, amino acid diversity at each position of VHH may be more extensively sampled in the camelid immune system compared with that in the conventional antibodies where many more positions are diversified. Better sampling may lead to the selection of better-fitted CDR residues.
By extensively sampling CDR sequencing, we were able to enhance the affinity of the VHH to 150 pM. The affinity enhancement was not achieved by simply expanding the interface area. Rather, the enhancement seems to be the results of the formation of cooperative network among the interface residues that collectively improve various types of biophysical factors including packing, hydrophobic interaction and H-bonding. This view is consistent with the conclusion from comprehensive studies on the mechanism of affinity maturation for the T cell receptor Vβ domain-superantigen interface where packing, hydrophobic burial, bridging water molecules and electrostatic complementarity are all improved by mutations.22; 23
As the affinity was improved, four “cold spot” positions became hot spots (Figure 4). The hot spot residues constitute 76% of the paratope surface, and this expansion of hot spot residues completely eliminated the “O-ring” of energetically inert residues. The affinity-matured VHH retained a very high level of binding specificity, as it did not interact with E. coli proteins (Figure 2). Together, These results indicate that the O-ring architecture is not a prerequisite either for high affinity or high specificity.
Recently, Thanos et al.24 demonstrated that a small molecule, SP4206, binding to a flat surface of interleukin 2 (IL2) has a much higher “ligand efficiency” (the binding free energy per contact atom)25 than that of IL2 receptor that binds to the same surface. Their study thus has set a benchmark for two distinct types of protein-ligand interactions. Interestingly, we found that the ligand efficiency of wild-type cAb-RN05 was identical to that of SP4206 (Table 1), indicating that the VHH is a highly efficient ligand. The affinity-matured VHH essentially maintained the ligand efficiency, and a large increase in contacting atoms resulted in much higher affinity (Table 1). Furthermore, we found that some of other VHHs have even higher ligand efficiency values (Table 1). Thus, these results clearly demonstrate that a flat interface built from a single-domain β-sandwich scaffold can be as efficient as drug-like small molecules.
Implications to interface engineering
The characteristics of the distribution of binding energies in the wild-type and affinity matured interfaces provide important insights into factors governing protein-protein interaction interfaces in general and a guideline for engineering a synthetic interface.
The large effects of replacing two “scaffolding” residues in CDR3 (L99 and Y100d) illustrate the importance of the CDR conformation in generating a high-affinity interface. They form hydrophobic interactions with the VHH scaffold, which are critical in defining the fold of CDR3. It is likely that these scaffolding residues are also important for the high specificity of the wild-type VHH. In the wild-type paratope, only three side chains are involved in critical contacts. Consequently, interactions mediated by the VHH backbone atoms must make large contribution to high-affinity interaction with the antigen and at the same time to prevent binding to unrelated molecules. Our results strongly suggest that L99 and Y100d play the central role in defining the conformation of CDR backbone that is suitable for binding to the RNaseA epitope, but incompatible with other surfaces. This notion in turn suggests that the positions and directions of peptide backbone functional groups can create a large ensemble of high-affinity and -specificity interfaces. This view is consistent with recent examples of “minimalist chemical diversity” interfaces that have demonstrated the importance of conformational diversity over chemical diversity in generating protein-protein interaction interfaces.4; 8; 26. The position equivalent to Y100d near the C-terminus of CDR3 is usually occupied by an aromatic amino acid among VHHs, and thus, residues from this position to the C-terminus of CDR3 do not exhibit a large degree of conformational diversity. In contrast, L99 at the center of CDR3 is not conserved. Thus, CDR3 has a capacity to generate diverse conformations by simply moving a scaffolding residue along the loop and also by changing the loop length.
CDR1 and CDR3 of the VHH studied in this work contribute approximately equally to binding. In the conventional antibodies, CDR-H3 is located near the center of the paratope, it has the greatest sequence diversity among the CDRs because of the V(D)J recombination mechanism, and usually CDR-H3 residues make the largest contribution to binding. The natural VHH sequence diversity is also produced with the same recombination mechanism6 and thus, CDR3 has the largest sequence diversity. The hot spot residues of CDR1 and CDR3 of the anti-RNaseA VHH are located near the center of the paratope, as seen in other interfaces.27; 28 Thus, there is a mismatch between the interface architecture and the focus of sequence diversity. We predict that synthetic VHH libraries containing approximately equal distribution of amino acid diversity between CDR1 and CDR3 would outperform the CDR3-focused diversity of the natural repertoire. In other words, the geometry of the interface should dictate the design of synthetic libraries, rather than the idiosyncrasy associated with the heritage of a scaffold.
