

Abstract
The Helicobacter pylori toxin VacA causes vacuolar degeneration in mammalian cell lines in vitro and plays a key role in peptic ulcer disease. Two alleles, m1 and m2, of the mid-region of the vacA gene have been described, and the m2 cytotoxin always has been described as inactive in the in vitro HeLa cell assay. However, the m2 allele is associated with peptic ulcer and is prevalent in populations in which peptic ulcer and gastric cancer have high incidence. In this paper, we show that, despite the absence of toxicity on HeLa cells, the m2 cytotoxin is able to induce vacuolization in primary gastric cells and in other cell lines such as RK-13. The absence of Hela cell activity is due to an inability to interact with the cell surface, suggesting a receptor-mediated interaction. This result is consistent with the observation that the m2 allele is found in a population that has a high prevalence of peptic ulcer disease and gastric cancer. VacA is the first bacterial toxin described for which the same active subunit can be delivered by different receptor binding domains.
Keywords: mosaic structure
Helicobacter pylori produces a secreted cytotoxin that induces cytoplasmic vacuolation in eukaryotic cells (1–3) and epithelial erosion when administered orally to mice (4). Biological and structural data suggest similarities to the AB family of dichain toxins, which contain an enzymatically active moiety (A) and a receptor binding and translocation moiety (B). VacA is produced as a 140-kDa precursor that is cleaved at the C-terminal domain and released into the extracellular milieu as a 95-kDa mature protein that assembles into large oligomeric structures with hexameric or heptameric radial symmetry (5–6). Each monomer can be cleaved proteolytically at a specific site into two fragments of 37 kDa and 58 kDa that remain associated after cleavage, suggesting that they may represent two distinct cytotoxin subunits (7–8).
It has been shown recently that the cytotoxin is able to bind to, and to be internalized by, the target cell (9–10), and a potential receptor has been identified as a membrane-associated protein of 140 kDa (11). Intracellular expression of a transfected vacA gene results in cell vacuolation, indicating activity of the toxin in the cytoplasm (12). The toxicity affects fluid phase endocytosis causing osmotic unbalance and the accumulation of a postendosomal compartment (13–14). Moreover, VacA interferes with antigen presentation by B cells by impairing processing and maturation of antigens by the antigen-presenting cell (15–16).
Only ≈50% of clinical isolates of H. pylori produce detectable cytotoxic activity in a HeLa cell vacuolation assay. However, most isolates (>80%) have a functionally expressed vacA gene. Toxicity has been associated with mosaicism in vacA genes in toxic and nontoxic isolates. Three different signal–peptide sequences (s1a, s1b, and s2) and two variants of the mid-region (m1 and m2) have been described (17–18). Isolates with the s1–m1 forms are toxic, whereas the s2–m2 forms are essentially nontoxic. The m region spans ≈300 amino acids at the carboxyl terminus of the P58 subunit. m1 and m2 forms of the toxin have only 55% amino acid identity in this region. The s1–m1 vacA alleles encode products with relatively high levels of toxicity for HeLa cells compared with the products of a s1–m2 alleles, which are essentially nontoxic in this assay. In one study, infection with type m1 alleles was associated with more epithelial damage than type m2 allele (19). However, Go et al. (20) have shown that s1–m2 is associated with duodenal ulcer. In support of this, the m2 allele is prevalent in the Chinese population, where there is a high incidence of peptic ulcer and gastric cancer.
In this paper, we analyzed an s1–m2 form of the cytotoxin that is secreted and assembled correctly into the oligomeric structure but that completely lacks cytotoxicity in vitro in the HeLa cell assay. We show that the m2 toxin is toxic when added to other cell types such as primary cultured human gastric cells or a different epithelial cell line such as RK-13. In addition, we show that the toxin is active on HeLa cells when expressed intracellularly and that the absence of toxicity on this cell line is likely to be due to the failure to bind HeLa cells. Taken together, these data demonstrate that the m2 form of VacA is fully toxic, which is consistent with epidemiological data. The vacuolating cytotoxin is a bacterial toxin showing two different receptor binding sites.
MATERIALS AND METHODS
Bacterial Strains and Toxin Production.
H. pylori CCUG 17874 and SPM 326 were used as the source of m1 VacA whereas H. pylori strain 95–54 was used as the source of the m2 VacA. Colonies of H. pylori grown on blood agar plates (Brucella agar with 5% horse blood) were inoculated into Brucella broth containing 0.2% β-cyclodextrin and were cultured for 3–4 days at 150 rpm in a controlled micro-aerophilic atmosphere of 8% O2 and 10% CO2. Fermentor facilities were used for volumes larger than 2 liters.
Protein Purification.
VacA from H. pylori strain CCUG 17874 and strain 95–54 were purified from the broth culture supernatants as described (21). Purified toxin was stored in PBS at 4°C.
Deep Etch Electron Microscopy.
VacA molecules were prepared for microscopy by a procedure of absorption to mica followed by quick-freeze deep etching (22). The samples were processed according to Lupetti et al. (5).
Cell Vacuolation Assay.
Cytotoxic activity was tested on HeLa and RK13 cells as described (23). Water extracts from H. pylori strains were prepared from cells harvested in water, vortexed for 90 s at room temperature, centrifuged for 3 min at 13,000 rpm in a microfuge, and low pH activated (24). The suspensions or purified VacA were incubated with Hela cells or RK13 cells for at least 6 h. The extent of vacuolation was determined quantitatively by measuring the uptake of neutral red (23).
PCR Amplification DNA Analysis.
Chromosomal DNA from H. pylori strain 95–54 was prepared as described (25). Three pairs of oligonucleotides, dEX1 (5′-AAAATCGCTTTGATGGACACCCCA-3′), rIGT (5′-GCCACAAATCCAGTGTGCCAATAT-3′), dVWM (5′-GTGTGGATGGGCCGTCTTGCAATAC-3′), rKYL (5′-AAGATACTTGTAATTGTTCAGGGTT-3′), dALY (5′-GCCCTTTATAACAACAATCAACCGC-3′), and rEX2 (5′-GGGTTAAAGTTTTGTAAGATTT -3′) were used by PCR to amplify three overlapping fragments, each ≈1,500 bp. The oligonucleotides were used at a concentration of 6 mM with 100 ng of chromosomal DNA in a PCR using Taq polymerase (Boehringer Mannheim). The reaction was cycled through 1 min at 94°C, 1 min at 50°C, and 1 min at 72°C 25 times. The products were purified by QIAquick (Qiagen) and directly sequenced by using a primer walking methodology. Sequences were determined by using the Taq Dye Deoxy Terminator Cycle Sequencing Kit (Applied Biosystems) on a Gene Amp PCR system 9600 (Perkin—Elmer) and a run on a DNA Analysis System-Model 373 (Applied Biosystems). The sequence was assembled and processed using Gene Navigator (Applied Biosystems).
Nucleotide Sequence Accession Number.
The nucleotide sequence of vacA from the H. pylori 95–54 strain has been deposited in the Genome Sequence Database (GSDB) under accession number U95971.
Expression of VacA 95–54 in HeLa Cells.
The gene encoding the 95–54 toxin was cloned into a expression vector according to de Bernard et al. (12). In brief, the sequence coding for amino acids 34–885 was amplified by PCR and cloned into plasmid pGEM7Zf(+) (Promega). PCR was carried out using the specific synthetic oligonucleotides 5′-AAATAGTCTAGAATGGCCTTTTTCACAACCGTGATCATT-3′ and 5′-AAACCGGAATTCTTAGAAACGCGCATTGTTCGTGGTGTT-3′, which were engineered to contain the start codon (5′-ATG underlined), the stop codon (5′-TTA underlined), and the recognition sequence for XbaI and EcoRI (bold), respectively. The PCR product was purified from agarose gel, digested by XbaI and EcoRI, and subsequently cloned into the corresponding site of pGEM7Zf(+). This resulting plasmid (pGEMp88J) was used to transfect HeLa cells as described (12).
Binding Assay.
Binding assays to Hela cells and RK13 cells were performed as described (10) with minor modifications. In brief, cells were incubated with increasing concentrations of VacA for 1 h at 4°C, washed thoroughly, and incubated with saturating concentration of rabbit immune polyclonal IgG (15 μg/ml) raised against either the m1 or m2 form of the toxin. Binding was revealed by flow cytometer using fluorescein isothiocyanate-labeled goat anti-rabbit antibodies.
Immunofluorescence Microscopy.