Our results clearly indicate that a small, flat interface consisting of only two surface loops is capable of producing flat binding interfaces with high affinity, high specificity and high spatial efficiency. The interface has relatively small number of essential positions, and thus engineering this type of interface from combinatorial libraries should not be particularly difficult. With demonstrated ability of this class of molecules to target a preformed cleft, our results strongly suggest the versatility of this class of scaffolds in generating diverse interfaces. These interfaces require the synergy of optimized backbone conformation and optimized placement of contacting side chains. Because both of these parameters are controlled by the amino acid sequences, a successful strategy should maximize the shape diversity and enrich amino acids that are poised to form molecular contacts in the loop sequences. This view is also supported by recent successes of the “restricted amino acid code” strategy for engineering protein-interaction interfaces.4
Experimental Procedures
VHH gene construction and yeast surface display
The synthetic gene for cAb-RN05 VHH was constructed by PCR using seven synthesized oligonucleotides. The PCR product was digested with BamHI and XhoI, and ligated between the BamHI and XhoI sites of the pGalAga vector that had been constructed by removing the Xpress tag from pYD1 (Invitrogen). We removed the Xpress tag because of its cross-reactivity with anti-FLAG tag antibodies (Sigma). The resulting plasmid, pGalAgaCamR, encodes a fusion protein of Aga2-VHH-V5 tag-His tag I. The GAL1 promoter regulates the expression of the fusion protein. Mutants with alanine substitution for alanine scanning were constructed using standard PCR methods.
RNaseA, monoclonal anti-V5 IgG and anti-mouse IgG-fluorescein isothiocyanate (FITC) conjugate were purchased from Sigma. Streptavidin-PE conjugate was purchased from Molecular Probes. RNaseA was biotinylated using EZ-Link NHS-LC-Biotin from Pierce.
The expression level of the VHH and the binding to RNaseA were measured by flow cytometry using a FACScan instrument (Beckton Dickinson) according to Boder et al.11 Yeast cells harboring the pGalAgaCamR plasmid or its variant were grown at 30 °C for 18 to 28 hours in media that contains 2% galactose. The cells were incubated with monoclonal anti-V5 antibody, followed by FITC-conjugated anti-mouse antibody. The binding was detected by positive signal of PE after incubating cells with biotinylated RNaseA, followed by PE-conjugated streptavidin.
Library sorting
The phage display vector for VHH, pCamR-V5p3, was constructed by replacing the gene for the fibronectin type III domain in the phage display vector, pAS38,29 with the DNA segment encoding VHH and the V5 epitope tag taken form the VHH yeast surface display vector described above. Phage particles were prepared in the presence of 0.2mM IPTG using previously described procedures 30.
Combinatorial phage display libraries were made using a high-efficiency Kunkel mutagenesis method.31 RNaseA was biotinylated with sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (EZ-Link Sulfo-NHS-SS-Biotin; Pierce Biotechnologies). Phage display libraries were sorted as follows. A library solution was incubated with 50 nM of biotinylated RNaseA for 30 minutes, and then the biotinylated RNaseA was captured with streptavidin-coated magnetic beads (Streptavidin MagneSphere Pramagnetic Particles; Promega). After washing the beads three times with TBST (150 mM NaCl, 50 mM Tris-HCl, pH7.5 and 0.5% Tween20), the phages were eluted by cleaving a linker within the biotinylation reagent with 100 mM DTT dissolved in 20 mM Tris-HCl, pH 8.0. Eluted phages were amplified and library sorting was repeated using 25 and 10 nM of RNaseA for the second and third rounds, respectively.
The enriched sequences for CDR1 and CDR3 were then combined using PCR methods, and the resulting ensemble of VHH genes was inserted into the yeast surface vector using the in vivo recombination method.11
Specificity test
The wild-type and affinity matured VHH proteins each containing an N-terminal His6 tag (80 μg) were mixed with 250 μl of E. coli lysate corresponding to a cell density of 25 OD600nm, and this mixture was purified in a 1 ml Ni-affinity cartridge (HisTrap column; Amersham). Fractions were analyzed using SDS-PAGE. As a control, human SUMO4 with an N-terminal His6 tag (a gift of Drs. I. Dementieva and S. Goldstein, the University of Chicago) was treated in the same manner.