HeLa cells were transfected with the plasmid pGEMp88J and processed as described (12). Anti-native VacA was diluted 1:500 in the permeabilization buffer and incubated with cells for 1 h. After several washes, fluoresceinated anti-rabbit Ig were diluted 1:2,000 in the same buffer and added for 30 min and washed. Samples were mounted on 90% (vol/vol) glycerol, 0.2% (wt/vol) N-propylgallate in PBS, and observed with a fluorescence microscope (Zeiss Axioplan).
Source of Anti-Cytotoxin Sera.
VacA from strain 95–54 was purified as described (6). A New Zealand White rabbit was multiply immunized with the purified VacA by using glyceryl monooleate and n-hexadecane as adjuvants (9). Rabbit serum against CCUG 17874 VacA has been described (21).
Primary Culture of Humans Gastric Cells.
Gastric biopsies taken during routine endoscopy were incubated in Hank’s medium containing 50 units/ml collagenase (Sigma) at 37°C for 4 h, dissociated with vigorous pipetting, washed, and plated in DMEM and 10% fetal calf serum. After 4 days, nonadherent cells and debris were removed, and VacA was added at a concentration of 40 μg/ml. Vacuolation was visible after 6 h of incubation at 37°C.
RESULTS
H. pylori Strain 95–54 Produces a s1–m2 VacA Protein that Is Nontoxic in the HeLa Cell Assay.
Strain 95–54, a clinical isolate of H. pylori from a patient with peptic ulcer disease, was assessed for cytotoxic activity by using the Hela cell assay. For this purpose, water extracts were prepared from toxigenic H. pylori SPM 326 and from H. pylori 95–54 and were added to HeLa cells. These two strains were shown to produce equivalent quantities of cytotoxin as judged by immunoblot (Fig. 1). Extract from the toxigenic strain induced extensive vacuolation of HeLa cells after 6 h of incubation, whereas incubation with H. pylori 95–54 extract did not induce any vacuoles even after prolongated incubation. The extent of vacuolation was quantified by using the neutral red uptake assay. As seen in Fig. 1, water extracts from the toxigenic strain induced neutral red uptake by HeLa cells whereas the water extract from the 95–54 strain did not induce any significant increase in neutral red uptake. The level of neutral red uptake in cells treated with water extract from the 95–54 strain was comparable to that induced by an extract of the toxigenic strain in which the vacA gene had been interrupted by insertion of a kanamycin resistance gene, thus demonstrating that this strain was devoid of cytotoxicity for HeLa cells.
Figure 1.

Vacuolation of HeLa cells in response to water extracts of H. pylori. SPM 326 (open-square), H. pylori SPM 326vacA∷Km (open-circle), or H. pylori 95–54 (closed-square) as determined by the neutral red uptake assay. The insert shows an immunoblot of VacA in the water extracts of H. pylori SPM 326 (lane a) or 95–54 (lane b).
Sequencing of the gene from strain 95–54 revealed the presence of a 3,969-bp ORF encoding a product of 1,323 amino acids. Analysis of the predicted amino acid sequence show that this protein consists of a combination of the s1 signal-peptide and the m2 mid-region (18). Hence, the lack of toxicity was not due to a defective signal–peptide sequence. The 95–54 VacA protein sequence showed 83% amino acid identity with the well characterized VacA sequence from toxigenic strain CCUG 17874. As seen in Fig. 2, the signal sequence, the P37 subunit, the loop, and the putative outer membrane exporter are >90% identical with the VacA sequence from the toxigenic strain, whereas the m region located in the the P58 subunit is only 53% identical to the toxigenic strain. Thus, the main region of difference between the toxigenic form and the nontoxic form is in the middle region (m region).
Figure 2.
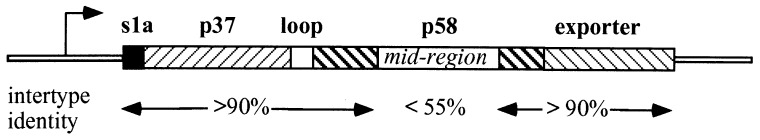
Schematic representation of the vacA gene of H. pylori 95–54. The amino acid identity between the deduced amino acid sequence of each region of the protein with that of a toxigenic strain is indicated.
The 95–54 Cytotoxin Is Produced, Secreted, and Assembled into a Multimeric Structure.
To exclude that the absence of activity of the water extract was due to a defect in the secretion or the processing of the protein, we analyzed the localization of the 95–54 cytotoxin in the H. pylori culture. Immunoblot of proteins in cell extract and culture supernatant revealed that strain 95–54 produced a 95-kDa polypeptide predominantly in the supernatant (Fig. 3A). The efficiency of secretion was similar to the secretion of m1-producing strains (data not shown). Probing the same material with antibodies raised against cytoplasmic heat-shock protein confirmed that the VacA was not released by cell lysis (Fig. 3B). This experiment shows that the 95–54 cytotoxin is capable of being expressed, secreted, and processed correctly. To assess structural integrity, VacA from strain 95–54 was purified from a culture supernatant and analyzed by quick-freeze deep etch electron microscopy, and as seen in Fig. 3C, the inactive 95–54 protein is able to form regular hexa- and heptameric structures, as already shown by Cover and coworkers (6). These data demonstrate that the lack of toxicity toward HeLa cells cannot be explained by defects in production, secretion, processing, or assembly of the cytotoxin.
Figure 3.

(A) Immunoblot of denaturing SDS/PAGE analysis of the localization of VacA and HspB. (B) S., supernatant, P., bacterial pellet. (C) Electron micrographs of purified 95–54 VacA. (Bar = 30 nm.)
The 95–54 Cytotoxin Is Active Intracellularly in Hela Cells.
de Bernard et al. (12) have shown that intracellular expression of a transfected m1 form of the vacA gene results in massive vacuolation of HeLa cells. To establish whether the absence of toxicity of 95–54 VacA is due to a defect in the active or delivery subunits, we transfected HeLa cells with the gene coding for 95–54 VacA. As seen in Fig. 4, transfection of the vacA gene from strain 95–54 also resulted in massive vacuolation. Vacuolized cells were colocalized with cells expressing the cytotoxin, as assessed by immunofluorescence microscopy, demonstrating that the vacuolation observed was due to the intracellular production of the cytotoxin. A neutral red uptake assay was performed to quantitate the extent of vacuolation, and as shown on Fig. 4C, the cytotoxic activity of the m2 vacA gene expressed by plasmid pGENp88J was similar to that of the m1 vacA gene expressed by plasmid pGEMp95. Hence, the m2 vacA gene from strain 95–54 encodes a potentially active cytotoxin. These data suggest that the lack of activity is due to inability to reach its cytoplasmic site of activity because of a defect of toxin binding or internalization.
Figure 4.

m2 vacA expression in HeLa cells transfected by pGEMp88J. (A) Phase–contrast microscopy. (B) Immunofluorescence microscopy with anti-VacA serum. (C) Neutral red uptake of HeLa cell transfected with pGEMp95 (CCUG 17874 VacA) or with pGEMp88J (95–54 VacA).
RK-13 Cells and Primary Cultured Human Gastric Cells Are Sensitive to m2-Type Cytotoxin.
Kamiya et al. (26) have shown that 21% of H. pylori strains were able to vacuolate HeLa cells whereas 73% of the strains were toxic for RK-13 cells, a rabbit kidney epithelial cell line, suggesting that this cell line might be sensitive to both m1 and m2 forms of the cytotoxin (26). As seen in Fig. 5, both the m1 and m2 forms of the cytotoxin induced neutral red uptake in RK-13 cells. Maximum neutral red uptake was similar with both toxins, but the dose–response curves revealed that the m2 form was ≈4-fold less active than the m1 form in these cells. In contrast, the m2 form induced no detectable neutral red uptake in HeLa cells even at the highest concentration used. The defect in activity was however not restricted to HeLa cells; no vacuolating activity was found in the human embryonic kidney cell line HEK 293 (data not shown). The 95–54 VacA did however induce vacuolation of adherent human cells cultured from human gastric biopsies (Fig. 5C).
Figure 5.

(A) Vacuolating activity of purified 95–54 VacA or purified CCUG 17874 cytotoxin on RK-13 cells or on HeLa cells (B) as determined by the neutral red uptake assay. (C) Phase–contrast microscopy of primary cultured human gastric cells vacuolated by 40 μg/ml 95–54 cytotoxin.
The Lack of Toxicity Toward HeLa Cells Is due to a Defect in Cell Interaction.