Crystallization
RNaseA was dissolved in 20mM TBS at a concentration of 100 mg/ml. The purified VHH proteins were dialyzed against 20mM TBS, concentrated to ∼20mg/ml and mixed with RNaseA protein at a 1:2 ratio to form a complex. The complex was purified with Superdex75 gel filtration column. The wild-type complex was eluted in 20mM Na-phosphate buffer, pH 8.0, 150mM NaCl and concentrated to 20mg/ml using Amicon ultrafiltration devices with 5kDa cutoff membranes. The affinity-matured VHH complex was eluted in 10mM Tris-HCl, pH 8.0, 150mM NaCl and concentrated to 15-20mg/ml.
Crystals of VHH complexes with RNaseA were grown by vapor diffusion in hanging drops at 19°C. The crystals of wild-type complex were obtained from solution containing 0.1 M Tris-HCl, pH 8.5, 0.2 M MgCl2, 25% PEG 3350. The single crystals (0.3×0.15×0.1 mm3) formed within 1-2 days and typically grew to the final dimensions in one week. The affinity matured complex was crystallized from solution containing 0.1 M Bis-Tris, pH 5.5, 0.2 M (NH4)2SO4, 25% PEG 3350 and produced thin plate clusters. Our attempts in optimizing the crystallization conditions to obtain single crystals were unsuccessful. For x-ray data collection the crystal clusters were broken into several pieces (0.1 mm × 0.1 mm). All crystals were flash-frozen in liquid nitrogen from the mother liquid supplemented with 20% of glycerol.
X-ray diffraction data collection and structure determination
X-ray diffraction data were collected at 100 K using the Advanced Photon Source (Argonne National Laboratory). The x-ray data collection for wild type complex was performed on one single crystal at the BioCARS 14BM beam line (wavelength l=0.900 Å) using 2 degrees (5 sec exposure) and 3 degrees (1 sec exposure) oscillation width per frame in high and low resolution diffraction experiments, respectively. The x-ray data for the affinity-matured complex were obtained from two crystal pieces at GM/CA 23ID beam line using 0.97936 Å wavelength, 2-second exposure time and 0.5 degrees oscillation width. The X-Ray data were processed with HKL2000.32 The structures were solved by molecular replacement using MOLREP 33 and x-ray data high-resolution cutoff of 3 Å. The PDB entry 1BZQ 9 was used as a trial model in the rotation and translation search for the wild-type complex. To solve the affinity-matured structure we used the refined wild-type structure. Similar refinement protocols were used for both complexes. Rfree was monitored by setting aside 5% of the reflection as a test set. Rigid-body, positional refinement and stimulated annealing were performed in CNS1.1.34 The resolution of x-ray data was increased gradually and all data were included in the last cycles. The models were checked manually against a composite omit map calculated in CNS1.1. The positional and temperature factor refinement was completed with the program REFMAC5 35 incorporated in CCP4 36. The SigmaA-weighted 2Fobs-Fcalc and Fobs-Fcalc Fourier maps were calculated using CCP4. The search for solvent molecules was performed with help of ARP-WARP program.37 The Fourier maps were displayed and examined in TURBO-FRODO.38 The crystal data, data collection and refinement statistics are summarized in Table 2. A Ramachandran plot calculated with PROCHECK39 indicates that 99.5% of the non-Gly and non-Pro residues in both complexes lie in the most favored and additional allowed regions. Figures showing the electron density map and three-dimensional structures were prepared using TURBO-FRODO and RIBBONS.40 The coordinates and structure factors have been deposited in the Protein Data Bank with an entry code 2P49 and 2P4A for the wild-type and affinity-matured complexes, respectively.
Table 2.
X-Ray data collection and refinement statistics
| Wild-type complex | Affinity matured complex | |
|---|---|---|
| Data collectiona) | ||
| Space group | P 21 | C 2 2 2 1 |
| Cell dimensions | ||
| a, b, c (Å) | 40.13, 54.39, 48.04 | 70.03, 131.62, 137.16 |
| α, β, γ (°) | 90, 108.5, 90 | 90, 90, 90 |
| Resolutionb) (Å) | 50-1.38 (1.43-1.38) | 50- 1.90 (1.97-1.90) |
| Rmerge | 0.05 (0.50) | 0.10 (0.39) |
| I/σI | 56.3 (6.1) | 17.4 (5.2) |
| Completeness (%) | 99.6 (99.3) | 99.1 (93.7) |
| Redundancy | 14.1 (13.5) | 7.1 (4.9) |
| Refinement | ||
| Resolution (Å) | 20-1.38 | 20 – 1.90 |
| No. reflections | 38,187 | 48,836 |
| Rwork/ Rfree | 15.7 / 18.9 | 18.5 / 21.7 |
| No. residues | ||
| VHH | 121 | 2×121 |
| RNaseA | 124 | 2×124 |
| Ions | PO4 | 2 SO4 |
| Water | 366 | 357 |
| B-factors | ||
| VHH | 15.1 | 35.3 |
| RNaseA | 14.6 | 29.0 |
| Ions | 20.1 | 71.6 |
| Water | 27.6 | 37.2 |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.014 | 0.008 |
| Bond angles (°) | 1.74 | 1.15 |
| Ramachandran plot statistics | ||
| Most favored | 90.3% (187) | 91.5% (386) |
| Additional allowed | 9.2% (19) | 8.1% (34) |
| Generously allowed | 0.5% (1) | 0.5% (2) |
| Disallowed | 0 | 0 |
Two crystals were used to obtain the complete data set in case of the affinity matured complex. Only one crystal was used for wild-type complex.
Data in parentheses correspond to two last resolution shells.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grant R01-GM072688 and by the University of Chicago Cancer Research Center. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38 and DE-AC02-06CH11357. Portions of this work were performed at the BioCARS and GM/CA -CAT located at Sector 14BM and 23ID, respectively, of the Advanced Photon Source. Use of the BioCARS was supported by the National Institutes of Health, National Center for Research Resources, under grant number RR07707. Use of the GM/CA-CAT was funded with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Binz HK, Amstutz P, Pluckthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–68. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 2.Binz HK, Pluckthun A. Engineered proteins as specific binding reagents. Curr Opin Biotechnol. 2005;16:459–69. doi: 10.1016/j.copbio.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Hosse RJ, Rothe A, Power BE. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 2006;15:14–27. doi: 10.1110/ps.051817606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sidhu SS, Kossiakoff AA. Exploring and designing protein function with restricted diversity. Curr Opin Chem Biol. 2007 doi: 10.1016/j.cbpa.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446–8. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 6.Muyldermans S, Cambillau C, Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem Sci. 2001;26:230–5. doi: 10.1016/s0968-0004(01)01790-x. [DOI] [PubMed] [Google Scholar]
- 7.De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, Muyldermans S, Wyns L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc Natl Acad Sci U S A. 2006;103:4586–91. doi: 10.1073/pnas.0505379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S. High-affinity single-domain binding proteins with a binary-code interface. Proc Natl Acad Sci U S A. 2007;104:6632–7. doi: 10.1073/pnas.0700149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Decanniere K, Desmyter A, Lauwereys M, Ghahroudi MA, Muyldermans S, Wyns L. A single-domain antibody fragment in complex with RNase A: noncanonical loop structures and nanomolar affinity using two CDR loops. Structure Fold Des. 1999;7:361–70. doi: 10.1016/s0969-2126(99)80049-5. [DOI] [PubMed] [Google Scholar]
- 10.Dumoulin M, Last AM, Desmyter A, Decanniere K, Canet D, Larsson G, Spencer A, Archer DB, Sasse J, Muyldermans S, Wyns L, Redfield C, Matagne A, Robinson CV, Dobson CM. A camelid antibody fragment inhibits the formation of amyloid fibrils by human lysozyme. Nature. 2003;424:783–8. doi: 10.1038/nature01870. [DOI] [PubMed] [Google Scholar]
- 11.Boder ET, Wittrup KD. Yeast surface display for directed evolution of protein expression, affinity, and stability. Methods Enzymol. 2000;328:430–44. doi: 10.1016/s0076-6879(00)28410-3. [DOI] [PubMed] [Google Scholar]
- 12.Frenken LG, van der Linden RH, Hermans PW, Bos JW, Ruuls RC, de Geus B, Verrips CT. Isolation of antigen specific llama VHH antibody fragments and their high level secretion by Saccharomyces cerevisiae. J Biotechnol. 2000;78:11–21. doi: 10.1016/s0168-1656(99)00228-x. [DOI] [PubMed] [Google Scholar]