As demonstrated above, the m2 vacA gene encodes a potentially functional toxin, but the purified cytotoxin was only toxic toward RK-13 cells. VacA, as other AB type toxins, binds to and is internalized by the cell before exerting its toxic activity (9–10). We have assayed the capacity of VacA from 95–54 to bind to target cells by indirect immunofluorescence and flow cytometry (Fig. 6). Whereas dose-dependent binding to RK-13 cells was observed, no binding to HeLa cells could be detected. m1 VacA from H. pylori CCUG 17874 bound to both HeLa and RK-13 cells. The fluorescence shift observed with 95–54 VacA on RK-13 cells was significantly less than that observed with VacA from H. pylori CCUG 17874. However, because antiserum to m1 VacA recognizes m2 VacA poorly and vice versa, different sera against each specific form were used. It is thus not possible to compare directly the levels of binding.
Figure 6.

Binding of CCUG 17874 (A) and 95–54 VacA (B) to HeLa and RK-13 cells. Cells were incubated at 0°C with increasing concentrations of cytotoxin, and the binding was revealed by flow cytometry by using m1- or m2-specific antiserum and fluorescein isothiocyanate-labeled anti-rabbit Ig. CCUG 17874 VacA (open-square), 95–54 VacA (closed-square). MFI, mean fluorescence intensity.
DISCUSSION
It has been reported that only ≈50% of H. pylori isolates produce vacuolating cytotoxin as assessed by the HeLa cell assay. Full activity has been associated in part with the presence of the s1 signal peptide, but combinations of the s1 signal peptide and the m2 mid-region have been reported to have little or no toxic activity for HeLa cells. We have demonstrated, by comparing crude extracts of strain 95–54 with a vacA knockout strain and by using highly purified proteins that an s1/m2 strain is completely devoid of vacuolating activity in HeLa cells. Nevertheless, this protein is active if expressed intracellularly in HeLa cells or extracellularly in other cells line such as RK-13. Furthermore, this toxin is active in primary cultured human gastric cells, suggesting that m2 forms of the toxin are toxic in vivo.
We have shown that the absence of activity of the m2 cytotoxin in HeLa cells is due to a lack of interaction with the cell, indicating that the two forms of the toxin differ in their binding domains. The major difference between the two proteins is in the mid-region, where there is only ≈55% amino acid identity. Hence, this region is likely to be responsible for binding although the involvement of sequences outside the region cannot be excluded. The fact that the m2 cytotoxin interacts with RK-13 cells but not with HeLa cells is not compatible with an interaction through nonspecific membrane insertion because biological membranes are highly similar between different cell lines. The differential binding can be explained best by the presence of a specific receptor on the surface of the target cell, as already suggested by Yahiro et al. (11). It is possible that there is a single receptor with a different affinity for each toxin type. However, the m2 cytotoxin was only 4- to 5-fold less active than the m1 form on RK-13 cells whereas there was at least a 15-fold difference in activity on HeLa cells. Furthermore, no binding of the m2 form to HeLa cells could be detected, even at the highest concentration of toxin used. This suggests the existence of a specific receptor for each toxin type. In this case, both RK-13 and HeLa cells would express the m1 receptor whereas the m2 receptor would be lacking in HeLa cells. There is no formal evidence for two different receptors, but the difference in binding between m1 and m2 toxins indicates that the binding domain of the toxin is different.
Recently, we have found that 75% of a group of patients in Shangai (China) were infected with strains expressing the m2 form of the toxin and that there was no significant difference in severity of peptic ulcer disease between patients infected with m1- or m2 best -expressing strains (unpublished data). The discovery that the m2 form is likely to be toxic in vivo may explain this apparent paradox. In contrast, 96% of strains isolated in Fukai (Japan) were found to be s1–m1 (27). These observations may reflect evolutionary bottlenecking. However, an interesting possible explanation may be polymorphism in expression of the VacA receptor between populations.
A mosaic organization also has been observed for the cagA gene encoding a immunodominant surface-exposed protein of H. pylori (28), and DNA sequencing may reveal mosaicism in other H. pylori genes. The phenomenon of mosaic structure also has been described for the IgA proteases of Neisseria gonorrhoeae and Haemophilus influenzae and penicillin binding proteins from Neisseria meningitidis and Streptococcus pneumoniae (29–33). It has been proposed that DNA uptake and recombination may have given rise to this allelic variation (17–18).