- 13.Wells JA. Systematic mutational analyses of protein-protein interfaces. Methods Enzymol. 1991;202:390–411. doi: 10.1016/0076-6879(91)02020-a. [DOI] [PubMed] [Google Scholar]
- 14.Bond CJ, Marsters JC, Sidhu SS. Contributions of CDR3 to V H H domain stability and the design of monobody scaffolds for naive antibody libraries. J Mol Biol. 2003;332:643–55. doi: 10.1016/s0022-2836(03)00967-7. [DOI] [PubMed] [Google Scholar]
- 15.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 16.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 17.Lang S, Xu J, Stuart F, Thomas RM, Vrijbloed JW, Robinson JA. Analysis of antibody A6 binding to the extracellular interferon gamma receptor alpha-chain by alanine-scanning mutagenesis and random mutagenesis with phage display. Biochemistry. 2000;39:15674–85. doi: 10.1021/bi000838z. [DOI] [PubMed] [Google Scholar]
- 18.Pons J, Rajpal A, Kirsch JF. Energetic analysis of an antigen/antibody interface: alanine scanning mutagenesis and double mutant cycles on the HyHEL-10/lysozyme interaction. Protein Sci. 1999;8:958–68. doi: 10.1110/ps.8.5.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vajdos FF, Adams CW, Breece TN, Presta LG, de Vos AM, Sidhu SS. Comprehensive functional maps of the antigen-binding site of an anti-ErbB2 antibody obtained with shotgun scanning mutagenesis. J Mol Biol. 2002;320:415–28. doi: 10.1016/S0022-2836(02)00264-4. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Urrutia M, Smith-Gill SJ, Mariuzza RA. Dissection of binding interactions in the complex between the anti-lysozyme antibody HyHEL-63 and its antigen. Biochemistry. 2003;42:11–22. doi: 10.1021/bi020589+. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–50. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 22.Cho S, Swaminathan CP, Yang J, Kerzic MC, Guan R, Kieke MC, Kranz DM, Mariuzza RA, Sundberg EJ. Structural basis of affinity maturation and intramolecular cooperativity in a protein-protein interaction. Structure. 2005;13:1775–87. doi: 10.1016/j.str.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Swaminathan CP, Huang Y, Guan R, Cho S, Kieke MC, Kranz DM, Mariuzza RA, Sundberg EJ. Dissecting cooperative and additive binding energetics in the affinity maturation pathway of a protein-protein interface. J Biol Chem. 2003;278:50412–21. doi: 10.1074/jbc.M306848200. [DOI] [PubMed] [Google Scholar]
- 24.Thanos CD, DeLano WL, Wells JA. Hot-spot mimicry of a cytokine receptor by a small molecule. Proc Natl Acad Sci U S A. 2006;103:15422–7. doi: 10.1073/pnas.0607058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuntz ID, Chen K, Sharp KA, Kollman PA. The maximal affinity of ligands. Proc Natl Acad Sci U S A. 1999;96:9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fellouse FA, Li B, Compaan DM, Peden AA, Hymowitz SG, Sidhu SS. Molecular recognition by a binary code. J Mol Biol. 2005;348:1153–62. doi: 10.1016/j.jmb.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 27.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–98. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 28.DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
- 29.Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998;284:1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- 30.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods in Enzymology. 2000;328:333–63. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 31.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods Enzymol. 2000;328:333–63. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–5. [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Collaborative_Computational_Project. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 37.Zwart PH, Langer GG, Lamzin VS. Modelling bound ligands in protein crystal structures. Acta Crystallogr D Biol Crystallogr. 2004;60:2230–9. doi: 10.1107/S0907444904012995. [DOI] [PubMed] [Google Scholar]
- 38.Cambillau C, Roussel A. Turbo Frodo, version OpenGL.1. University Aix-Marseille II; Mrseille: 1997. [Google Scholar]
- 39.Laskowski RA, MacArthur MW, Moss D, Thornton JM. PROCHECK: a program to check the sterochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 40.Carson M. Ribbons. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]
- 41.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.