Immunization with VacA has been shown to confer protection to mice against subsequent experimental colonization by H. pylori. Our observations that anti-sera raised against one mid-region form poorly recognizes the other form and that both forms are likely to be toxic in vivo should be taken into consideration when designing potential vaccines or for diagnostics assays based on this antigen. For example, a vaccination based on one type would not necessarily protect against infection by the other type.
In conclusion, it is likely that the HeLa cell assay does not fully reflect the potential pathogenicity of clinical isolates of H. pylori, and previous conclusions based on these assays will require reconsideration. The results of this study indicate that the m2 form of VacA plays a more important role in the pathogenesis of H. pylori infection than has been recognized.
Acknowledgments
We thank A. Muzzi for synthesis of the oligonucleotides and S. Guidotti for automated sequencing of the vacA locus. We acknowledge C. Ulivieri for her availability and help with the flow cytometer. We are very grateful to M. Kantaku for her expertise in fermentation. We thank V. Pelicic for very helpful discussions. G. Corsi is gratefully acknowledged for artwork. We thank S. Pasquini, L. Fini, and S. Ciabattini for bacterial medium preparation. We gratefully acknowledge N. Norais for the generous gift of type m1 VacA and Remo Vermillo and N. Figura for gastric biopsies. This work was carried out in partial fulfillment of the doctorate of C.P. on molecular and cellular biology and pathology (University of Padova). J.M.R. is a recipient of a grant from the European Union (BIO4-CT-96-5027). This work was supported by EU Grants IC18CT95-0024, BMH4-97-2410, and TMRFMRXCT 96-0004 and National Institutes of Health Grants AI-39657 and DK-53623.
References
- 1.Cover T L, Blaser M J. J Biol Chem. 1992;267:10570–10575. [PubMed] [Google Scholar]
- 2.Telford J L, Ghiara P, Dell’Orco M, Comanducci M, Burroni D, Bugnoli M, Tecce M F, Censini S, Covacci A, Xiang Z, et al. J Exp Med. 1994;179:1653–1658. doi: 10.1084/jem.179.5.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cover T L. Mol Microbiol. 1996;20:241–246. doi: 10.1111/j.1365-2958.1996.tb02612.x. [DOI] [PubMed] [Google Scholar]
- 4.Marchetti M, Aricò B, Burroni D, Figura N, Rappuoli R, Ghiara P. Science. 1995;267:1655–1658. doi: 10.1126/science.7886456. [DOI] [PubMed] [Google Scholar]
- 5.Lupetti P, Heuser J E, Manetti R, Massari P, Lanzavecchia S, Bellon P L, Dallai R, Rappuoli R, Telford J L. J Cell Biol. 1996;133:801–806. doi: 10.1083/jcb.133.4.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cover T L, Hanson P I, Heuser J E. J Cell Biol. 1997;138:759–769. doi: 10.1083/jcb.138.4.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Telford J L, Covacci A, Ghiara P, Montecucco C, Rappuoli R. Trends Biotechnol. 1994;12:420–426. doi: 10.1016/0167-7799(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 8.Lanzavecchia S, Lupetti P, Bellon P L, Dallai R, Rappuoli R, Telford J L. J Struct Biol. 1998;121:9–18. doi: 10.1006/jsbi.1997.3941. [DOI] [PubMed] [Google Scholar]
- 9.Garner J A, Cover T L. Infect Immun. 1996;64:4197–4203. doi: 10.1128/iai.64.10.4197-4203.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massari, P., Manetti, R., Burroni, D., Nuti, S., Norais, N., Rappuoli, R. & Telford, J. L. (1998) Infect. Immun., in press. [DOI] [PMC free article] [PubMed]
- 11.Yahiro K, Niidome T, Hatakeyama T, Aoyagi H, Kurazono H, Padilla P A, Wada A, Hirayama T. BBRC. 1997;238:629–632. doi: 10.1006/bbrc.1997.7345. [DOI] [PubMed] [Google Scholar]
- 12.de Bernard M, Arico B, Papini E, Rizzuto R, Grandi G, Rappuoli R, Montecucco C. Mol Microbiol. 1997;4:665–674. doi: 10.1046/j.1365-2958.1997.5881952.x. [DOI] [PubMed] [Google Scholar]
- 13.Papini E, de Bernard M, Milia E, Bugnoli M, Zerial M, Rappuoli R, Montecucco C. Proc Natl Acad Sci USA. 1994;91:9720–9724. doi: 10.1073/pnas.91.21.9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molinari M, Galli C, Norais N, Telford J L, Rappuoli R, Luzio J P, Montecucco C. J Biol Chem. 1997;272:25339–25344. doi: 10.1074/jbc.272.40.25339. [DOI] [PubMed] [Google Scholar]
- 15.Satin B, Norais N, Telford J, Rappuoli R, Murgia M, Montecucco C, Papini E. J Biol Chem. 1997;272:25022–25028. doi: 10.1074/jbc.272.40.25022. [DOI] [PubMed] [Google Scholar]
- 16.Molinari M, Salio M, Galli C, Norais N, Rappuoli R, Lanzavecchia A, Montecucco C. J Exp Med. 1998;187:135–140. doi: 10.1084/jem.187.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cover T L, Tummuru M, Cao P, Thompson S, Blaser M J. J Biol Chem. 1994;269:10566–10573. [PubMed] [Google Scholar]
- 18.Atherton J C, Cao P, Peek R M, Tummuru M, Blaser J, Cover T L. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 19.Atherton J C, Peek R M, Tham K T, Cover T L, Blaser M J. Gastroenterology. 1997;112:92–99. doi: 10.1016/s0016-5085(97)70223-3. [DOI] [PubMed] [Google Scholar]
- 20.Go M F, Cissell L, Grayham D Y. Scand J Gastroenterol. 1998;33:132–136. doi: 10.1080/00365529850166842. [DOI] [PubMed] [Google Scholar]
- 21.Manetti R, Massari P, Burroni D, de Bernard M, Marchini A, Olivieri O, Papini E, Montecucco C, Rappuoli R, Telford J L. Infect Immun. 1995;63:4476–4480. doi: 10.1128/iai.63.11.4476-4480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heuser J E. J Mol Biol. 1983;169:155–195. doi: 10.1016/s0022-2836(83)80179-x. [DOI] [PubMed] [Google Scholar]
- 23.Cover T L, Puryear W, Perez-Perez G I, Blaser M J. Infect Immun. 1991;59:1264–1270. doi: 10.1128/iai.59.4.1264-1270.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Bernard M, Papini E, de Filippis V, Gottardi E, Telford J L, Manetti R, Fontana A, Rappuoli R, Montecucco C. J Biol Chem. 1995;270:23937–23940. doi: 10.1074/jbc.270.41.23937. [DOI] [PubMed] [Google Scholar]
- 25.Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N, Rappuoli R. Proc Natl Acad Sci USA. 1993;90:5791–5795. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamiya, S., Kai, M., Ozawa, A., Kobayashi, H., Shirai, T., Harasawa, S. & Miwa, T. (1994) Eur. J. Gastroenterol. Hepatol. 6, Suppl. 1, S23–S27. [PubMed]
- 27.Ito Y, Azuma T, Ito S, Miyaji H, Hirai M, Yamazaki Y, Sato F, Kato T, Kohli Y, Kuriyama M. J Clin Microbiol. 1997;35:1710–1714. doi: 10.1128/jcm.35.7.1710-1714.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miehlke S, Kibler K, Kim J G, Figura N, Small S M, Graham D Y, Go M F. Am J Gastroenterol. 1996;91:1322–1325. [PubMed] [Google Scholar]
- 29.Smith J M, Dowson C G, Spratt B G. Nature (London) 1991;349:29–31. doi: 10.1038/349029a0. [DOI] [PubMed] [Google Scholar]
- 30.Halter R, Pohlner J, Meyer T F. EMBO J. 1989;8:2737–2744. doi: 10.1002/j.1460-2075.1989.tb08415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spratt B G, Zhang Q Y, Jones D M, Hutchison A, Branningan J A, Dowson C G. Proc Natl Acad Sci USA. 1989;86:8988–8992. doi: 10.1073/pnas.86.22.8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dowson C J, Hutchison A, Branningan J A, George R C, Hansman D, Linares J, Tomasz A, Smith J M, Spratt B G. Proc Natl Acad Sci USA. 1989;86:8842–8846. doi: 10.1073/pnas.86.22.8842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poulsen K, Reinholdt J, Kilian M. J Bacteriol. 1992;174:2913–2921. doi: 10.1128/jb.174.9.2913-2921.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]